
A New Potent Inhibitor against α-Glucosidase Based on an In Vitro Enzymatic Synthesis Approach
 ,
,
Abstract
:1. Introduction
2. Results and Discussion
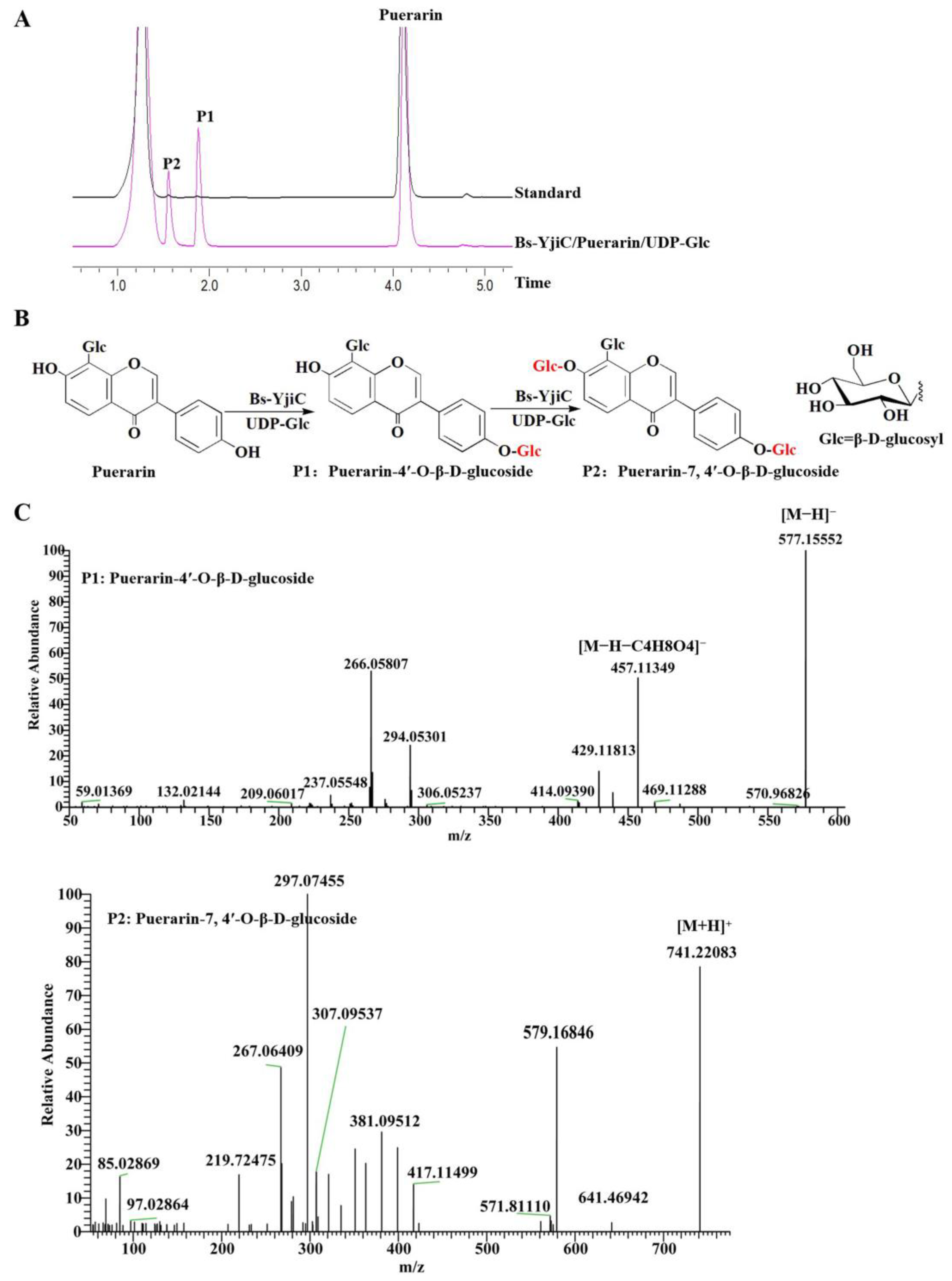
2.1. Enzymatic Synthesis of Puerarin Derivatives with Bs-YjiC
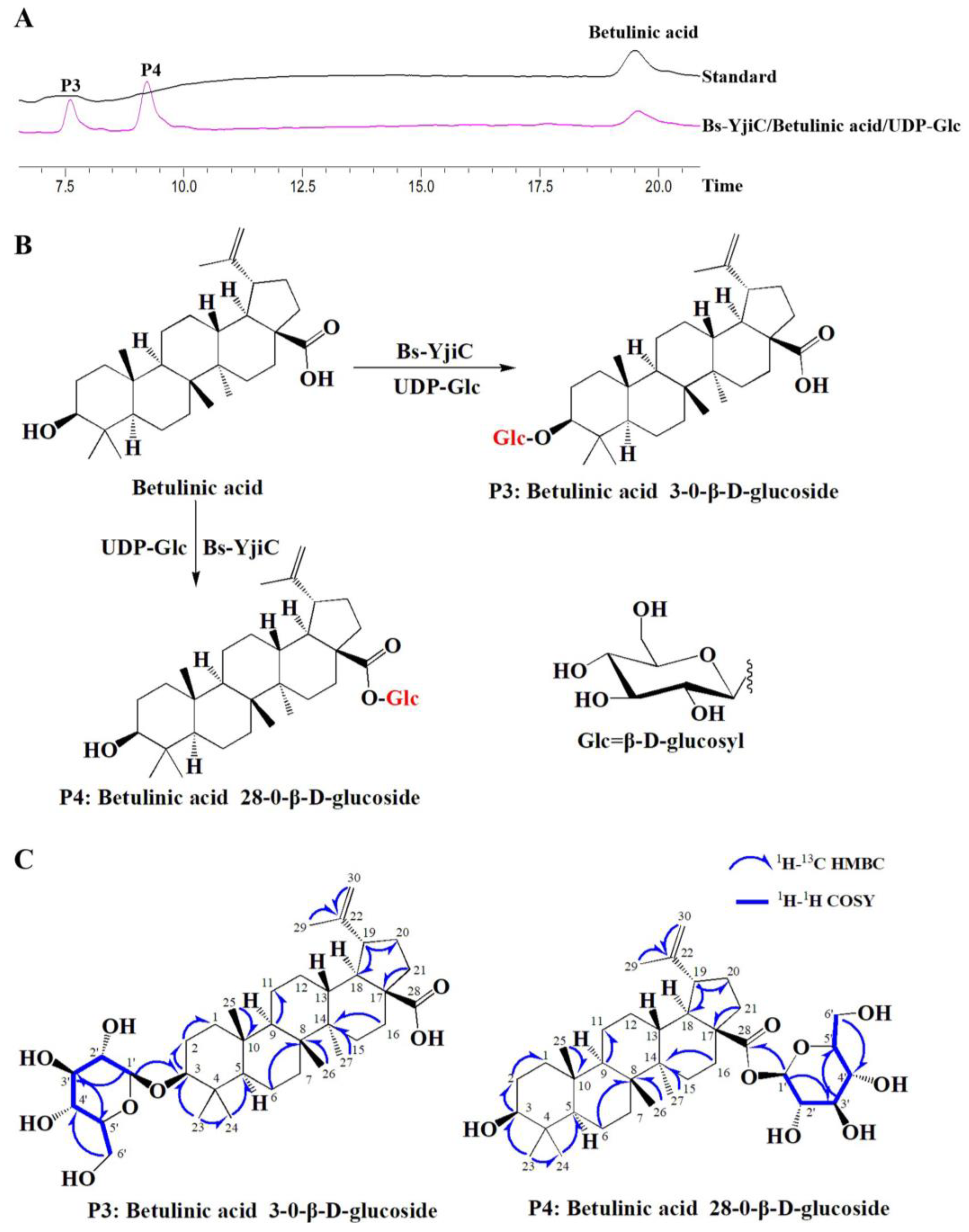
2.2. Enzymatic Synthesis of Betulinic Acid Derivatives with Bs-YjiC
2.3. Common Targets Prediction
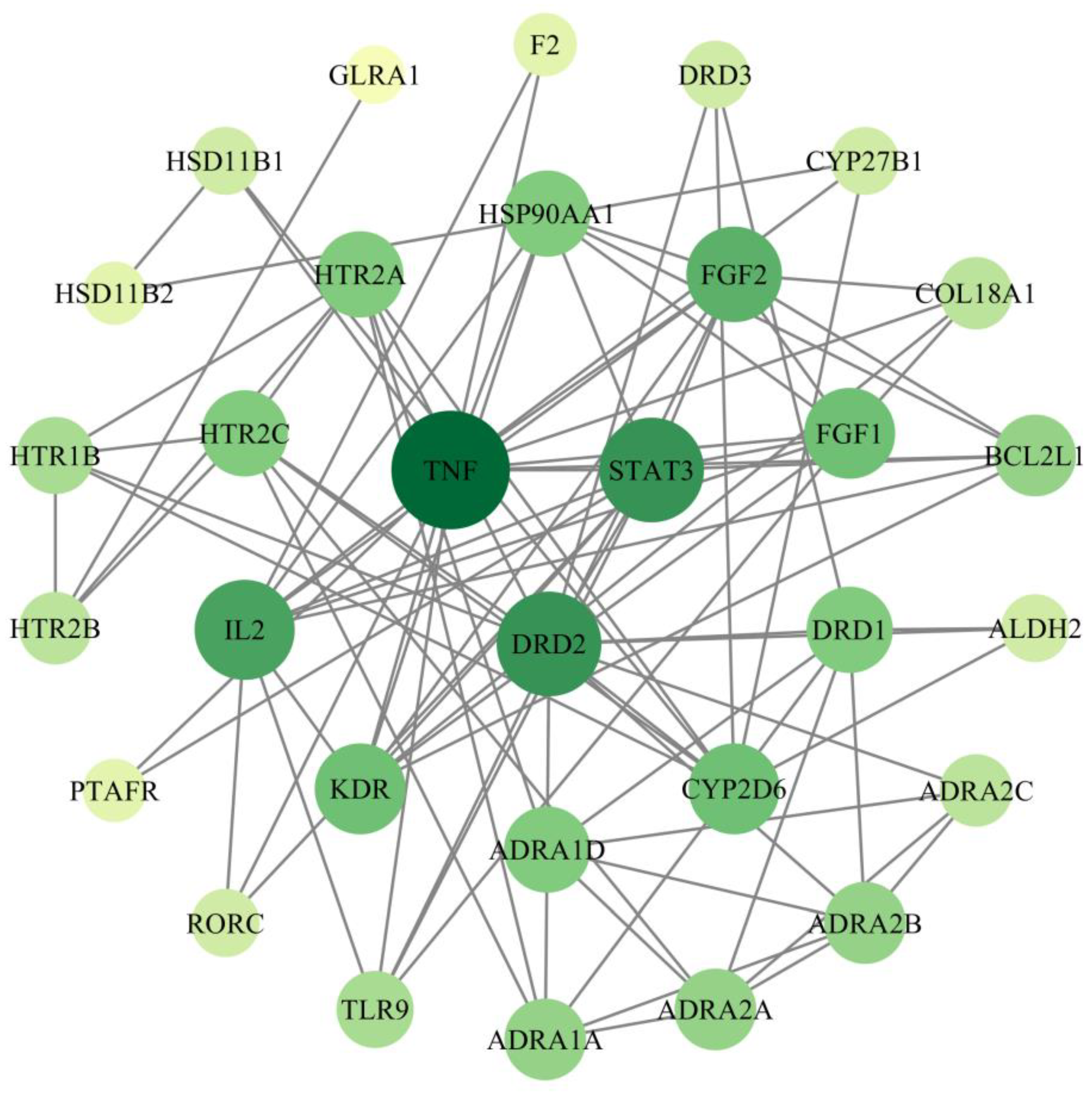
2.4. Screening of PPI Networks and Core Targets
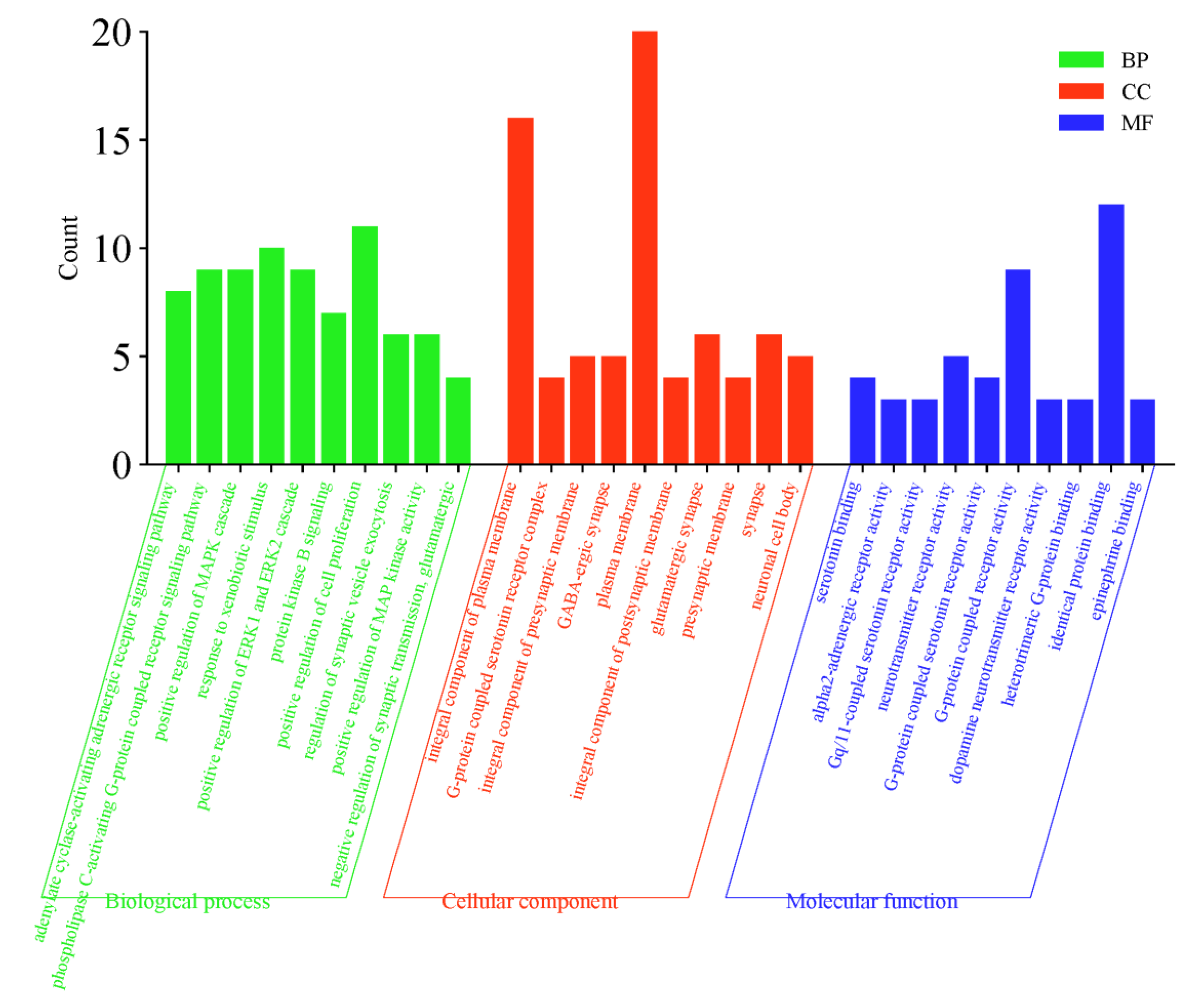
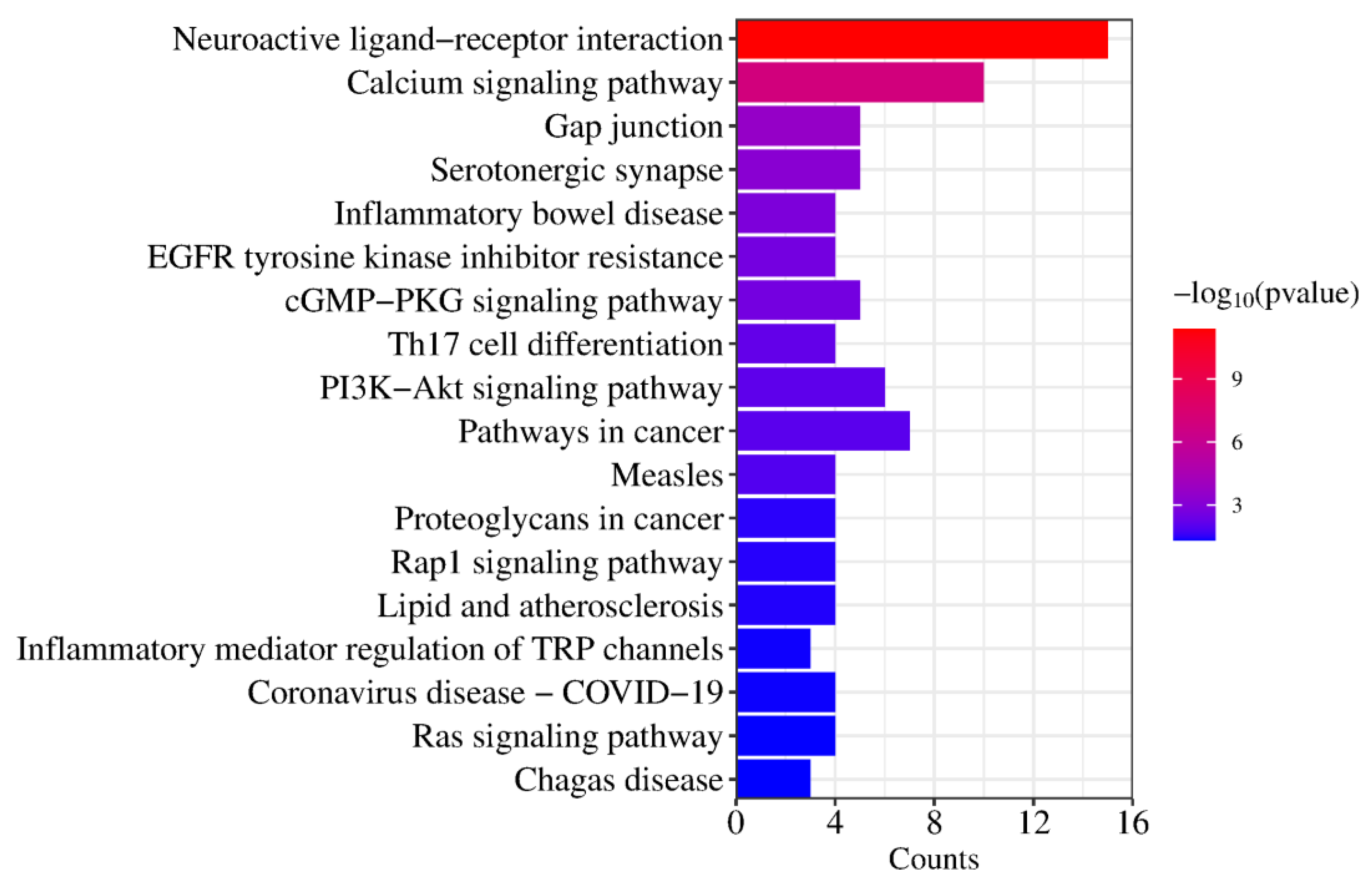
2.5. GO Function and KEGG Pathway Enrichment Analysis
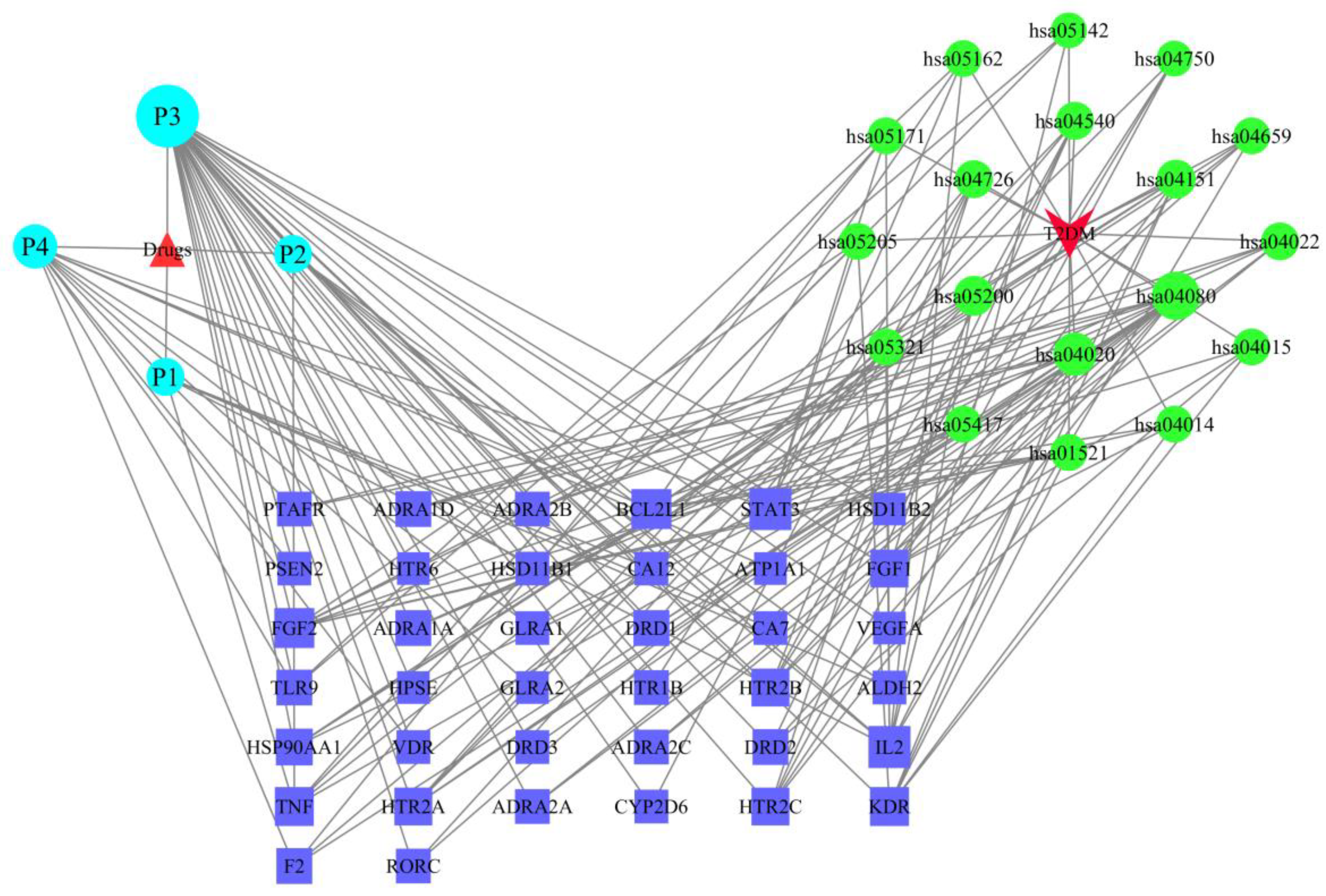
2.6. The Drug–Target–Pathway–Disease Network
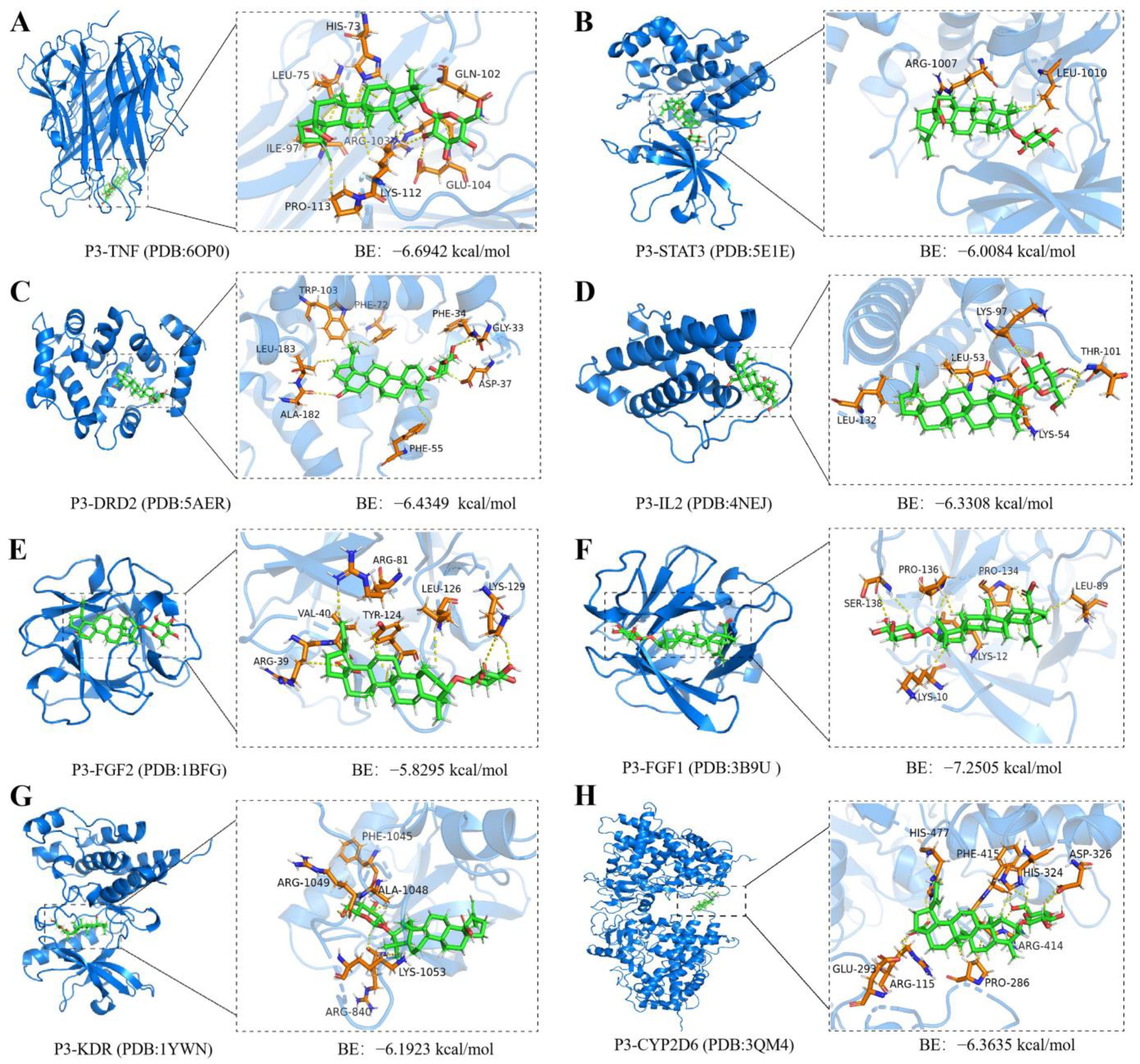
2.7. Molecular Docking and Molecular Dynamics Simulations of P3 with Core Targets
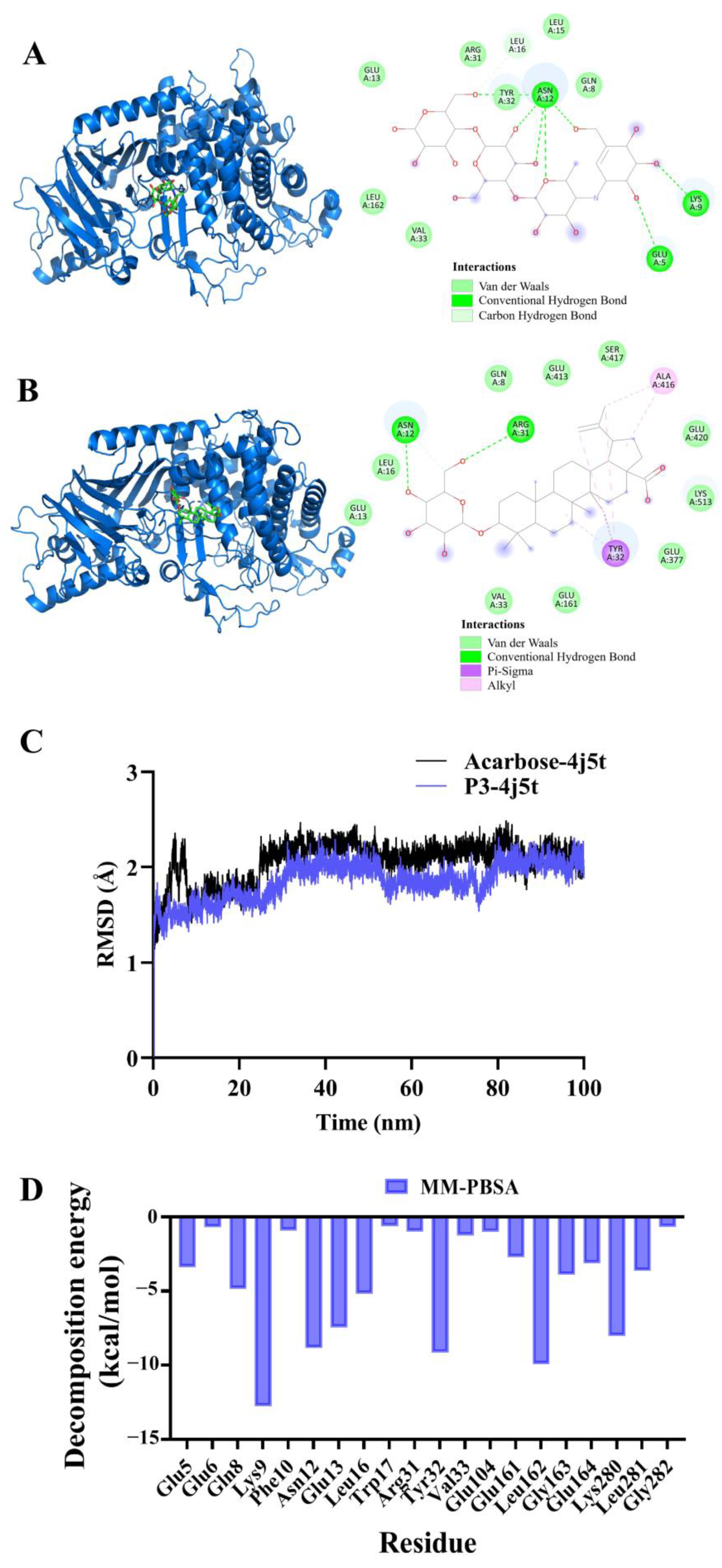
2.8. Molecular Docking and Molecular Dynamics Simulation of P3 and Acarbose with α-Glucosidase
2.9. Pharmacokinetic Prediction
2.10. Site-Directed Mutagenesis for the Directed Synthesis of Compound P3
3. Method
3.1. Materials and Reagents
3.2. Heterogonous Expression and Purification of Bs-YjiC
3.3. Enzyme Activity Assay
3.4. Structural Analysis of the Glycosylated Products
3.5. Common Targets Prediction of Four Compounds and Type II Diabetes
3.6. Construction of the Protein–Protein Interaction Network and Screening of Key Targets
3.7. GO Function and KEGG Pathway Enrichment Analysis of Key Targets
3.8. Construction of the Drug–Target–Pathway–Disease Network
3.9. Molecular Docking, Molecular Dynamic Simulations and Calculation of Binding Free Energy
3.10. Pharmacokinetic Prediction
3.11. Site-Directed Mutagenesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whiting, D.R.; Guariguata, L.; Weil, C.; Shaw, J. IDF Diabetes Atlas: Global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res. Clin. Pract. 2011, 94, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Khursheed, R.; Singh, S.K.; Wadhwa, S.; Gulati, M.; Awasthi, A. Therapeutic potential of mushrooms in diabetes mellitus: Role of polysaccharides. Int. J. Biol. Macromol. 2020, 164, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Yaghootkar, H.; Whitcher, B.; Bell, J.D.; Thomas, E.L. Ethnic differences in adiposity and diabetes risk—Insights from genetic studies. J. Intern. Med. 2020, 288, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Halim, M.; Halim, A. The effects of inflammation, aging and oxidative stress on the pathogenesis of diabetes mellitus (type 2 diabetes). Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Pennells, L.; Kaptoge, S.; Ostergaard, H.B.; Read, S.H.; Carinci, F.; Franch-Nadal, J.; Petitjean, C.; Taylor, O.; Hageman, S.H.J.; Xu, Z.; et al. SCORE2-Diabetes: 10-year cardiovascular risk estimation in type 2 diabetes in Europe. Eur. Heart J. 2023, 44, 2544–2556. [Google Scholar]
- Mu, X.D.; Wu, A.H.; Hu, H.Y.; Zhou, H.; Yang, M. Prediction of Diabetic Kidney Disease in Newly Diagnosed Type 2 Diabetes Mellitus. Diabetes Metab. Syndr. Obes. 2023, 16, 2061–2075. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-L.; Wang, L.-N.; Li, Y.-J.; Fan, Q.-F.; Huang, Q.-H.; Chen, J.-J. Anti-hyperglycaemic effect of labdane diterpenes isolated from the rhizome of Amomum maximum Roxb. an edible plant in Southwest China. Nat. Prod. Res. 2022, 36, 2570–2574. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H.; Fu, X.H.; Zhang, T.Y.; Li, H.; Chen, T.P.; Liu, X.Q. Isolation and identification of an α-glucosidase inhibitory peptide from extruded soybean protein and its hypoglycemic activity in T2DM mice. Food Funct. 2023, 14, 4288–4301. [Google Scholar] [CrossRef]
- Shah, M.A.; Khalil, R.; Ul-Haq, Z.; Panichayupakaranant, P. α-Glucosidase inhibitory effect of rhinacanthins-rich extract from Rhinacanthus nasutus leaf and synergistic effect in combination with acarbose. J. Funct. Foods 2017, 36, 325–331. [Google Scholar] [CrossRef]
- Wang, X.-L.; Jiao, F.-R.; Yu, M.; Lin, L.-B.; Xiao, J.; Zhang, Q.; Wang, L.; Duan, D.-Z.; Xie, G. Constituents with potent α-glucosidase inhibitory activity from Pueraria lobata (Willd.) ohwi. Bioorg. Med. Chem. Lett. 2017, 27, 1993–1998. [Google Scholar] [CrossRef]
- Chen, S.; Lin, B.; Gu, J.; Yong, T.; Gao, X.; Xie, Y.; Xiao, C.; Zhan, J.Y.; Wu, Q. Binding Interaction of Betulinic Acid to α-Glucosidase and Its Alleviation on Postprandial Hyperglycemia. Molecules 2022, 27, 2517. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qian, L.; Wang, B.; Zhang, Z.; Liu, H.; Zhang, Y.; Liu, J. Synergistic Hypoglycemic Effects of Pumpkin Polysaccharides and Puerarin on Type II Diabetes Mellitus Mice. Molecules 2019, 24, 955. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zhao, S.; Wei, G.; Zhao, H.; Qu, Q. Functional regulation of ginsenoside biosynthesis by RNA interferences of a UDP-glycosyltransferase gene in Panax ginseng and Panax quinquefolius. Plant Physiol. Biochem. 2017, 111, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Liu, C.; Li, J.; Dong, C.; Yang, J.; Dai, Z.; Zhang, X.; Sun, Y. One-Pot Synthesis of Ginsenoside Rh2 and Bioactive Unnatural Ginsenoside by Coupling Promiscuous Glycosyltransferase from Bacillus subtilis 168 to Sucrose Synthase. J. Agr. Food Chem. 2018, 66, 2830–2837. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Qin, L.; Hu, Y.; Huang, J.-W.; Hu, Z.; Min, J.; Sun, Y.; Guo, R.-T. Structural dissection of unnatural ginsenoside-biosynthetic UDP-glycosyltransferase Bs-YjiC from Bacillus subtilis for substrate promiscuity. Biochem. Biophys. Res. Commun. 2021, 534, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Li, J.; Yang, J.; Zhu, Y.; Men, Y.; Zeng, Y.; Cai, Y.; Dong, C.; Dai, Z.; Zhang, X.; et al. Use of a Promiscuous Glycosyltransferase from Bacillus subtilis 168 for the Enzymatic Synthesis of Novel Protopanaxatriol-Type Ginsenosides. J. Agr. Food Chem. 2018, 66, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Aijijiyah, N.P.; Wati, F.A.; Rahayu, R.; Srilistiani, A.; Mahzumi, F.; Aulia, T.; Santoso, L.; Pamela, E.; Ramadhani, E.Y.; Ilfahmi, Y.A.; et al. Synthesis, α-glucosidase inhibitory activity, and molecular docking of cinnamamides. Med. Chem. Res. 2023, 32, 723–735. [Google Scholar] [CrossRef]
- Jiang, X.; Zhang, X.H.; Li, Y.X.; Chen, K.; Lin, B.; Lu, T.Q.; Yang, M.; Chen, G.T.; Fan, B.Y.; Wang, W.L. Chemical constituents from the leaves of Cinnamomum camphora and their α-glucosidase inhibitory activities. Phytochem. Lett. 2023, 57, 101–105. [Google Scholar] [CrossRef]
- Wei, Y.; Yu, N.; Wang, Z.; Hao, Y.; Wang, Z.; Yang, Z.; Liu, J.; Wang, J. Analysis of the multi-physiological and functional mechanism of wheat alkylresorcinols based on reverse molecular docking and network pharmacology. Food Funct. 2022, 13, 9091–9107. [Google Scholar] [CrossRef]
- Luo, W.; Deng, J.; He, J.; Yin, L.; You, R.; Zhang, L.; Shen, J.; Han, Z.; Xie, F.; He, J.; et al. Integration of molecular docking, molecular dynamics and network pharmacology to explore the multi-target pharmacology of fenugreek against diabetes. J. Cell. Mol. Med. 2023, 27, 1959–1974. [Google Scholar] [CrossRef]
- Ma, W.; Zhao, L.; Ma, Y.; Li, Y.; Qin, S.; He, B. Oriented efficient biosynthesis of rare ginsenoside Rh2 from PPD by compiling UGT-Yjic mutant with sucrose synthase. Int. J. Biol. Macromol. 2020, 146, 853–859. [Google Scholar] [CrossRef]
- Liu, B.; Zhao, C.; Xiang, Q.; Zhao, N.; Luo, Y.; Bao, R. Structural and biochemical studies of the glycosyltransferase Bs-YjiC from Bacillus subtilis. Int. J. Biol. Macromol. 2021, 166, 806–817. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. B 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A.; Open Source Drug Discovery Consortium. A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KEGG Pathways | Target Count | Targets |
|---|---|---|
| hsa04080: Neuroactive ligand–receptor interaction | 15 | PTAFR, HTR2B, HTR1B, HTR2C, ADRA1D, HTR2A, F2, ADRA2C, ADRA1A, ADRA2B, ADRA2A, GLRA1, DRD1, DRD2, DRD3 |
| hsa04020: Calcium signaling pathway | 10 | PTAFR, HTR2B, KDR, HTR2C, ADRA1D, DRD1, HTR2A, FGF1, ADRA1A, FGF2 |
| hsa04540: Gap junction | 5 | HTR2B, HTR2C, DRD1, HTR2A, DRD2 |
| hsa04726: Serotonergic synapse | 5 | CYP2D6, HTR2B, HTR1B, HTR2C, HTR2A |
| hsa05321: Inflammatory bowel disease | 4 | STAT3, RORC, TNF, IL2 |
| hsa01521: EGFR tyrosine kinase inhibitor resistance | 4 | STAT3, KDR, FGF2, BCL2L1 |
| hsa04022: cGMP-PKG signaling pathway | 5 | ADRA1D, ADRA2C, ADRA1A, ADRA2B, ADRA2A |
| hsa04659: Th17 cell differentiation | 4 | HSP90AA1, STAT3, RORC, IL2 |
| hsa04151: PI3K-Akt signaling pathway | 6 | HSP90AA1, KDR, FGF1, FGF2, IL2, BCL2L1 |
| hsa05200: Pathways in cancer | 7 | HSP90AA1, STAT3, F2, FGF1, FGF2, IL2, BCL2L1 |
| hsa05162: Measles | 4 | STAT3, TLR9, IL2, BCL2L1 |
| hsa05205: Proteoglycans in cancer | 4 | STAT3, KDR, TNF, FGF2 |
| hsa04015: Rap1 signaling pathway | 4 | KDR, DRD2, FGF1, FGF2 |
| hsa05417: Lipid and atherosclerosis | 4 | HSP90AA1, STAT3, TNF, BCL2L1 |
| hsa04750: Inflammatory mediator regulation of TRP channels | 3 | HTR2B, HTR2C, HTR2A |
| hsa05171: Coronavirus disease—COVID-19 | 4 | STAT3, F2, TNF, IL2 |
| hsa04014: Ras signaling pathway | 4 | KDR, FGF1, FGF2, BCL2L1 |
| hsa05142: Chagas disease | 3 | TLR9, TNF, IL2 |
| Compound | P3 |
|---|---|
| Human Intestinal Absorption | 0.37 |
| Oral Bioavailability | 0.46 |
| Aqueous Solubility | −6.62 log mol/L |
| Lipophilicity | 2.17 log-ratio |
| Hydration Free Energy | −7.72 kcal/mol |
| Cell Effective Permeability | −6.24 cm/s |
| PAMPA Permeability | 0.33 |
| P-glycoprotein Inhibition | 0.43 |
| Blood–Brain Barrier Penetration | 0.23 |
| Plasma Protein Binding Rate | 96.92% |
| Volume of Distribution at Steady State | −7.8 L/kg |
| CYP1A2 Inhibition | 0.00124 |
| CYP2C19 Inhibition | 0.01 |
| CYP2C9 Substrate | 0.02 |
| CYP2C9 Inhibition | 0.01 |
| CYP2D6 Substrate | 0.02 |
| CYP2D6 Inhibition | 0.01 |
| CYP3A4 Substrate | 0.62 |
| CYP3A4 Inhibition | 0.01 |
| Half Life | 62.98 h |
| hERG Blocking | 0.44 |
| Clinical Toxicity | 0.09 |
| Mutagenicity | 0.1 |
| Drug Induced Liver Injury | 0.2 |
| Carcinogenicity | 0.01 |
| Acute Toxicity LD50 | 3.39 log(1/(mol/kg)) |
| Skin Reaction | 0.31 |
| Androgen Receptor (Full Length) | 0.07 |
| Androgen Receptor (Ligand Binding Domain) | 0.04 |
| Aryl Hydrocarbon Receptor | 0.0047 |
| Aromatase | 0.12 |
| Estrogen Receptor (Full Length) | 0.22 |
| Estrogen Receptor (Ligand Binding Domain) | 0.08 |
| Peroxisome Proliferator-Activated Receptor Gamma | 0.17 |
| Nuclear Factor (Erythroid-Derived 2)-Like 2/Antioxidant Responsive Element | 0.37 |
| ATPase Family AAA Domain-Containing Protein 5 (ATAD5) | 0.03 |
| Heat Shock Factor Response Element | 0.05 |
| Mitochondrial Membrane Potential | 0.15 |
| Tumor Protein p53 | 0.13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Che, X.; Jing, H.; Su, Y.; Yang, W.; Wang, R.; Zhang, G.; Meng, J.; Yuan, W.; Wang, J.; et al. A New Potent Inhibitor against α-Glucosidase Based on an In Vitro Enzymatic Synthesis Approach. Molecules 2024, 29, 878. https://doi.org/10.3390/molecules29040878
Zhang H, Che X, Jing H, Su Y, Yang W, Wang R, Zhang G, Meng J, Yuan W, Wang J, et al. A New Potent Inhibitor against α-Glucosidase Based on an In Vitro Enzymatic Synthesis Approach. Molecules. 2024; 29(4):878. https://doi.org/10.3390/molecules29040878
Chicago/Turabian StyleZhang, Huanyu, Xiance Che, Hongyan Jing, Yaowu Su, Wenqi Yang, Rubing Wang, Guoqi Zhang, Jie Meng, Wei Yuan, Juan Wang, and et al. 2024. "A New Potent Inhibitor against α-Glucosidase Based on an In Vitro Enzymatic Synthesis Approach" Molecules 29, no. 4: 878. https://doi.org/10.3390/molecules29040878