
Regioselective Cyclic Iminium Formation of Ugi Advanced Intermediates: Rapid Access to 3,4-Dihydropyrazin-2(1H)-ones and Other Diverse Nitrogen-Containing Heterocycles


Abstract
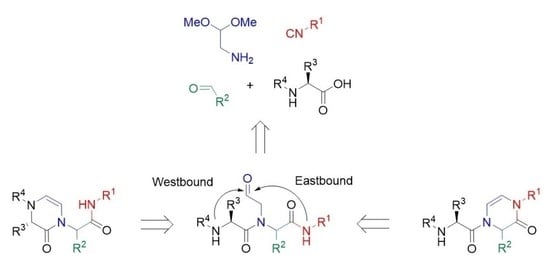
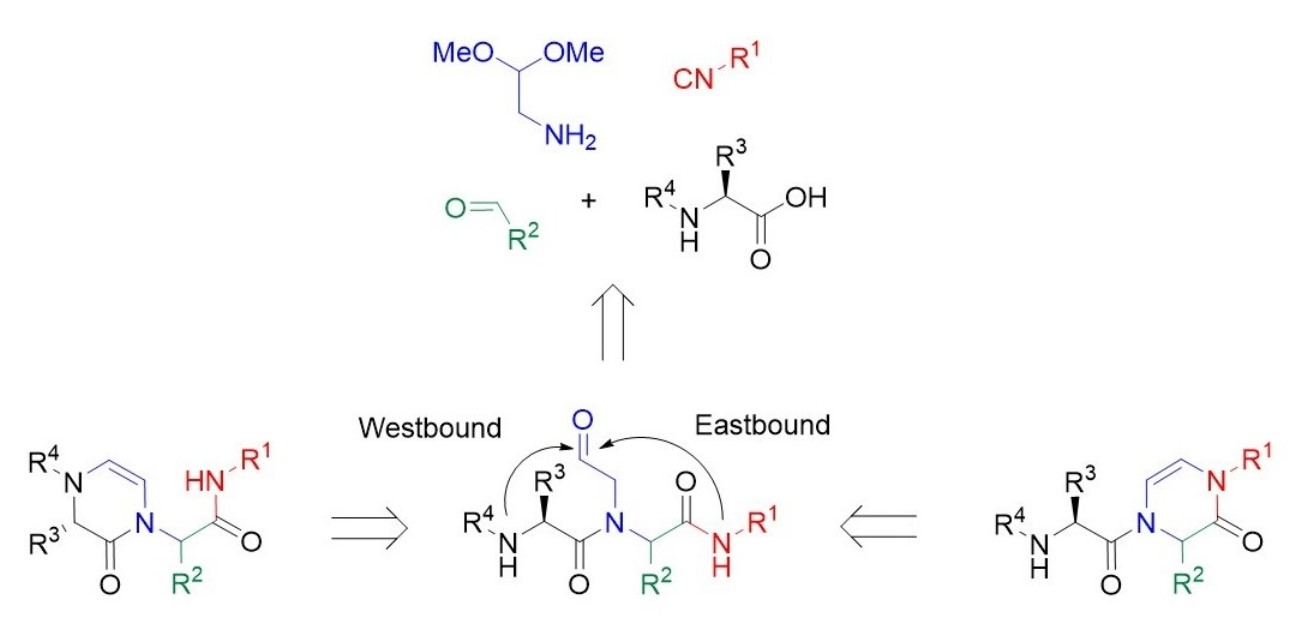
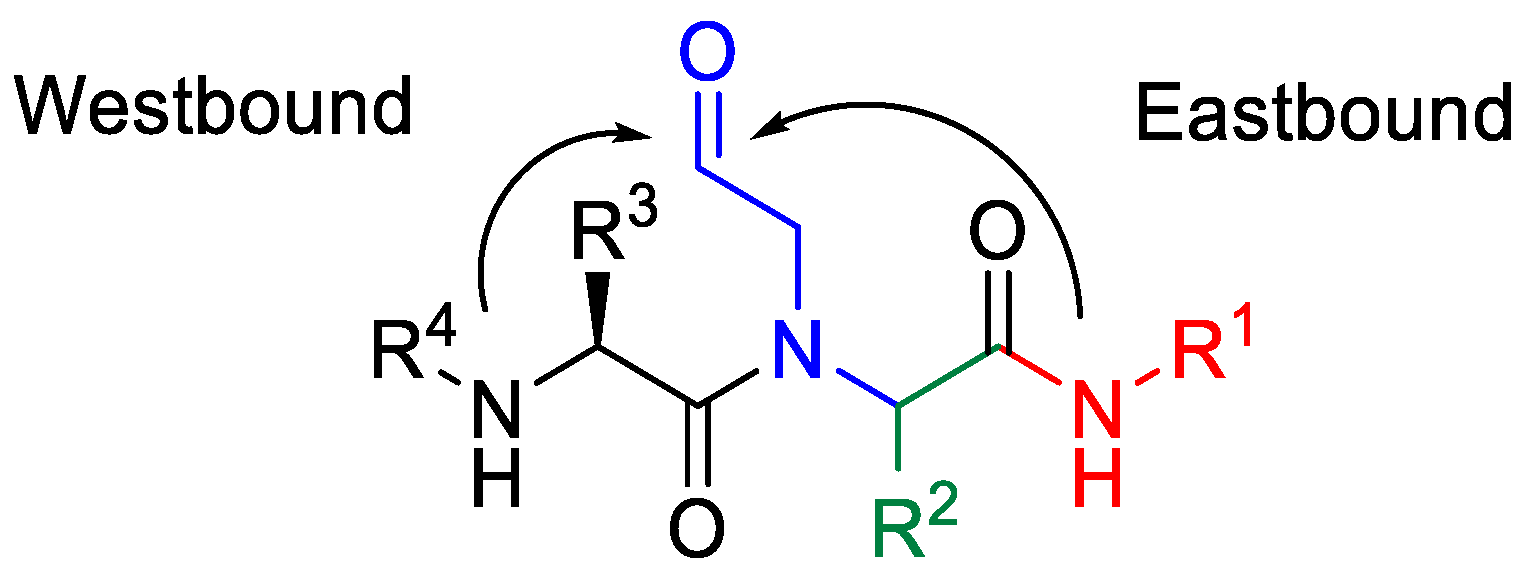
:
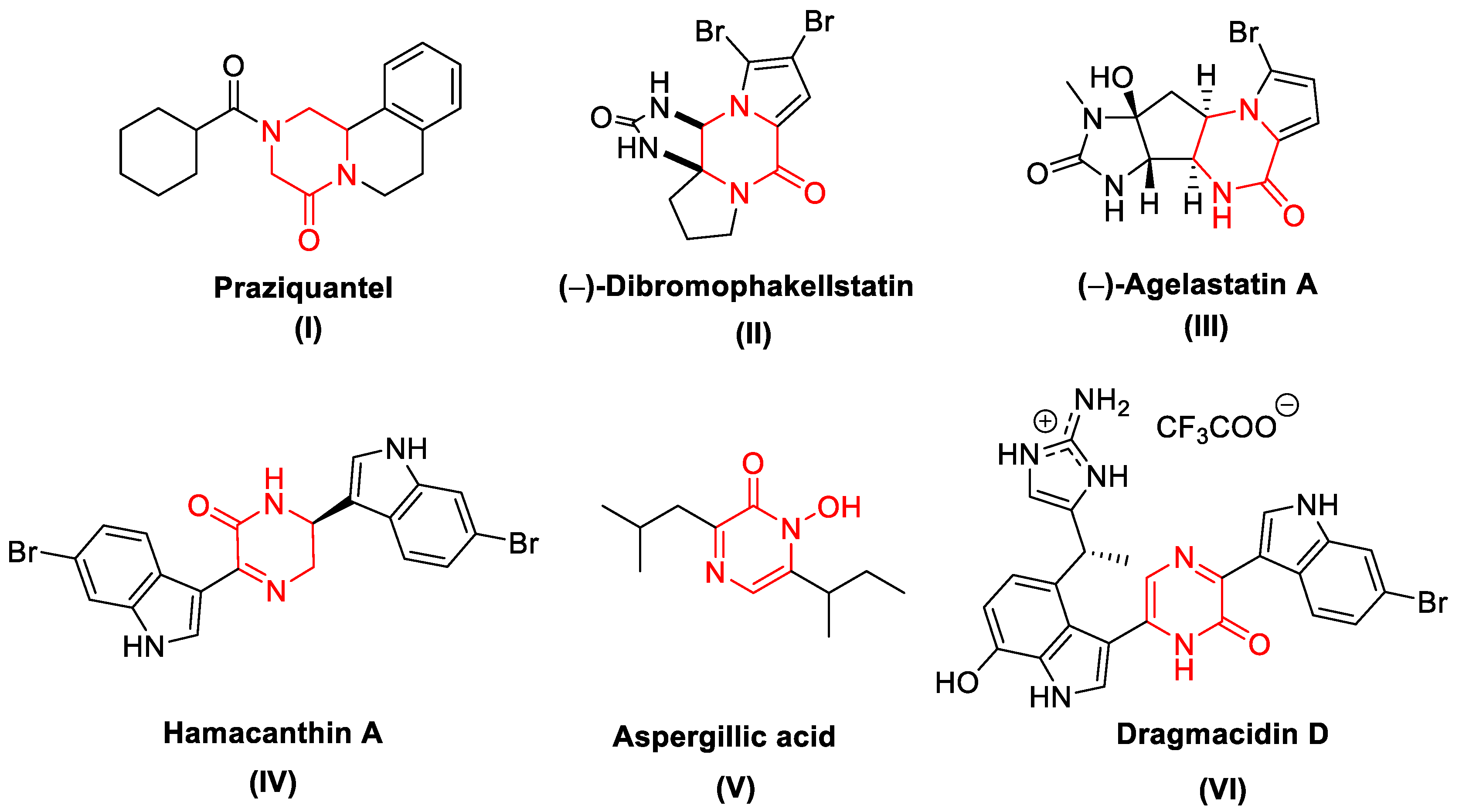
1. Introduction
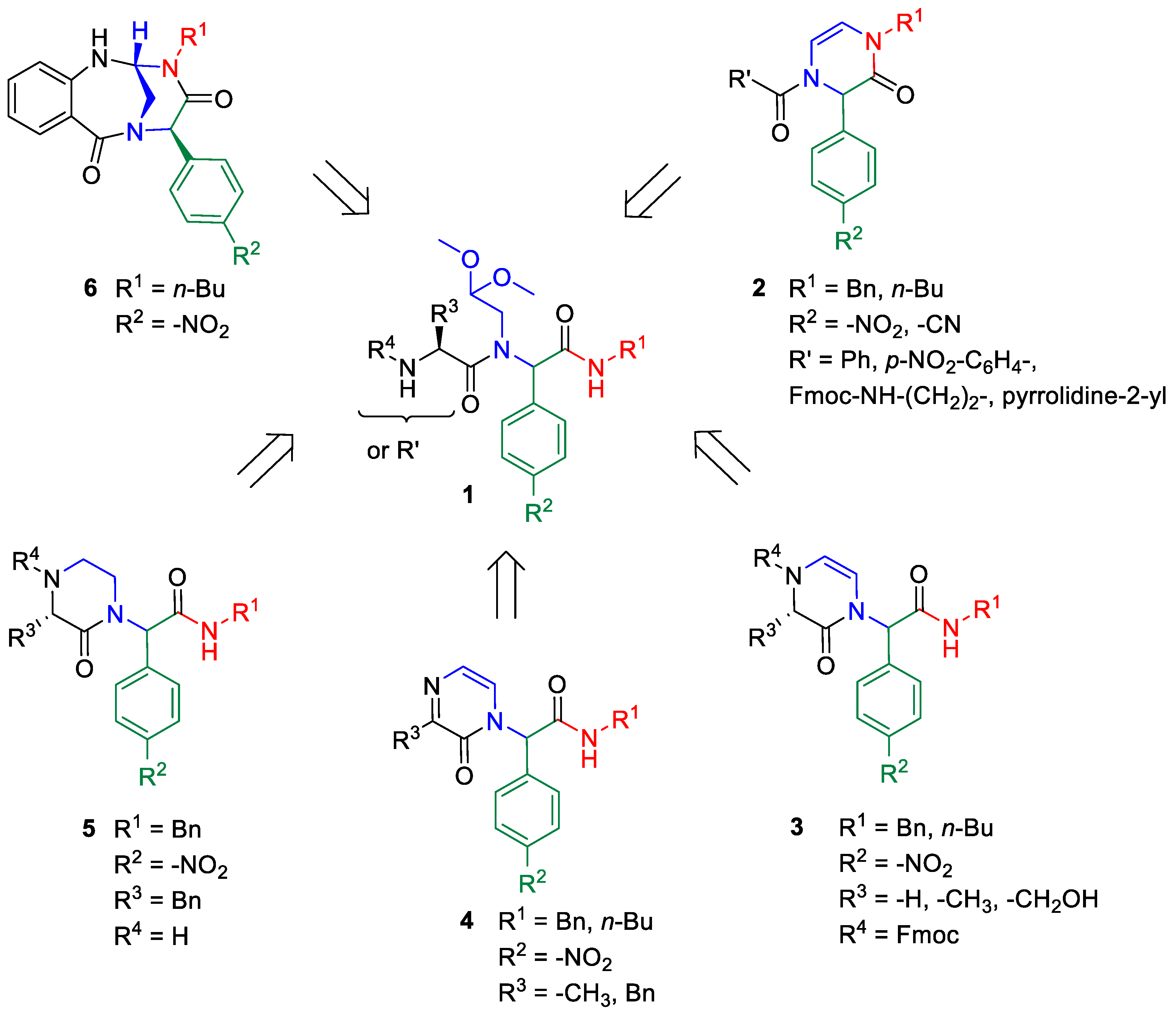
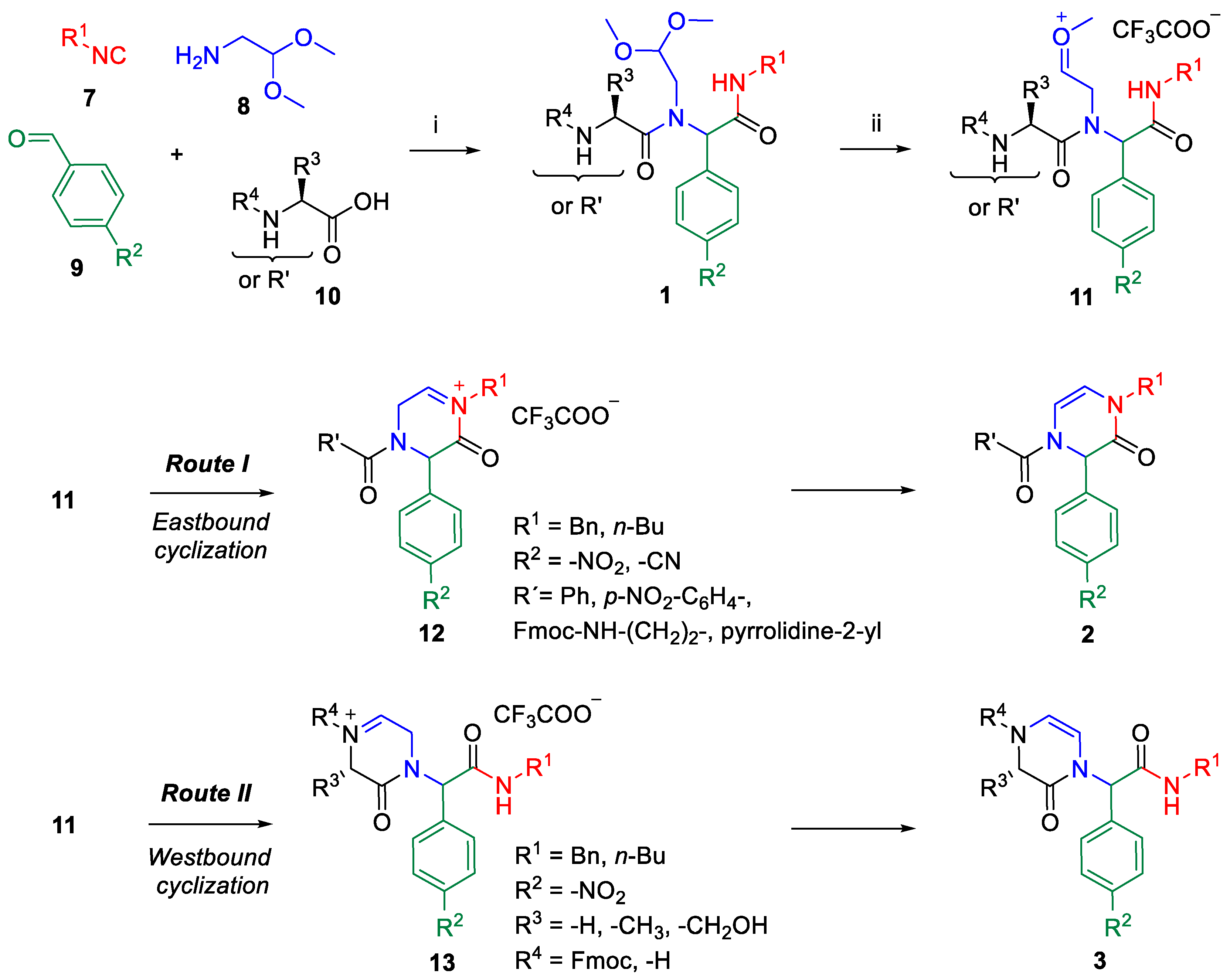
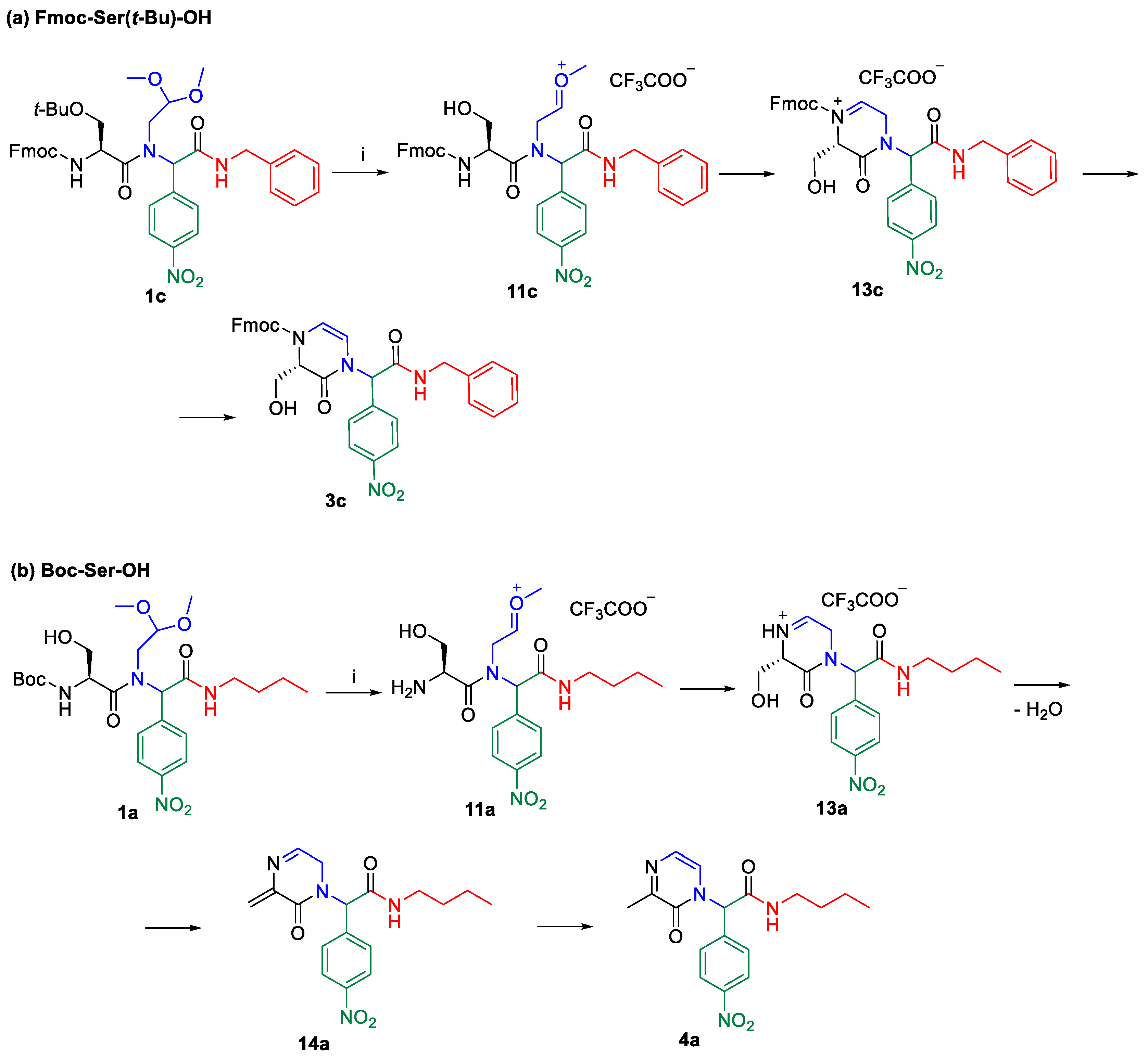
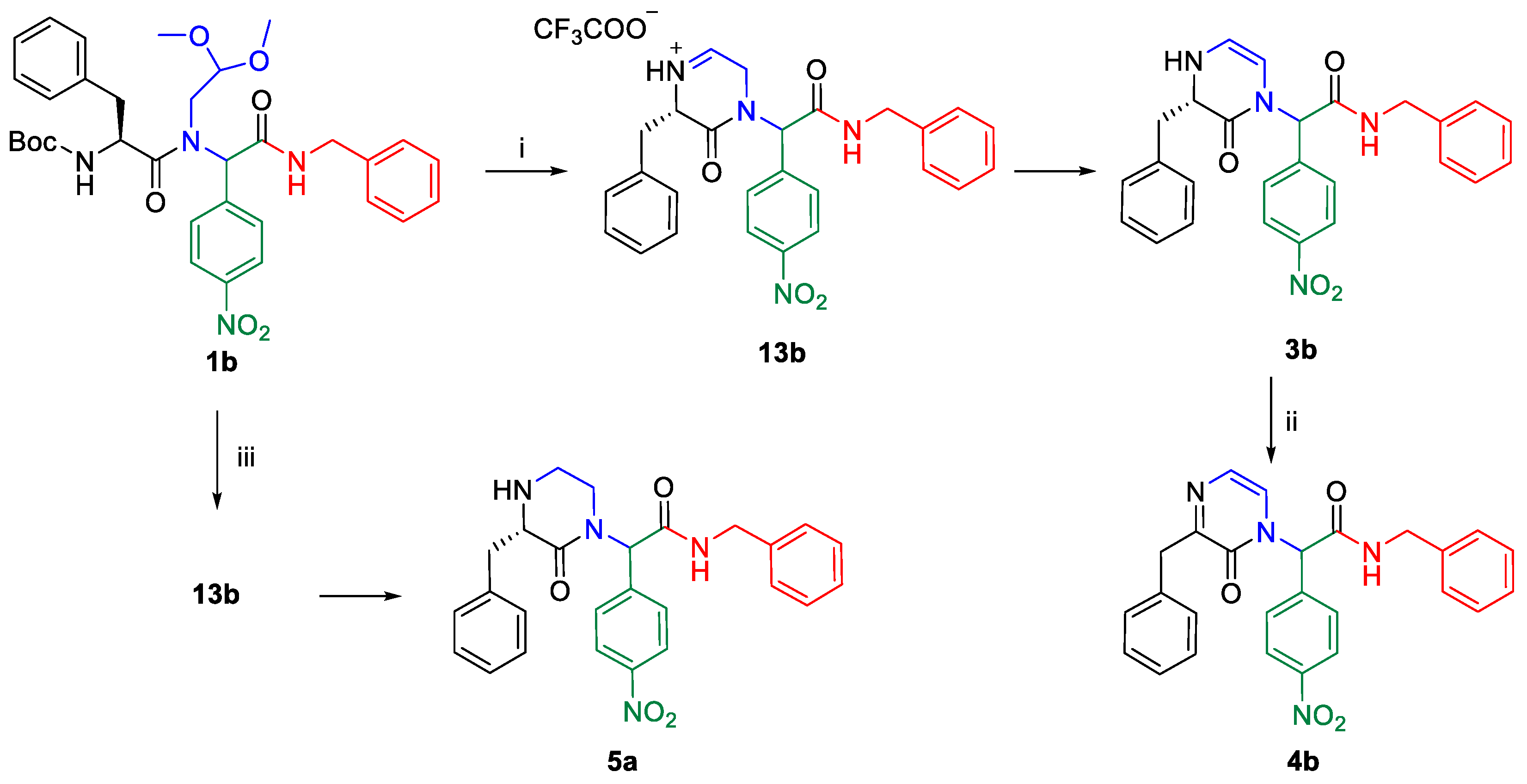
2. Results
3. Materials and Methods
3.1. General Information
3.2. Analytical Data of Individual Compounds














4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Weber, L. The Application of Multi-Component Reactions in Drug Discovery. Curr. Med. Chem. 2003, 9, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Hulme, C.; Gore, V. “Multi-component Reactions: Emerging Chemistry in Drug Discovery” ‘From Xylocain to Crixivan’. Curr. Med. Chem. 2003, 10, 51–80. [Google Scholar] [CrossRef]
- Cankařová, N.; Krchňák, V. Isocyanide Multicomponent Reactions on Solid Phase: State of the Art and Future Application. Int. J. Mol. Sci. 2020, 21, 9160. [Google Scholar] [CrossRef] [PubMed]
- Li Petri, G.; Di Martino, S.; De Rosa, M. Peptidomimetics: An Overview of Recent Medicinal Chemistry Efforts toward the Discovery of Novel Small Molecule Inhibitors. J. Med. Chem. 2022, 65, 7438–7475. [Google Scholar] [CrossRef] [PubMed]
- Smyslová, P.; Kisseljova, K.; Krchňák, V. Base-Mediated Intramolecular C- and N-Arylation of N,N-Disubstituted 2-Nitrobenzenesulfonamides: Advanced Intermediates for the Synthesis of Diverse Nitrogenous Heterocycles. ACS Comb. Sci. 2014, 16, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Zoghroban, H.S.; El-Kowrany, S.I.; Aboul Asaad, I.A.; El Maghraby, G.M.; El-Nouby, K.A.; Abd Elazeem, M.A. Niosomes for enhanced activity of praziquantel against Schistosoma mansoni: In vivo and in vitro evaluation. Parasit. Res. 2019, 118, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Poullennec, K.G.; Romo, D. Enantioselective Total Synthesis of (+)-Dibromophakellstatin. J. Am. Chem. Soc. 2003, 125, 6344–6345. [Google Scholar] [CrossRef] [PubMed]
- Mason, C.K.; McFarlane, S.; Johnston, P.G.; Crowe, P.; Erwin, P.J.; Domostoj, M.M.; Campbell, F.C.; Manaviazar, S.; Hale, K.J.; El-Tanani, M. Agelastatin A: A novel inhibitor of osteopontin-mediated adhesion, invasion, and colony formation. Mol. Cancer Ther. 2008, 7, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Gunasekera, S.P.; McCarthy, P.J.; Kelly-Borges, M. Hamacanthins A and B, New Antifungal Bis Indole Alkaloids from the Deep-Water Marine Sponge, Hamacantha Sp. J. Nat. Prod. 1994, 57, 1437–1441. [Google Scholar] [CrossRef]
- Miller, M.J. Syntheses and therapeutic potential of hydroxamic acid based siderophores and analogs. Chem. Rev. 1989, 89, 1563–1579. [Google Scholar] [CrossRef]
- Khattak, S.U.; Lutfullah, G.; Iqbal, Z.; Ahmad, J.; Rehman, I.U.; Shi, Y.; Ikram, S. Aspergillus flavus originated pure compound as a potential antibacterial. BMC Microbiolog. 2021, 21, 322. [Google Scholar] [CrossRef] [PubMed]
- Longley, R.E.; Beach, V.; Isbrucker, R.A.; Wright, A.E. Use of Imidazole and Indole Compounds as Inhibitors of Nitric Oxide Synthase. Patent 6087363, 7 November 2000. [Google Scholar]
- Thorns, V.; Hansen, L.; Masliah, E. nNOS Expressing Neurons in the Entorhinal Cortex and Hippocampus Are Affected in Patients with Alzheimer’s Disease. Exp. Neurol. 1998, 150, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.A.; Jiménez-Jiménez, F.J.; Ortí-Pareja, M.; Navarro, J.A. The Role of Nitric Oxide in Neurodegeneration. Drugs Aging 1998, 12, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.K.; Sarpong, R.; Stoltz, B.M. The First Total Synthesis of Dragmacidin D. J. Am. Chem. Soc. 2002, 124, 13179–13184. [Google Scholar] [CrossRef] [Green Version]
- La Venia, A.; Lemrová, B.; Krchňák, V. Regioselective Incorporation of Backbone Constraints Compatible with Traditional Solid-Phase Peptide Synthesis. ACS Comb. Sci. 2013, 15, 59–72. [Google Scholar] [CrossRef]
- Vaňková, B.; Brulíková, L.; Wu, B.; Krchňák, V. Synthesis of Piperazinones, Piperazines, Tetrahydropyrazines, and Dihydropyrazinones from Polymer-Supported Acyclic Intermediates via N-Alkyl- and N-Acyliminiums. Eur. J. Org. Chem. 2012, 2012, 5075–5084. [Google Scholar] [CrossRef]
- Lee, S.C.; Park, S.B. Practical Solid-Phase Parallel Synthesis of D5-2-Oxopiperazines via N-Acyliminium Ion Cyclization. J. Comb. Chem. 2007, 9, 828–835. [Google Scholar] [CrossRef]
- Lenci, E.; Innocenti, R.; Menchi, G.; Faggi, C.; Trabocchi, A. Two-step one-pot synthesis of dihydropyrazinones as Xaa-Ser dipeptide isosteres through morpholine acetal rearrangement. Org. Biomol. Chem. 2015, 13, 7013–7019. [Google Scholar] [CrossRef]
- Broggini, G.; Galli, S.; Rigamonti, M.; Sottocornola, S.; Zecchi, G. Entry to nitrogen-containing heterocycles by based-promoted heterocyclization on allenylamides of L-α-aminoacids. Tetrahedron Lett. 2009, 50, 1447–1449. [Google Scholar] [CrossRef]
- Dömling, A.; Ugi, I. Multicomponent Reactions with Isocyanides. Angew. Chem. Int. Ed. 2000, 39, 3168–3210. [Google Scholar] [CrossRef]
- Dömling, A.; Wang, W.; Wang, K. Chemistry and Biology of Multicomponent Reactions. Chem. Rev. 2012, 112, 3083–3135. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Sello, J.K.; Schreiber, S.L. Pairwise Use of Complexity-Generating Reactions in Diversity-Oriented Organic Synthesis. Org. Lett. 2000, 2, 709–712. [Google Scholar] [CrossRef]
- Cheng, J.F.; Chen, M.; Arrhenius, T.; Nadzan, A. A convenient solution and solid-phase synthesis of D5-2-oxopiperazines via N-acyliminium ions cyclization. Tetrahedron Lett. 2002, 43, 6293–6295. [Google Scholar] [CrossRef]
- Azuaje, J.; El Maatougui, A.; Pérez-Rubio, J.M.; Coelho, A.; Fernández, F.; Sotelo, E. Multicomponent Assembly of Diverse Pyrazin-2(1H)-one Chemotypes. J. Org. Chem. 2013, 78, 4402–4409. [Google Scholar] [CrossRef] [PubMed]
- Zoll, A.J.; Molas, J.C.; Mercado, B.Q.; Ellman, J.A. Imine Directed Cp*RhIII-Catalyzed N-H Functionalization and Annulation with Amino Amides, Aldehydes, and Diazo Compounds. Angew. Chem. Int. Ed. 2023, 62, e202210822. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Tripathi, S.; Yadav, A.; Kant, R.; Srivastava, H.K.; Srivastava, A.K. Synthesis of β- and γ-lactam fused dihydropyrazinones from Ugi adducts via a sequential ring construction strategy. Chem. Commun. 2020, 56, 12789–12792. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Kumar, A.; Kant, R.; Srivastava, A.K. Regioselective Synthesis of Functionalized Pyrrolo[1,2-a]pyrazine-3,6(2H,4H)-diones via Tandem Post-Ugi Cyclization and Gold(I)-Catalyzed Annulation. J. Org. Chem. 2022, 87, 12799–12815. [Google Scholar] [CrossRef] [PubMed]
- Icelo-Ávila, E.; Amador-Sánchez, Y.A.; Polindara-García, L.A.; Miranda, L.D. Synthesis of 6-methyl-3,4-dihydropyrazinones using an Ugi 4-CR/allenamide cycloisomerization protocol. Org. Biomol. Chem. 2017, 15, 360–372. [Google Scholar] [CrossRef]
- Amador-Sánchez, Y.A.; Hernández-Vázquez, E.; González-Mojica, N.; Ramírez-Apan, M.T.; Miranda, L.D. Diversity-oriented synthesis and cytotoxic screening of fused dihydropyrazin-2(1H)-ones through a Ugi 4-CR/deprotection/Heck sequence. Tetrahedron 2020, 76, 131383. [Google Scholar] [CrossRef]
- Schreiber, S.L. Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef] [Green Version]
- Burke, M.D.; Schreiber, S.L. A Planning Strategy for Diversity-Oriented Synthesis. Angew. Chem. Int. Ed. 2004, 43, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Galloway, W.R.J.D.; Isidro-Llobet, A.; Spring, D.R. Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun. 2010, 1, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vojkovský, T.; Weichsel, A.; Pátek, M. Solid-Phase Synthesis of Heterocycles Containing an 1-Acyl-3-oxopiperazine Skeleton. J. Org. Chem. 1998, 63, 3162–3163. [Google Scholar] [CrossRef]
- Royer, J.; Bonin, M.; Micouin, L. Chiral Heterocycles by Iminium Ion Cyclization. Chem. Rev. 2004, 104, 2311–2352. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E. Scaffold Diversity from N-Acyliminium Ions. Chem. Rev. 2017, 117, 7811–7856. [Google Scholar] [CrossRef]
- Cankařová, N.; Krchňák, V. Polymer-Supported Stereoselective Synthesis of Benzimidazolinopiperazinones. J. Org. Chem. 2012, 77, 5687–5695. [Google Scholar] [CrossRef]
- Cankařová, N.; La Venia, A.; Krchňák, V. Polymer-Supported Stereoselective Synthesis of Tetrahydrobenzopyrazino-thiadiazinone Dioxides via N-Sulfonyl Iminiums. ACS Comb. Sci. 2014, 16, 293–302. [Google Scholar] [CrossRef]
- Cankařová, N.; La Venia, A.; Krajčovičová, S.; Krchňák, V. Configuration-Dependent Medium-Sized Ring Formation: Chiral Molecular Framework with Three-Dimensional Architecture. J. Org. Chem. 2019, 84, 636–644. [Google Scholar] [CrossRef]
- Ventosa-Andrés, P.; Hradilová, L.; Krchňák, V. Privileged Structures as Peptide Backbone Constraints: Polymer-Supported Stereoselective Synthesis of Benzimidazolinopiperazinone Peptides. ACS Comb. Sci. 2014, 16, 359–366. [Google Scholar] [CrossRef]
- Ventosa-Andrés, P.; Barea Ripoll, C.A.; La-Venia, A.; Krchňák, V. Solid-phase synthesis of fused 1,4-diazepanone peptidomimetics via tandem N-iminium ion cyclization-nucleophilic addition. Tetrahedron Lett. 2015, 56, 5424–5428. [Google Scholar] [CrossRef]
- Schütznerová, E.; Oliver, A.G.; Zajíček, J.; Krchňák, V. Polymer-Supported Stereoselective Synthesis of (1S,5S)-6-Oxa-3,8-diazabicyclo[3.2.1]octanes. Eur. J. Org. Chem. 2013, 2013, 3158–3165. [Google Scholar] [CrossRef]
- La Venia, A.; Ventosa-Andrés, P.; Hradilová, L.; Krchňák, V. From Amino Acids to Nature-Inspired Molecular Scaffolds: Incorporation of Medium-Sized Bridged Heterocycles into a Peptide Backbone. J. Org. Chem. 2014, 79, 10378–10389. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
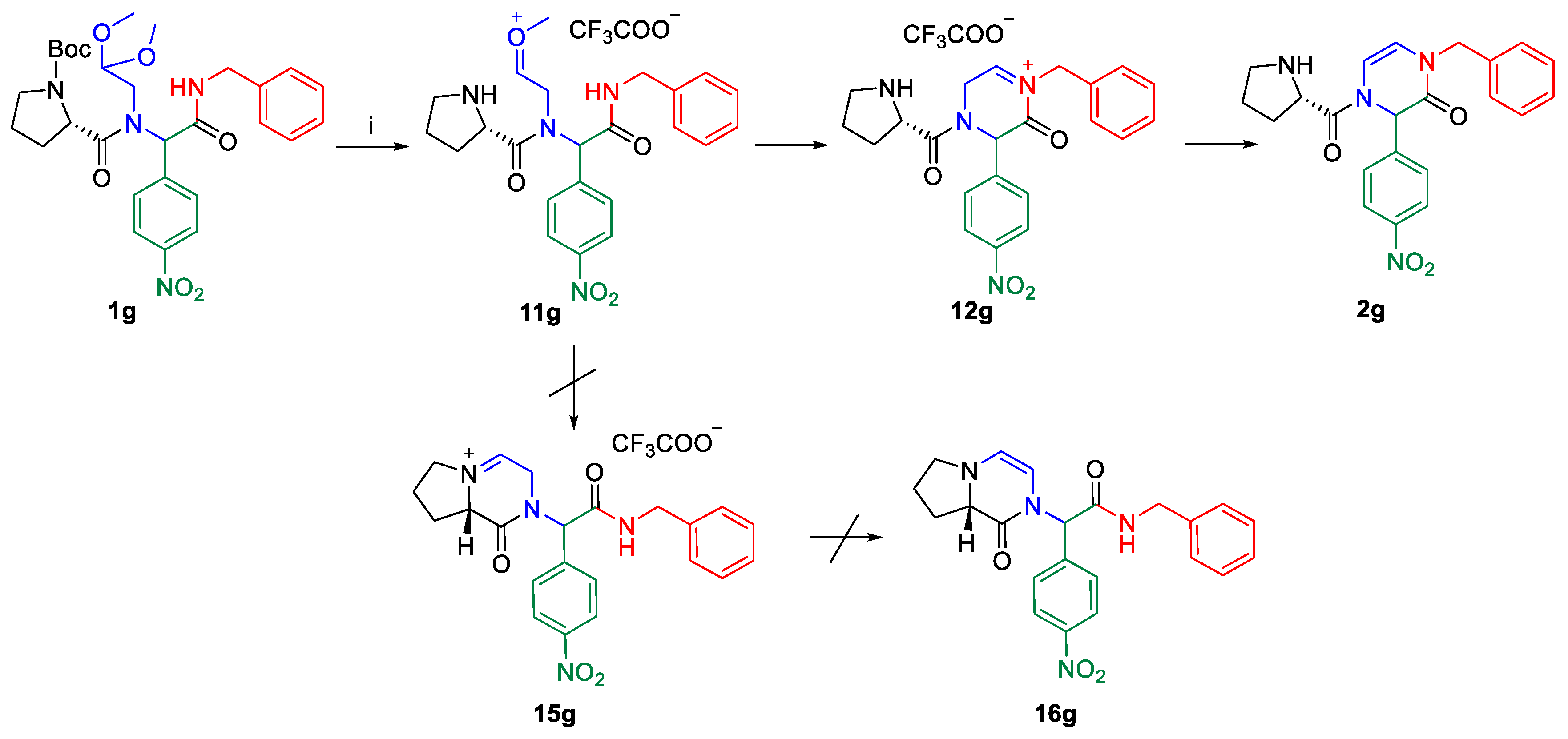{kind=link}
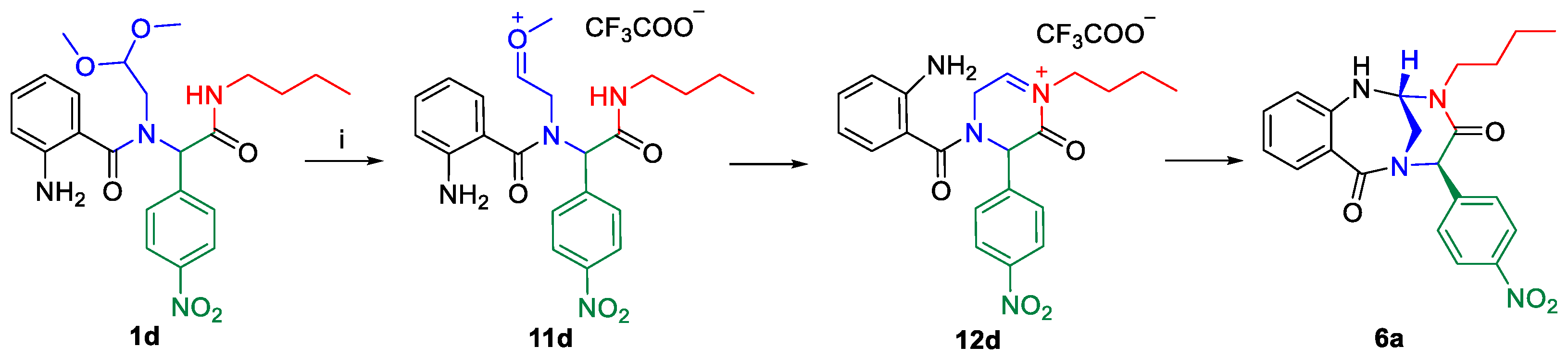{kind=link}
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Product | R1 | R2 | R’ | R3 | R4 | Purity (%) a | Purity (%) b | Yield (%) c |
| 2a | Bn | –NO2 | Ph | NA d | NA d | 93 | 95 | 69 |
| 2b | Bn | –CN | Ph | NA d | NA d | 90 | 96 | 69 |
| 2c | Bn | –NO2 | p-NO2–C6H4– | NA d | NA d | 71 | 99 | 61 |
| 2d | n-Bu | –NO2 | Ph | NA d | NA d | 95 | 98 | 40 |
| 2e | n-Bu | –NO2 | p-NO2–C6H4– | NA d | NA d | 48 | 99 | 29 |
| 2f | n-Bu | –NO2 | Fmoc–NH–(CH2)2– | NA d | NA d | 50 | 99 | 40 |
| 2g | Bn | –NO2 | Pyrrolidine-2-yl | NA d | NA d | 83 | 97 | 60 |
| 3a | n-Bu | –NO2 | NA d | –H | Fmoc | 51 | 89 | 44 |
| 3b | n-Bu | –NO2 | NA d | –CH3 | Fmoc | 65 | 98 | 45 |
| 3c | Bn | –NO2 | NA d | –CH2OH | Fmoc | 32 | 99 | 31 |
| 4a | n-Bu | –NO2 | NA d | –CH3 | NA d | 31 | 99 | 11 |
| 4b | Bn | –NO2 | NA d | Bn | NA d | 40 | 97 | 27 |
| 5a | Bn | –NO2 | NA d | Bn | –H | 87 | 97 | 58 |
| 6a | n-Bu | –NO2 | NA d | NA d | NA d | 37 | 99 | 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cankařová, N.; Krchňák, V. Regioselective Cyclic Iminium Formation of Ugi Advanced Intermediates: Rapid Access to 3,4-Dihydropyrazin-2(1H)-ones and Other Diverse Nitrogen-Containing Heterocycles. Molecules 2023, 28, 3062. https://doi.org/10.3390/molecules28073062
Cankařová N, Krchňák V. Regioselective Cyclic Iminium Formation of Ugi Advanced Intermediates: Rapid Access to 3,4-Dihydropyrazin-2(1H)-ones and Other Diverse Nitrogen-Containing Heterocycles. Molecules. 2023; 28(7):3062. https://doi.org/10.3390/molecules28073062
Chicago/Turabian StyleCankařová, Naděžda, and Viktor Krchňák. 2023. "Regioselective Cyclic Iminium Formation of Ugi Advanced Intermediates: Rapid Access to 3,4-Dihydropyrazin-2(1H)-ones and Other Diverse Nitrogen-Containing Heterocycles" Molecules 28, no. 7: 3062. https://doi.org/10.3390/molecules28073062