
Cleistocalyx nervosum var. paniala Berry Seed Protects against TNF-α-Stimulated Neuroinflammation by Inducing HO-1 and Suppressing NF-κB Mechanism in BV-2 Microglial Cells


 , and
, and 
Abstract
:1. Introduction
2. Results
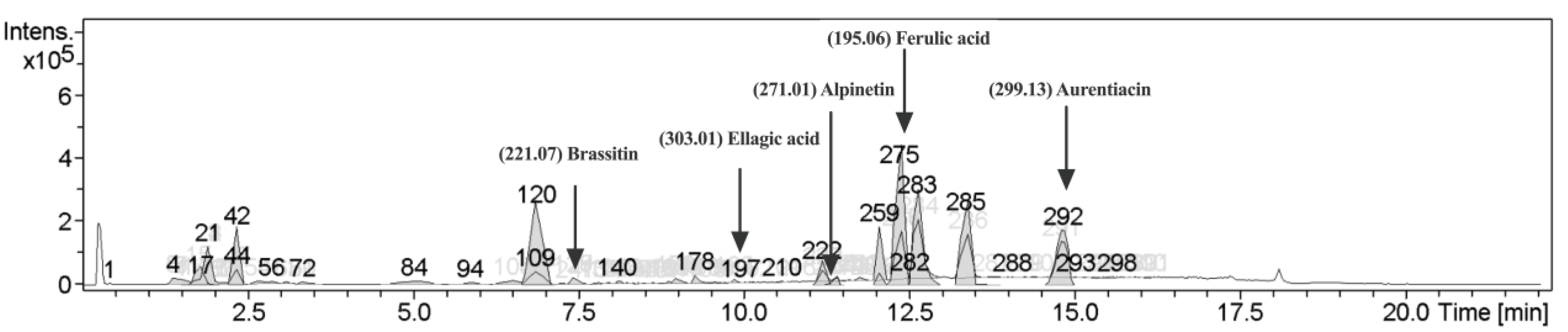
2.1. Characterization of Phytochemical Components in CNSE
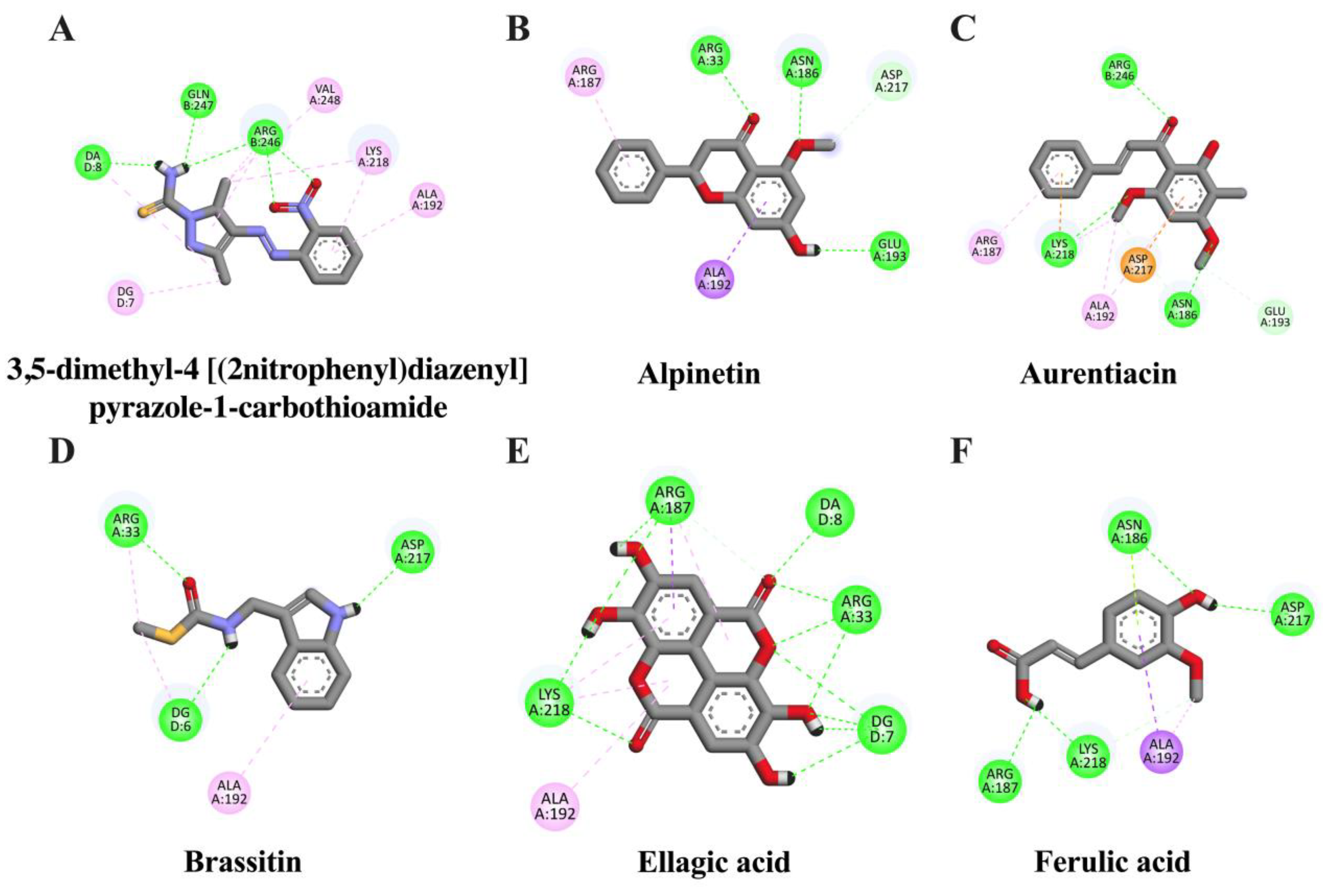
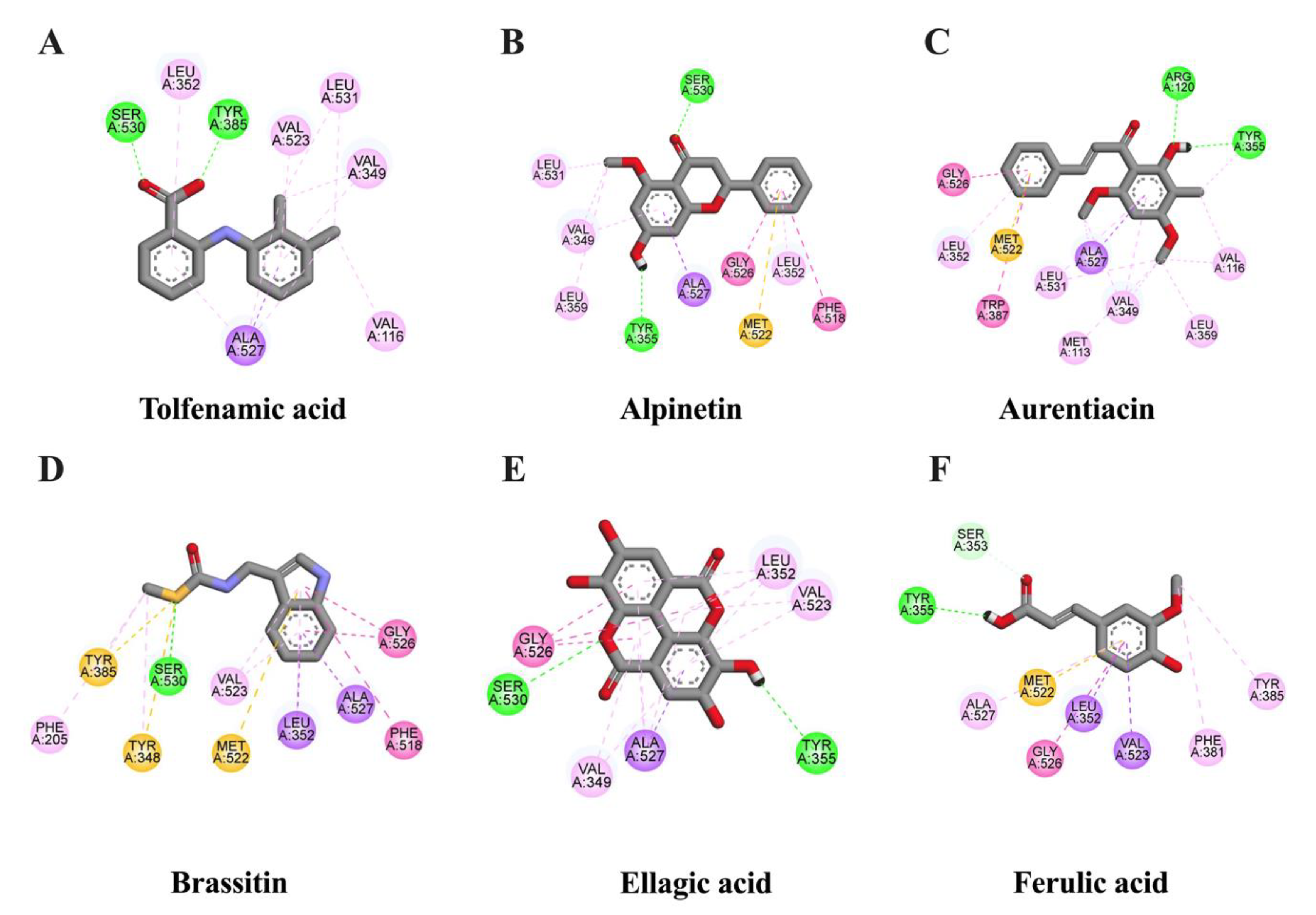
2.2. In Silico Evaluation of Identified Compounds in CNSE against Inflammatory-Related Transcription Factors
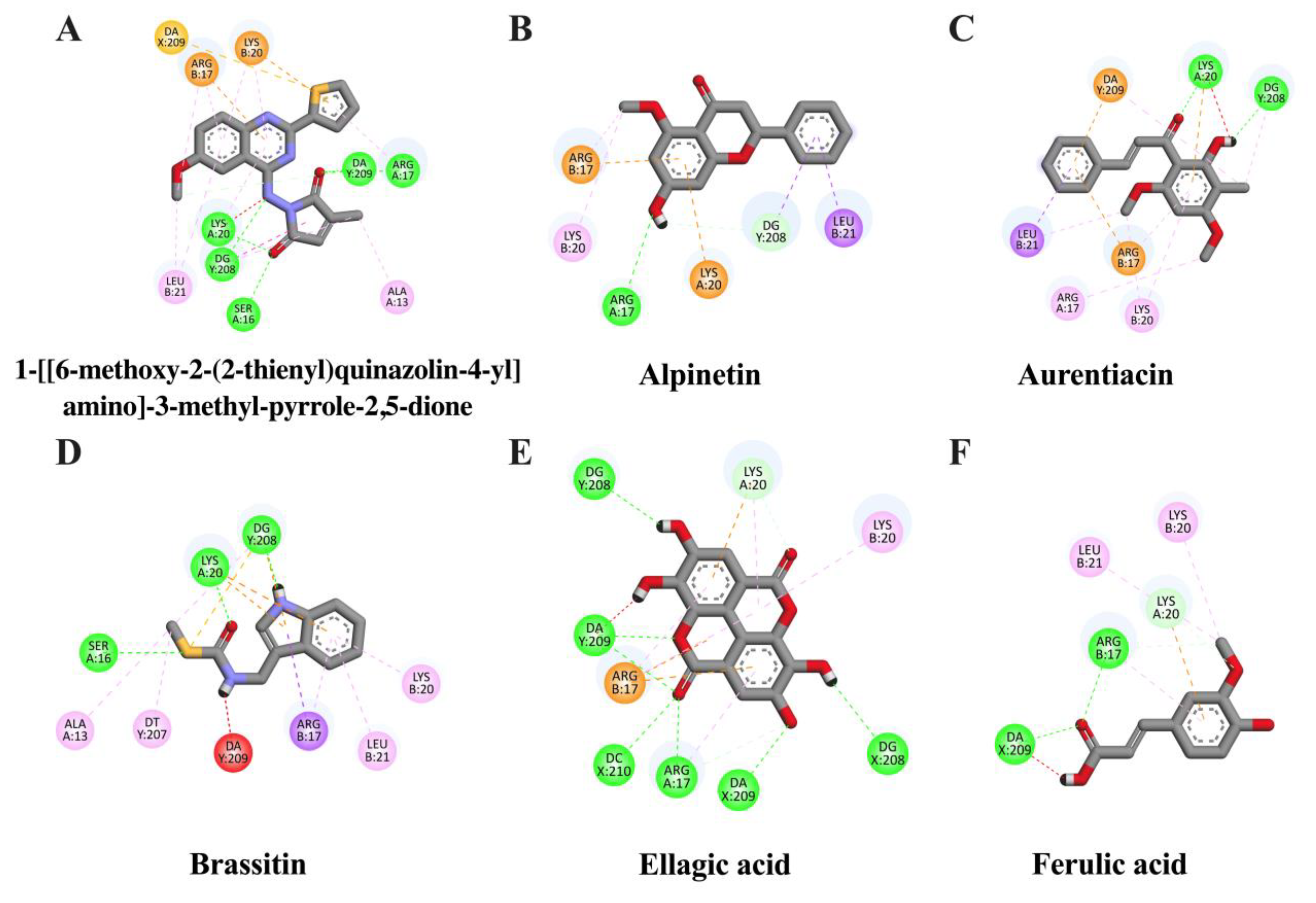
2.3. In Silico Evaluation of Identified Compounds in CNSE against iNOS Protein
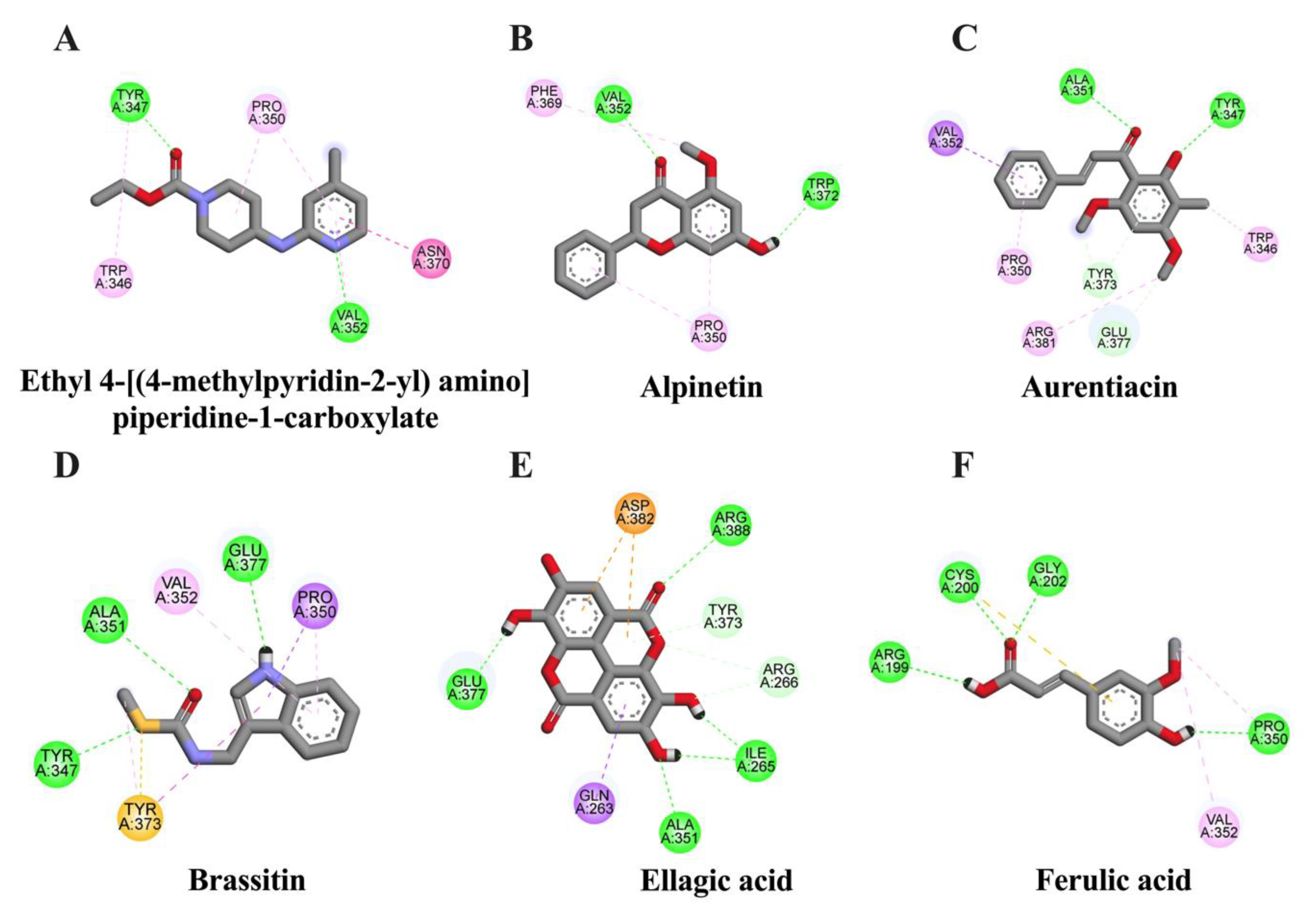
2.4. In Silico Evaluation of Identified Compounds in CNSE against COX-2 Protein
2.5. Lipinski’s Rule of Five Parameters and ADMET Properties of Bioactive Components in CNSE
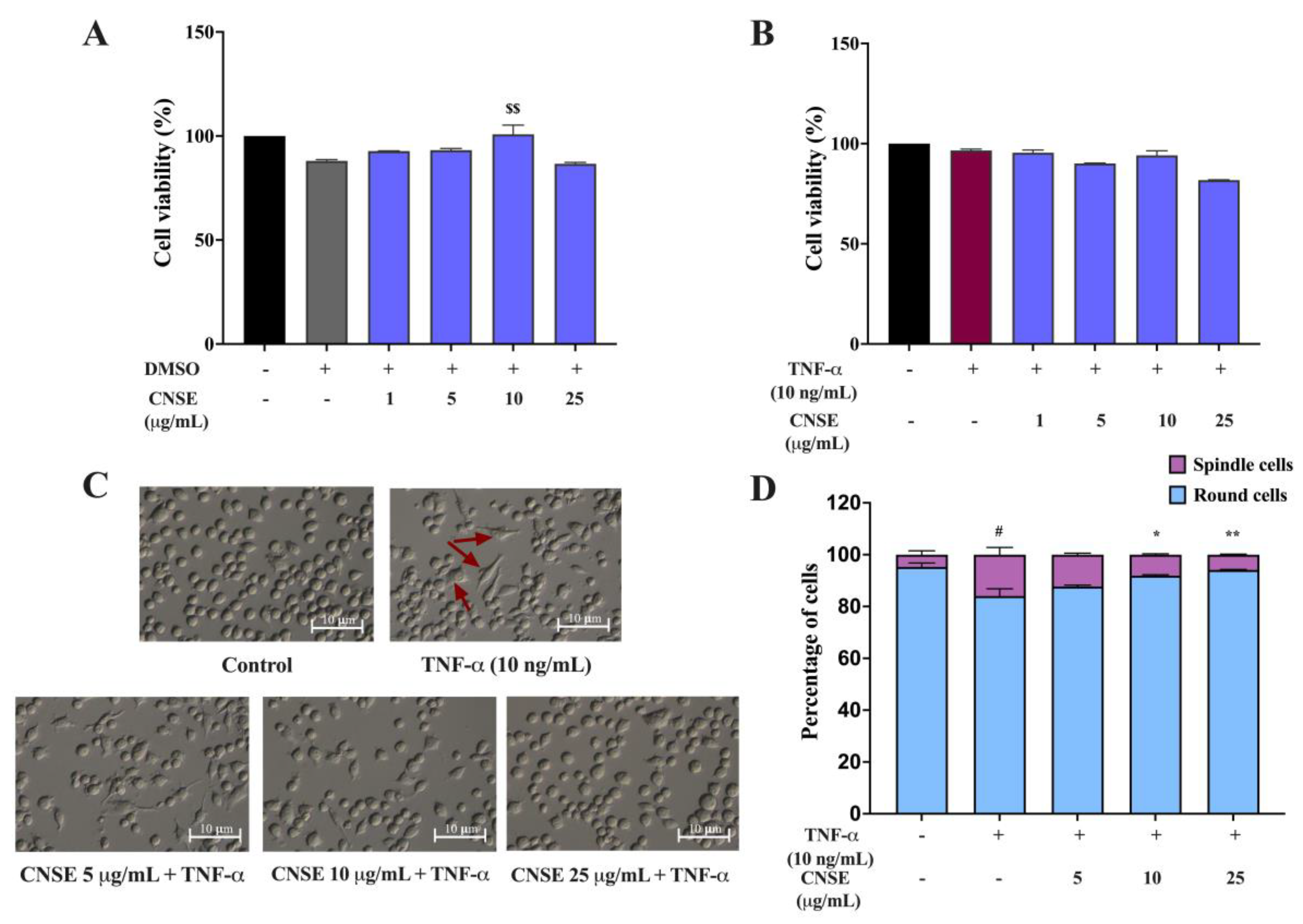
2.6. Effect of CNSE on the Viability of BV-2 Cells
2.7. Effect of CNSE on the Morphological Changes of BV-2 Cells
2.8. Inhibitory Effect of CNSE on Levels of Proinflammatory Cytokines
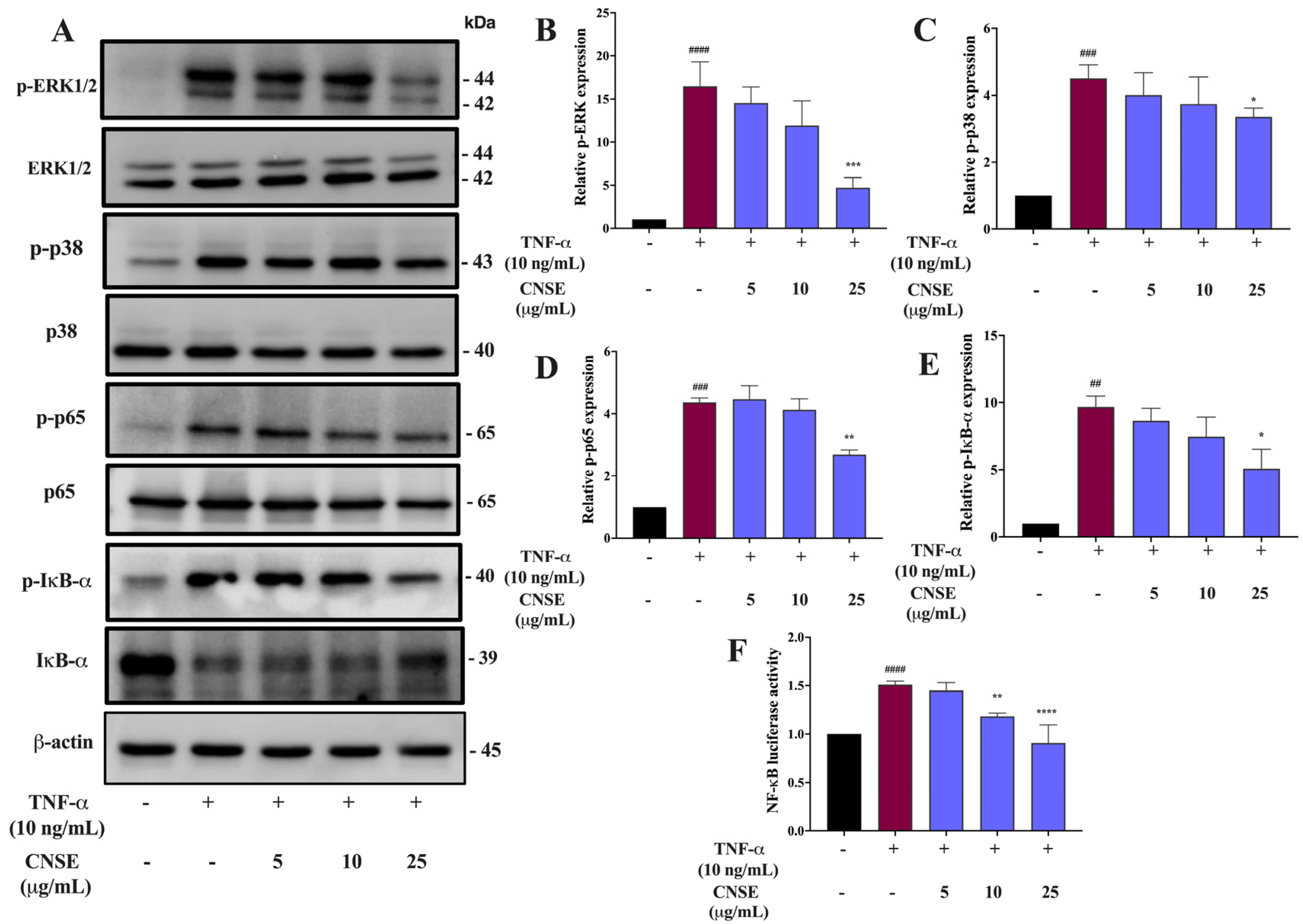
2.9. Effect of CNSE on MAPKs Signaling Activation
2.10. Effect of CNSE on NF-κB Signaling Activation
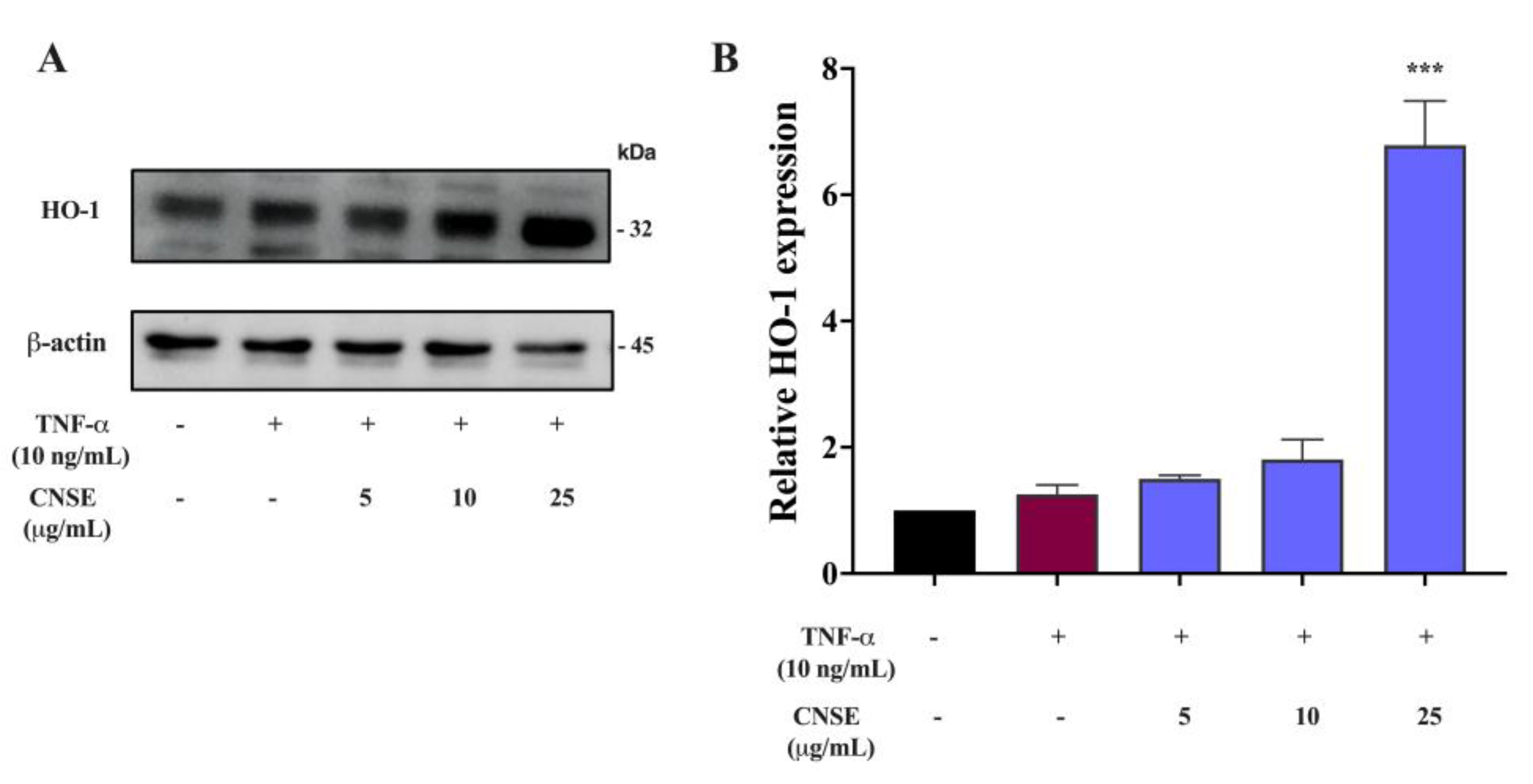
2.11. Effect of CNSE on HO-1 Activation
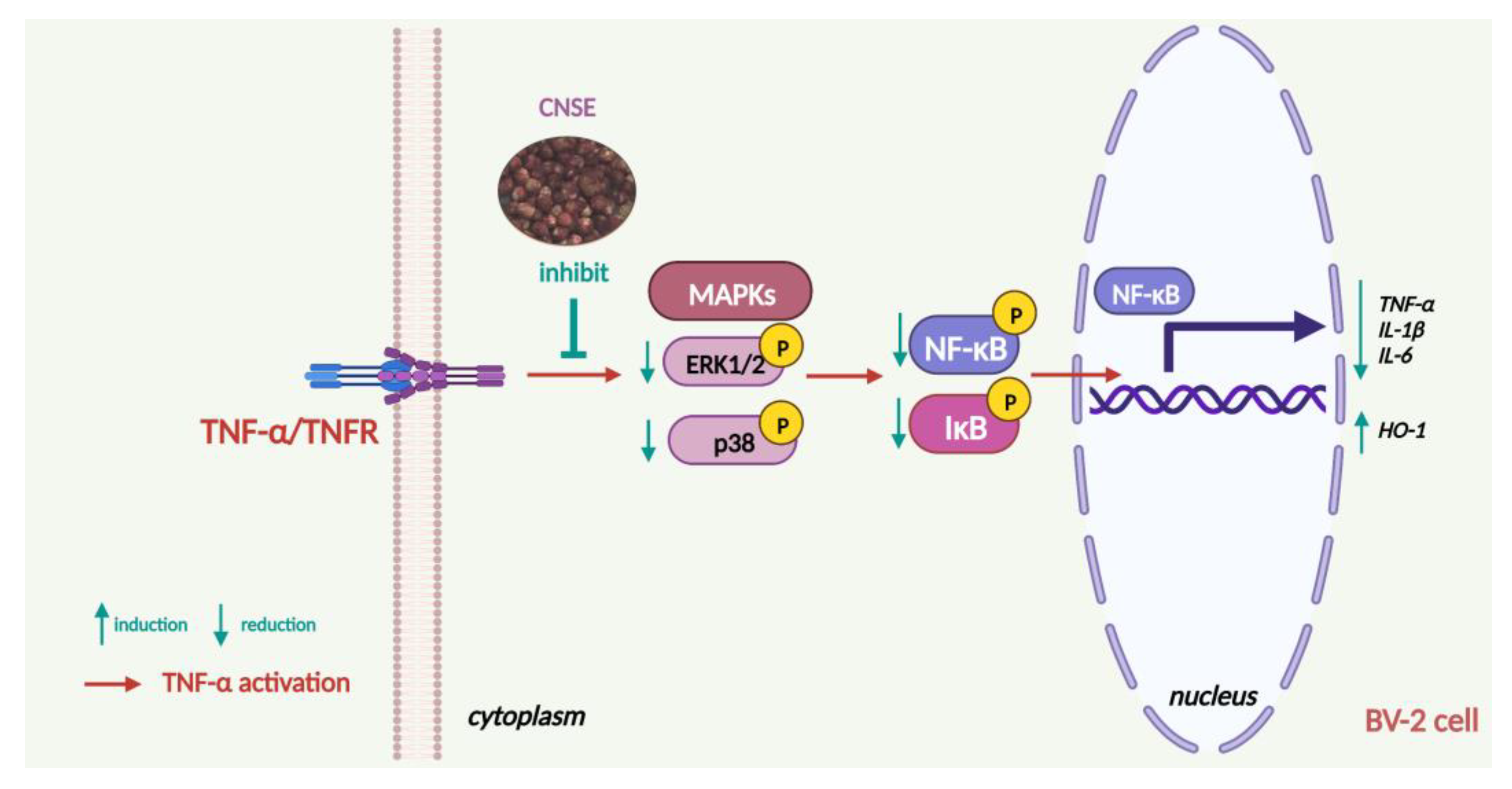
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. CNSE Extraction
4.3. Liquid Chromatography-Mass Spectrometry (LC-MS) Analysis
4.4. Molecular Docking
4.4.1. Protein Preparation
4.4.2. Ligand Preparation
4.4.3. Method Validation
4.4.4. Molecular Docking of Candidate Ligands
4.5. Lipinski’s Rule and Pharmacokinetic Property Analysis
4.6. Cell Culture
4.7. Cell Viability Assay
4.8. Cell Morphological Analysis
4.9. Quantitative Real-Time PCR Analysis
4.10. Immunoblotting Analysis
4.11. Enzyme-Linked Immunosorbent Assay (ELISA)
4.12. Dual-Luciferase Assay
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar] [PubMed]
- Rauf, A.; Badoni, H.; Abu-Izneid, T.; Olatunde, A.; Rahman, M.M.; Painuli, S.; Semwal, P.; Wilairatana, P.; Mubarak, M.S. Neuroinflammatory Markers: Key Indicators in the Pathology of Neurodegenerative Diseases. Molecules 2022, 27, 3194. [Google Scholar] [CrossRef] [PubMed]
- Yuste, J.E.; Tarragon, E.; Campuzano, C.M.; Ros-Bernal, F. Implications of glial nitric oxide in neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, P.K.; Gross, E.J.; Lianos, E.A. Interactions between inducible nitric oxide synthase and heme oxygenase-1 in glomerulonephritis. Kidney Int. 2002, 61, 847–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.-Y.; Shen, S.-C.; Cheng, K.-T.; Subbaraju, G.V.; Chien, C.-C.; Chen, Y.-C. Hispolon inhibition of inflammatory apoptosis through reduction of iNOS/NO production via HO-1 induction in macrophages. J. Ethnopharmacol. 2014, 156, 61–72. [Google Scholar] [CrossRef]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [Green Version]
- Gough, P.; Myles, I.A. Tumor Necrosis Factor Receptors: Pleiotropic Signaling Complexes and Their Differential Effects. Front. Immunol. 2020, 11, 585880. [Google Scholar] [CrossRef]
- Shi, J.H.; Sun, S.C. Tumor Necrosis Factor Receptor-Associated Factor Regulation of Nuclear Factor κB and Mitogen-Activated Protein Kinase Pathways. Front. Immunol. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Farrugia, M.; Baron, B. The role of TNF-α in rheumatoid arthritis: A focus on regulatory T cells. J. Clin. Transl. Res. 2016, 2, 84–90. [Google Scholar] [CrossRef]
- Jang, D.I.; Lee, A.H.; Shin, H.Y.; Song, H.R.; Park, J.H.; Kang, T.B.; Lee, S.R.; Yang, S.H. The Role of Tumor Necrosis Factor Alpha (TNF-α) in Autoimmune Disease and Current TNF-α Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zheng, Y.; Chen, X. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Front. Pharm. 2017, 8, 460. [Google Scholar] [CrossRef] [PubMed]
- Chariyakornkul, A.; Juengwiroj, W.; Ruangsuriya, J.; Wongpoomchai, R. Antioxidant Extract from Cleistocalyx nervosum var. paniala Pulp Ameliorates Acetaminophen-Induced Acute Hepatotoxicity in Rats. Molecules 2022, 27, 553. [Google Scholar] [CrossRef]
- Poontawee, W.; Natakankitkul, S.; Wongmekiat, O. Protective Effect of Cleistocalyx nervosum var. paniala Fruit Extract against Oxidative Renal Damage Caused by Cadmium. Molecules 2016, 21, 133. [Google Scholar] [CrossRef] [Green Version]
- Sukprasansap, M.; Chanvorachote, P.; Tencomnao, T. Cleistocalyx nervosum var. paniala berry fruit protects neurotoxicity against endoplasmic reticulum stress-induced apoptosis. Food Chem. Toxicol. 2017, 103, 279–288. [Google Scholar] [CrossRef]
- Nantacharoen, W.; Baek, S.J.; Plaingam, W.; Charoenkiatkul, S.; Tencomnao, T.; Sukprasansap, M. Cleistocalyx nervosum var. paniala Berry Promotes Antioxidant Response and Suppresses Glutamate-Induced Cell Death via SIRT1/Nrf2 Survival Pathway in Hippocampal HT22 Neuronal Cells. Molecules 2022, 27, 5813. [Google Scholar] [CrossRef] [PubMed]
- Chariyakornkul, A.; Inboot, N.; Taya, S.; Wongpoomchai, R. Low-polar extract from seed of Cleistocalyx nervosum var. paniala modulates initiation and promotion stages of chemically-induced carcinogenesis in rats. Biomed. Pharm. 2021, 133, 110963. [Google Scholar] [CrossRef] [PubMed]
- Tantratian, S.; Balmuang, N. Enriched makiang (Cleistocalyx nervosum var. paniala) seed extract and citric acid to control pathogenic bacteria and color of fresh cut cantaloupe. LWT 2021, 138, 110626. [Google Scholar] [CrossRef]
- Brimson, J.M.; Prasanth, M.I.; Isidoro, C.; Sukprasansap, M.; Tencomnao, T. Cleistocalyx nervosum var. paniala seed extracts exhibit sigma-1 antagonist sensitive neuroprotective effects in PC12 cells and protects C. elegans from stress via the SKN-1/NRF-2 pathway. Nutr. Healthy Aging 2021, 6, 131–146. [Google Scholar] [CrossRef]
- Murakami, A.; Ohigashi, H. Targeting NOX, INOS and COX-2 in inflammatory cells: Chemoprevention using food phytochemicals. Int. J. Cancer 2007, 121, 2357–2363. [Google Scholar] [CrossRef]
- Vaish, V.; Piplani, H.; Rana, C.; Sanyal, S.N. Angiostatic Properties of Sulindac and Celecoxib in the Experimentally Induced Inflammatory Colorectal Cancer. Cell Biochem. Biophys. 2013, 66, 205–227. [Google Scholar] [CrossRef] [PubMed]
- Herowati, R.; Widodo, G. Molecular Docking Analysis: Interaction Studies of Natural Compounds to Anti-inflammatory Targets. 2017. Quant. Struct.-Act. Relatsh. 2017. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Konsman, J.P. Cytokines in the Brain and Neuroinflammation: We Didn’t Starve the Fire! Pharmaceuticals 2022, 15, 140. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.N.; Jana, M.; Pahan, K. MAPK p38 regulates transcriptional activity of NF-kappaB in primary human astrocytes via acetylation of p65. J. Immunol. 2007, 179, 7101–7109. [Google Scholar] [CrossRef] [Green Version]
- Shih, R.H.; Wang, C.Y.; Yang, C.M. NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front. Mol. Neurosci. 2015, 8, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, N.; Patial, S. Tumor necrosis factor-α signaling in macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef]
- Bradley, J.R. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Frankola, K.A.; Greig, N.H.; Luo, W.; Tweedie, D. Targeting TNF-α to elucidate and ameliorate neuroinflammation in neurodegenerative diseases. CNS Neurol. Disord. Drug Targets 2011, 10, 391–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brás, J.P.; Bravo, J.; Freitas, J.; Barbosa, M.A.; Santos, S.G.; Summavielle, T.; Almeida, M.I. TNF-alpha-induced microglia activation requires miR-342: Impact on NF-kB signaling and neurotoxicity. Cell Death Dis. 2020, 11, 415. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Kim, J.; Min, D.Y.; Jung, E.; Lim, Y.; Shin, S.Y.; Lee, Y.H. Inhibition of TNFα-induced interleukin-6 gene expression by barley (Hordeum vulgare) ethanol extract in BV-2 microglia. Genes Genom. 2019, 41, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Ajmone-Cat, M.A.; Bernardo, A.; Greco, A.; Minghetti, L. Non-Steroidal Anti-Inflammatory Drugs and Brain Inflammation: Effects on Microglial Functions. Pharmaceuticals 2010, 3, 1949–1965. [Google Scholar] [CrossRef] [PubMed]
- Wongrakpanich, S.; Wongrakpanich, A.; Melhado, K.; Rangaswami, J. A Comprehensive Review of Non-Steroidal Anti-Inflammatory Drug Use in The Elderly. Aging Dis. 2018, 9, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani Iyer, P.; Sivamaruthi, B.; Sukprasansap, M.; Chuchawankul, S.; Tencomnao, T.; Chaiyasut, C. Functional properties and Bioactivities of Cleistocalyx nervosum var. paniala berry plant: A review. Food Sci. Technol. 2020, 40, 369–373. [Google Scholar]
- Narkprasom, N.; Narkprasom, K.; Upara, U. Optimization of Total Phenolic from Cleistocalyx nervosum by Microwave-Assisted Extraction. Am. J. Eng. Appl. Sci. 2015, 8, 302. [Google Scholar] [CrossRef] [Green Version]
- Ismail, E.; Jantan, I.; Azmi, N. Ellagic Acid Protects against Activation of Microglia by Inhibiting MAPKs and NF-κB Signalling. Indian J. Pharm. Educ. Res. 2020, 54, s529–s536. [Google Scholar] [CrossRef]
- Banc, R.; Rusu, M.E.; Filip, L.; Popa, D.-S. The Impact of Ellagitannins and Their Metabolites through Gut Microbiome on the Gut Health and Brain Wellness within the Gut–Brain Axis. Foods 2023, 12, 270. [Google Scholar] [CrossRef]
- Vallarino, G.; Salis, A.; Lucarini, E.; Turrini, F.; Olivero, G.; Roggeri, A.; Damonte, G.; Boggia, R.; Di Cesare Mannelli, L.; Ghelardini, C.; et al. Healthy Properties of a New Formulation of Pomegranate-Peel Extract in Mice Suffering from Experimental Autoimmune Encephalomyelitis. Molecules 2022, 27, 914. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, S.; He, L.; Wang, C.; Yang, L. Alpinetin Protects Chondrocytes and Exhibits Anti-Inflammatory Effects via the NF-κB/ERK Pathway for Alleviating Osteoarthritis. Inflammation 2020, 43, 1742–1750. [Google Scholar] [CrossRef] [PubMed]
- Huo, M.; Chen, N.; Chi, G.; Yuan, X.; Guan, S.; Li, H.; Zhong, W.; Guo, W.; Soromou, L.W.; Gao, R.; et al. Traditional medicine alpinetin inhibits the inflammatory response in Raw 264.7 cells and mouse models. Int. Immunopharmacol. 2012, 12, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kim, H.C.; Ko, H.; Amor, E.C.; Lee, J.W.; Yang, H.O. Inhibitory effects of aurentiacin from Syzygium samarangense on lipopolysaccharide-induced inflammatory response in mouse macrophages. Food Chem. Toxicol. 2012, 50, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Ali, T.; Alam, S.I.; Ullah, R.; Zeb, A.; Lee, K.W.; Rutten, B.P.F.; Kim, M.O. Ferulic Acid Rescues LPS-Induced Neurotoxicity via Modulation of the TLR4 Receptor in the Mouse Hippocampus. Mol. Neurobiol. 2019, 56, 2774–2790. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-M.; Shen, J.-D.; Xu, L.-P.; Li, H.-B.; Li, Y.-C.; Yi, L.-T. Ferulic acid inhibits neuro-inflammation in mice exposed to chronic unpredictable mild stress. Int. Immunopharmacol. 2017, 45, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Li, Y.; Pei, G. Polysaccharides from Ganoderma lucidum attenuate microglia-mediated neuroinflammation and modulate microglial phagocytosis and behavioural response. J. Neuroinflammation 2017, 14, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsini, A.; Di Benedetto, M.G.; Giacobbe, J.; Pariante, C.M. Pro- and Anti-Inflammatory Properties of Interleukin in Vitro: Relevance for Major Depression and Human Hippocampal Neurogenesis. Int. J. Neuropsychopharmacol. 2020, 23, 738–750. [Google Scholar] [CrossRef]
- Ávila, C.; Trindade, F.; Penteado, J.; Janke, F.; Schneider, J.; Uecker, J.; Alvarado Rincón, J.; Barros, C.; Andreazza, R.; Pieniz, S. Anti-inflammatory Effect of a Goji Berry Extract (Lycium barbarum) in Rats Subjected to Inflammation by Lipopolysaccharides (LPS). Braz. Arch. Biol. Technol. 2020, 63. [Google Scholar] [CrossRef]
- Webster, J.D.; Vucic, D. The Balance of TNF Mediated Pathways Regulates Inflammatory Cell Death Signaling in Healthy and Diseased Tissues. Front. Cell Dev. Biol. 2020, 8, 365. [Google Scholar] [CrossRef]
- Bernhardi, R.V. Neurodegenerative Diseases—MAPK Signalling Pathways in Neuroinflammation. In Encyclopedia of Neuroscience; Binder, M.D., Hirokawa, N., Windhorst, U., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 2614–2620. [Google Scholar]
- Dresselhaus, E.C.; Meffert, M.K. Cellular Specificity of NF-κB Function in the Nervous System. Front. Immunol. 2019, 10, 1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Ma, C.; Zhang, Z.; Zhang, H.; Hu, H. NF-κB signaling in inflammation and cancer. MedComm 2021, 2, 618–653. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farzaei, M.H.; Tewari, D.; Momtaz, S.; Argüelles, S.; Nabavi, S.M. Targeting ERK signaling pathway by polyphenols as novel therapeutic strategy for neurodegeneration. Food Chem. Toxicol. 2018, 120, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Bauer, I. Heme oxygenase-1: Redox regulation and role in the hepatic response to oxidative stress. Antioxid. Redox Signal 2002, 4, 749–758. [Google Scholar] [CrossRef]
- Takahashi, T.; Morita, K.; Akagi, R.; Sassa, S. Heme oxygenase-1: A novel therapeutic target in oxidative tissue injuries. Curr. Med. Chem. 2004, 11, 1545–1561. [Google Scholar] [CrossRef] [PubMed]
- Campbell, N.K.; Fitzgerald, H.K.; Dunne, A. Regulation of inflammation by the antioxidant haem oxygenase 1. Nat. Rev. Immunol. 2021, 21, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Hsieh, H.L. Roles of Heme Oxygenase-1 in Neuroinflammation and Brain Disorders. Antioxidants 2022, 11, 923. [Google Scholar] [CrossRef]
- Jeong, Y.H.; Li, W.; Go, Y.; Oh, Y.C. Atractylodis Rhizoma Alba Attenuates Neuroinflammation in BV2 Microglia upon LPS Stimulation by Inducing HO-1 Activity and Inhibiting NF-κB and MAPK. Int. J. Mol. Sci. 2019, 20, 4015. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.W.; Kim, H.J.; Sohn, J.H.; Yim, J.H.; Kim, Y.C.; Oh, H. Terrein suppressed lipopolysaccharide-induced neuroinflammation through inhibition of NF-κB pathway by activating Nrf2/HO-1 signaling in BV2 and primary microglial cells. J. Pharm. Sci. 2020, 143, 209–218. [Google Scholar] [CrossRef]
- Fatima, A.; Abdul, A.B.H.; Abdullah, R.; Karjiban, R.A.; Lee, V.S. Binding Mode Analysis of Zerumbone to Key Signal Proteins in the Tumor Necrosis Factor Pathway. Int. J. Mol. Sci. 2015, 16, 2747–2766. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Tan, S.; Fang, D.; Zhang, R.; Zhou, S.; Wu, W.; Zheng, K. Probing the binding mechanism of novel dual NF-κB/AP-1 inhibitors by 3D-QSAR, docking and molecular dynamics simulations. RSC Adv. 2015, 5, 81523–81532. [Google Scholar] [CrossRef]
- Orlando, B.J.; Malkowski, M.G. Substrate-selective Inhibition of Cyclooxygeanse-2 by Fenamic Acid Derivatives Is Dependent on Peroxide Tone. J. Biol. Chem. 2016, 291, 15069–15081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcin, E.D.; Arvai, A.S.; Rosenfeld, R.J.; Kroeger, M.D.; Crane, B.R.; Andersson, G.; Andrews, G.; Hamley, P.J.; Mallinder, P.R.; Nicholls, D.J.; et al. Anchored plasticity opens doors for selective inhibitor design in nitric oxide synthase. Nat. Chem. Biol. 2008, 4, 700–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sillapachaiyaporn, C.; Rangsinth, P.; Nilkhet, S.; Moungkote, N.; Chuchawankul, S. HIV-1 Protease and Reverse Transcriptase Inhibitory Activities of Curcuma aeruginosa Roxb. Rhizome Extracts and the Phytochemical Profile Analysis: In Vitro and In Silico Screening. Pharmaceuticals 2021, 14, 1115. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Binding Energy (kcal/mol) | Inhibition Constant (μM) | Amino Acid Interaction | ||
|---|---|---|---|---|---|
| Hydrogen Bond | Hydrophobic Bond | Other | |||
| 3,5-dimethyl-4-[(2-nitrophenyl)diazenyl]pyrazole-1-carbothioamide (native inhibitor) | −6.33 | 22.78 | ARG246 (3) GLN247 | LYS218 (2) VAL248 ARG246 (2) ALA192 | - |
| Ellagic acid | −7.31 | 4.41 | ARG33 (2) LYS218 (2) ARG187 | LYS218 (2) ARG187 (2) ALA192 | - |
| Alpinetin | −6.17 | 30.19 | ARG33 (2) ASN186 GLU193 ASP217 | ALA192 ARG187 | - |
| Aurentiacin | −6.01 | 39.06 | ASN186 (2) LYS218 (2) ARG246 GLU193 | ALA192 (2) LYS218 ARG187 | ASP217 |
| Brassitin | −5.11 | 178.17 | ARG33 (2) ASP217 | ARG33 ALA192 | - |
| Ferulic acid | −4.52 | 488.99 | ASN186 LYS218 (2) ASP217 ARG187 | ALA192 (2) LYS218 | ASN186 |
| Ligand | Binding Energy (kcal/mol) | Inhibition Constant (μM) | Amino Acid Interaction | ||
|---|---|---|---|---|---|
| Hydrogen Bond | Hydrophobic Bond | Other | |||
| 1-[[6-methoxy-2-(2-thienyl)quinazolin-4-yl]amino]-3-methyl-pyrrole-2,5-dione (native inhibitor) | −9.29 | 0.016 | SER16 ARG17 LYS20 (2) DG208 DA209 (3) | ALA13 ARG17 (4) LYS20 (3) LEU21 (2) DG208 (4) | ARG17 LYS20 DA209 (2) |
| Aurentiacin | −7.69 | 2.32 | LYS20 DG208 | ARG17 (3) LEU21 (2) LYS20 (2) DG208 DA209 | ARG17 LYS20 DA209 |
| Alpinetin | −7.22 | 5.06 | ARG17 DG208 | ARG17 (2) LYS20 LEU21 DG208 | ARG17 LYS20 |
| Ellagic acid | −7.06 | 6.67 | ARG17 (2) LYS20 DG208 (2) DA209 (3) DC210 | ALA192 (2) ARG17 (4) LYS20 (2) | ARG17 (2) LYS20 |
| Ferulic acid | −6.72 | 11.81 | ARG17 (2) LYS20 DA209 | ARG17 (2) LYS20 LEU21 | LYS20 |
| Brassitin | −6.7 | 12.26 | SER16 LYS20 DG208 | ALA13 ARG17 (3) LYS20 LEU21 DT207 DG208 (2) | LYS20 (2) DG208 (3) |
| Ligand | Binding Energy (kcal/mol) | Inhibition Constant (μM) | Amino Acid Interaction | ||
|---|---|---|---|---|---|
| Hydrogen Bond | Hydrophobic Bond | Other | |||
| Ethyl 4-[(4-methylpyridin-2-yl) amino] piperidine-1-carboxylate (native inhibitor) | −6.91 | 8.64 | TYR347 VAL352 | TRP346 (2) TYR347 PRO350 (2) VAL352 ASN370 | - |
| Ellagic acid | −6.32 | 23.46 | ILE265 (2) ARG266 (2) ALA351 TYR373 GLU377 ARG388 | GLN263 | ASP382 (2) |
| Aurentiacin | −6.13 | 32.37 | TYR347 ALA351 TYR373 GLU377 (2) | TRP346 (2) PRO350 VAL352 TYR373 ARG381 | - |
| Alpinetin | −6.11 | 33.02 | VAL352 TRP372 | PRO350 (2) PHE369 | - |
| Brassitin | −5.63 | 75.29 | TYR347 ALA351 GLU377 | PRO350 (2) VAL352 TYR373 (2) | TYR347 TYR373 |
| Ferulic acid | −4.71 | 323.29 | ARG199 CYS200 GLY202 PRO350 | PRO350 VAL352 | CYS200 |
| Ligand | Binding Energy (kcal/mol) | Inhibition Constant (μM) | Amino Acid Interaction | ||
|---|---|---|---|---|---|
| Hydrogen Bond | Hydrophobic Bond | Other | |||
| Tolfenamic acid (native inhibitor) | −8.13 | 1.1 | TYR385 SER530 | VAL116 VAL349 (2) LEU352 VAL523 ALA527 (4) LEU531 (2) | - |
| Alpinetin | −8.14 | 1.08 | TYR355 SER530 | VAL349 (2) LEU352 LEU359 PHE518 GLY526 ALA527 (2) LEU531 | MET522 |
| Aurentiacin | −8.03 | 1.3 | ARG120 TYR355 | MET113 VAL116 (2) VAL349 (3) LEU352 TYR355 LEU359 TRP387 GLY526 ALA527 (3) LEU531 (2) | MET522 |
| Ellagic acid | −6.89 | 8.94 | TYR355 SER530 | VAL349 (2) LEU352 (4) VAL523 (2) GLY526 (3) ALA527 (6) | - |
| Brassitin | −6.29 | 24.61 | SER530 | PHE205 TYR348 LEU352 (2) TYR385 PHE518 VAL523 (2) GLY526 (2) ALA527 (3) | TYR348 TYR385 MET522 |
| Ferulic acid | −5.07 | 193.72 | TYR355 SER353 | LEU352 PHE381 TYR385 MET522 VAL523 (2) GLY526 ALA527 (2) | MET522 |
| Compound | Molecular Weight (≤500) | #H-Bond Acceptors (≤10) | #H-Bond Donors (≤5) | MLOGP (≤4.15) | Lipinski #Violations (≤1) |
|---|---|---|---|---|---|
| Alpinetin | 194.18 | 4 | 2 | 1 | 0 |
| Aurentiacin | 298.33 | 4 | 1 | 2.31 | 0 |
| Brassitin | 302.19 | 8 | 4 | 0.14 | 0 |
| Ellagic acid | 220.29 | 1 | 2 | 1.4 | 0 |
| Ferulic acid | 270.28 | 4 | 1 | 1.52 | 0 |
| Pharmacokinetic Property | Alpinetin | Aurentiacin | Brassitin | Ellagic Acid | Ferulic Acid |
|---|---|---|---|---|---|
| GI absorption | High | High | High | High | High |
| Pgp substrate | Yes | No | No | No | No |
| log Kp (skin permeation) (cm/s) | −6.07 | −5.14 | −6.22 | −7.36 | −6.41 |
| BBB permeant | Yes | Yes | Yes | No | Yes |
| CYP1A2 inhibitor | Yes | Yes | Yes | Yes | No |
| CYP2C19 inhibitor | Yes | Yes | Yes | No | No |
| CYP2C9 inhibitor | No | Yes | No | No | No |
| CYP2D6 inhibitor | No | Yes | No | No | No |
| CYP3A4 inhibitor | Yes | Yes | No | No | No |
| Carcinogenicity (mouse) | Negative | Negative | Negative | Negative | Negative |
| hERG inhibition | Medium risk | Medium risk | Medium risk | Low risk | Medium risk |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janpaijit, S.; Sillapachaiyaporn, C.; Theerasri, A.; Charoenkiatkul, S.; Sukprasansap, M.; Tencomnao, T. Cleistocalyx nervosum var. paniala Berry Seed Protects against TNF-α-Stimulated Neuroinflammation by Inducing HO-1 and Suppressing NF-κB Mechanism in BV-2 Microglial Cells. Molecules 2023, 28, 3057. https://doi.org/10.3390/molecules28073057
Janpaijit S, Sillapachaiyaporn C, Theerasri A, Charoenkiatkul S, Sukprasansap M, Tencomnao T. Cleistocalyx nervosum var. paniala Berry Seed Protects against TNF-α-Stimulated Neuroinflammation by Inducing HO-1 and Suppressing NF-κB Mechanism in BV-2 Microglial Cells. Molecules. 2023; 28(7):3057. https://doi.org/10.3390/molecules28073057
Chicago/Turabian StyleJanpaijit, Sakawrat, Chanin Sillapachaiyaporn, Atsadang Theerasri, Somsri Charoenkiatkul, Monruedee Sukprasansap, and Tewin Tencomnao. 2023. "Cleistocalyx nervosum var. paniala Berry Seed Protects against TNF-α-Stimulated Neuroinflammation by Inducing HO-1 and Suppressing NF-κB Mechanism in BV-2 Microglial Cells" Molecules 28, no. 7: 3057. https://doi.org/10.3390/molecules28073057