5.1.9. Synthesis and Characterization of the New Compounds
2-(2′-Deoxy-2′-nitro-3′,4′,6′-tri-O-benzyl-β-d-glucopyranosyl)pyridine (2a)
Prepared from 2-nitroglucal
1 (0.50 g, 1.08 mmol) and 2-bromopyridine (0.21 mL, 0.34 g, 2.16 mmol, 2 eq.) according to general procedure I, method A. Reaction time: 1 h. Purified by column chromatography (1:4 EtOAc-hexane) to afford 0.30 g (52%) of a colorless syrup. R
f = 0.18 (1:4 EtOAc-hexane).
1H NMR (400 MHz, CDCl
3) δ (ppm): 8.57 (1H, ddd,
J = 4.9, 1.8, 0.9 Hz, H-6), 7.71 (1H, td,
J = 7.8, 1.8 Hz, H-4), 7.42 (1H, ddd,
J = 7.8, 1.8, 0.9 Hz, H-3), 7.32–7.18 (16H, m, Ar, H-5), 4.95 (1H, pt,
J = 9.8, 9.2 Hz, H-2′ or H-3′ or H-4′), 4.91 (1H, d,
J = 9.7 Hz, H-1′), 4.83, 4.61 (2 × 1H, 2 d,
J = 10.6 Hz in both, Ph
CH2), 4.81, 4.63 (2 × 1H, 2 d,
J = 10.9 Hz in both, Ph
CH2), 4.59, 4.53 (2 × 1H, 2 d,
J = 12.1 Hz in both, Ph
CH2), 4.46 (1H, pt,
J = 8.6, 8.5 Hz, H-2′ or H-3′ or H-4′), 3.82 (1H, pt,
J = 9.5, 8.5 Hz, H-2′ or H-3′ or H-4′), 3.81–3.77 (3H, m, H-5′, H-6′a,b);
13C NMR (90 MHz, CDCl
3) δ (ppm): 154.9 (C-2), 149.6 (C-6), 137.1 (C-4), 138.0, 137.7, 137.4, 128.6–127.9 (Ar), 124.1, 122.4 (C-3, C-5), 90.0, 83.2, 80.0, 77.9 (2) (C-1′–C-5′), 75.7, 75.4, 73.7 (3 × Ph
CH2), 68.7 (C-6′).
1H and
13C NMR data correspond to those reported in [
50].
3-(2′-Deoxy-2′-nitro-3′,4′,6′-tri-O-benzyl-β-d-glucopyranosyl)pyridazine (2b)
Prepared from 2-nitroglucal 1 (2.00 g, 4.33 mmol) and 3-bromopyridazine (0.83 g, 5.20 mmol, 1.2 eq.) according to general procedure I, method B. Purification of the crude product by column chromatography (1:1 EtOAc-hexane) afforded a syrup, which was triturated in a solvent mixture of EtOAc (0.5 mL) and diisopropyl ether (15 mL). The precipitated product was filtered off and washed with diisopropyl ether to afford 0.30 g (13%) of a white, amorphous solid. Rf = 0.21 (1:1 EtOAc-hexane). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.19 (1H, dd, J = 5.0, 1.7 Hz, H-6), 7.65 (1H, dd, J = 8.5, 1.7 Hz, H-4), 7.53 (1H, dd, J = 8.5, 5.0 Hz, H-5), 7.35–7.19 (15H, m, Ar), 5.19 (1H, d, J = 10.0 Hz, H-1′), 4.95 (1H, pt, J = 10.0, 9.9 Hz, H-2′ or H-3′ or H-4′), 4.85, 4.63 (2 × 1H, 2 d, J = 10.8 Hz in both, PhCH2), 4.83, 4.65 (2 × 1H, 2 d, J = 10.4 Hz in both, PhCH2), 4.58, 4.52 (2 × 1H, 2 d, J = 12.2 Hz in both, PhCH2), 4.51 (1H, pt, J = 9.8, 8.3 Hz, H-2′ or H-3′ or H-4′), 3.86 (1H, pt, J = 9.4, 8.3 Hz, H-2′ or H-3′ or H-4′), 3.83–3.74 (3H, m, H-5′, H-6′a,b); 13C NMR (100 MHz, CDCl3) δ (ppm): 158.0 (C-3), 151.9 (C-6), 137.9, 137.6, 137.2, 128.7–127.9 (Ar), 127.3, 125.5 (C-4, C-5), 89.5, 82.8, 80.0, 78.5, 77.6 (C-1′–C-5′), 75.9, 75.4, 73.7 (3 × PhCH2), 68.5 (C-6′). ESI-HRMS positive mode (m/z): calcd for C31H32N3O6+ [M+H]+ 542.2286; C31H31N3O6Na+ [M+Na]+ 564.2105; C62H62N6O12Na+ [2M+Na]+ 1105.4318. Found: [M+H]+ 542.2291; [M+Na]+ 564.2106; [2M+Na]+ 1105.4318.
2-(2′-Deoxy-2′-nitro-3′,4′,6′-tri-O-benzyl-β-d-glucopyranosyl)pyrimidine (2c)
Prepared from 2-nitroglucal 1 (1.00 g, 2.17 mmol) and 2-iodopyrimidine (0.54 g, 2.60 mmol, 1.2 eq.) according to general procedure I, method B. Purified by column chromatography (1:2 EtOAc-hexane) to afford 0.83 g (71%) of a white, amorphous solid. Rf = 0.29 (1:2 EtOAc-hexane). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.77 (2H, d, J = 4.9 Hz, H-4, H-6), 7.35–7.15 (16H, m, Ar, H-5), 5.24 (1H, pt, J = 10.1, 10.0 Hz, H-2′ or H-3′ or H-4′), 5.07 (1H, d, J = 10.0 Hz, H-1′), 4.82, 4.63 (2 × 1H, 2 d, J = 10.6 Hz in both, PhCH2), 4.82, 4.58 (2 × 1H, 2 d, J = 10.6 Hz in both, PhCH2), 4.58,4.49 (2 × 1H, 2 d, J = 12.2 Hz in both, PhCH2), 4.46 (1H, pt, J = 9.9, 9.5 Hz, H-2′ or H-3′ or H-4′), 3.86 (1H, pt, J = 9.9, 8.5 Hz, H-2′ or H-3′ or H-4′), 3.84–3.74 (3H, m, H-5′, H-6′a,b); 13C NMR (100 MHz, CDCl3) δ (ppm): 163.6 (C-2), 157.8 (C-4, C-6), 137.8, 137.6, 137.3, 128.7–127.9 (Ar), 121.3 (C-5), 88.4, 83.0, 80.6, 80.2, 77.7 (C-1′–C-5′), 75.7, 75.3, 73.6 (3 × PhCH2), 68.4 (C-6′). ESI-HRMS positive mode (m/z): calcd for C31H32N3O6+ [M+H]+ 542.2286; C31H31N3O6Na+ [M+Na]+ 562.2105. Found: [M+H]+ 542.2288; [M+Na]+ 562.2105.
2-(2′-Deoxy-2′-nitro-3′,4′,6′-tri-O-benzyl-β-d-glucopyranosyl)pyrazine (2d)
Prepared from 2-nitroglucal 1 (1.00 g, 2.17 mmol) and 2-iodopyrazine (0.54 g, 2.60 mmol, 1.2 eq.) according to general procedure I, method B. Purified by column chromatography (1:3 EtOAc-hexane) to afford 0.74 g (63%) of a white, amorphous solid. Rf = 0.54 (1:2 EtOAc-hexane). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.75 (1H, d, J = 1.4 Hz, H-3), 8.58 (1H, d, J = 2.5 Hz, H-6), 8.52 (1H, dd, J = 2.5, 1.4 Hz, H-5), 7.35–7.19 (15H, m, Ar) 5.00 (1H, d, J = 9.9 Hz, H-1′), 4.94 (1H, pt, J = 9.9, 9.7 Hz, H-2′ or H-3′ or H-4′), 4.84, 4.62 (2 × 1H, 2 d, J = 10.8 Hz in both, PhCH2), 4.82, 4.63 (2 × 1H, 2 d, J = 10.6 Hz in both, PhCH2), 4.59, 4.53 (2 × 1H, 2 d, J = 12.1 Hz in both, PhCH2), 4.45 (1H, pt, J = 8.7, 8.7 Hz, H-2′ or H-3′ or H-4′), 3.83 (1H, pt, J = 9.4, 8.7 Hz, H-2′ or H-3′ or H-4′), 3.82–3.76 (3H, m, H-5′, H-6′a,b); 13C NMR (100 MHz, CDCl3) δ (ppm): 150.7 (C-2), 145.3, 144.2, 143.9 (C-3, C-5, C-6), 137.8, 137.6, 137.2, 128.7–127.9 (Ar), 89.2, 83.0, 80.1, 77.8, 77.6 (C-1′–C-5′), 75.9, 75.4, 73.7 (3 × PhCH2), 68.5 (C-6′). ESI-HRMS positive mode (m/z): calcd for C31H31N3O6Na+ [M+Na]+ 564.2105. Found: 564.2107.
2-(2′-Deoxy-2′-nitro-3′,4′,6′-tri-O-benzyl-β-d-glucopyranosyl)quinoline (2e)
Prepared from 2-nitroglucal 1 (2.00 g, 4.33 mmol) and 2-bromoquinoline (1.83 g, 8.80 mmol, 2 eq.) according to general procedure I, method A. Reaction time: 2 h. Purified by column chromatography (1:9 EtOAc-hexane) to afford 1.53 g (60%) of a colorless syrup. Rf = 0.35 (1:4 EtOAc-hexane). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.18 (1H, d, J = 8.5 Hz, H-3 or H-4), 8.04 (1H, dd, J = 8.5, 1.0 Hz, H-5 or H-8), 7.79 (1H, dd, J = 8.2, 1.4 Hz, H-5 or H-8), 7.69 (1H, ddd, J = 8.5, 7.0, 1.4 Hz, H-6 or H-7), 7.57 (1H, d, J = 8.5 Hz, H-3 or H-4), 7.53 (1H, ddd, J = 8.2, 7.0, 1.0 Hz, H-6 or H-7), 7.35–7.20 (15H, m, Ar), 5.13 (1H, d, J = 9.7 Hz, H-1′), 5.10 (1H, pt, J = 9.8, 8.0 Hz, H-2′ or H-3′ or H-4′), 4.86, 4.65 (2 × 1H, 2 d, J = 10.9 Hz in both, PhCH2), 4.84, 4.64 (2 × 1H, 2 d, J = 10.6 Hz in both, PhCH2), 4.61, 4.53 (2 × 1H, 2 d, J = 12.2 Hz in both, PhCH2), 4.50 (1H, pt, J = 8.5, 8.4 Hz, H-2′ or H-3′ or H-4′), 4.88 (1H, pt, J = 9.5, 8.4 Hz, H-2′ or H-3′ or H-4′), 3.86–3.80 (3H, m, H-5′, H-6′a,b); 13C NMR (100 MHz, CDCl3) δ (ppm): 155.0, 147.3 (C-2, C-8a), 138.0, 137.7, 137.3, 137.4, 130.0, 129.9, 128.7–127.8, 127.6, 127.2, 119.4 (Ar, C-3–C-8, C-4a), 89.3, 83.3, 80.1, 80.0, 77.8 (C-1′–C-5′), 75.8, 75.4, 73.7 (3 × PhCH2), 68.6 (C-6′). ESI-HRMS positive mode (m/z): calcd for C36H34N2O6Na+ [M+Na]+ 613.2309. Found: 613.2309.
2-(2′-Amino-2′-deoxy-3′,4′,6′-tri-O-benzyl-β-d-glucopyranosyl)pyridine (3a)
Compound 2a (0.19 g, 0.35 mmol) and Zn powder (0.69 g, 10.55 mmol, 30 eq.) were suspended in a solvent mixture of THF (10 mL) and water (5 mL). This suspension was cooled down in an ice bath, and ccHCl solution was added (0.7 mL, 8.14 mmol, 23 eq.). The reaction mixture was stirred at rt until the TLC (1:1 EtOAc-hexane) showed total consumption of the starting material (1 h). The reaction was quenched by the addition of sat. aq. NaHCO3 solution (50 mL). The insoluble inorganic salts and the rest of the Zn were filtered off, and the remaining solution was extracted with CH2Cl2 (2 × 50 mL). The combined organic phase was extracted with water (50 mL), then with brine (50 mL), dried over MgSO4 and filtered. The solvent was removed under reduced pressure. The residue was purified by column chromatography (95:5 CHCl3-MeOH) to afford114 mg (64%) of a pale yellow amorphous solid. Rf = 0.34 (95:5 CHCl3-MeOH); [α]D = +30 (c 0.5, CHCl3). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.56 (1H, ddd, J = 4.9, 1.8, 1.0 Hz, H-6), 7.70 (1H, td, J = 7.7, 1.8 Hz, H-4), 7.47 (1H, ddd, J = 7.7, 1.8, 1.0 Hz, H-3), 7.37–7.20 (16H, m, Ar, H-5), 5.01, 4.80 (2 × 1H, 2 d, J = 11.4 Hz in both, PhCH2), 4.84, 4.63 (2 × 1H, 2 d, J = 10.7 Hz in both, PhCH2), 4.62, 4.56 (2 × 1H, 2 d, J = 12.2 Hz in both, PhCH2), 4.27 (1H, d, J = 9.6 Hz, H-1′), 3.80–3.75 (3H, m, H-4′, H-6′a,b), 3.70 (1H, m, H-5′), 3.62 (1H, pt, J = 9.2, 9.2 Hz, H-3′), 3.22 (1H, pt, J = 9.7, 9.6 Hz, H-2′), 1.65 (2H, s, NH2); 13C NMR (90 MHz, CDCl3) δ (ppm): 158.7 (C-2), 148.9 (C-6), 138.7, 138.3, 138.2 (Ar), 137.0 (C-4), 128.6–127.7 (Ar), 123.2, 122.7 (C-3, C-5), 87.0, 83.0, 79.8, 78.9 (C-1′, C-3′–C-5′), 75.5, 75.0, 73.6 (3 × PhCH2), 69.4 (C-6′), 57.2 (C-2′). ESI-HRMS positive mode (m/z): calcd for C32H34N2O4Na+ [M+Na]+ 533.2410. Found: 533.2411.
2-(2′-Amino-2′-deoxy-3′,4′,6′-tri-O-benzyl-β-d-glucopyranosyl)pyrazine (3d)
Compound 2d (0.10 g, 0.19 mmol) and Zn powder (0.12 g, 1.84 mmol, 10 eq.) were suspended in a solvent mixture of THF (5 mL) and water (2.5 mL). To this stirred mixture, a 1 M aq. solution of HCl (1.5 mL, 1.50 mmol, 8 eq.) was added dropwise over 1 h using a syringe pump. The reaction mixture was further stirred at rt until TLC (95:5 CHCl3-MeOH) indicated the completion of the reaction (2 h). The reaction was quenched by the addition of sat. aq. NaHCO3 solution (25 mL). The insoluble inorganic salts and the rest of the Zn were filtered off, and the remaining solution was extracted with CH2Cl2 (2 × 25 mL). The combined organic phase was extracted with water (25 mL), dried over MgSO4 and filtered. The solvent was removed under reduced pressure. Column chromatographic purification of the residue (95:5 CHCl3-MeOH) afforded 6.8 mg (7%) of a pale yellow syrup. Rf = 0.23 (95:5 CHCl3-MeOH); [α]D = +19 (c 0.1, CHCl3). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.77 (1H, d, J = 1.0 Hz, H-3), 8.53–8.52 (2H, m, H-5, H-6), 7.35–7.21 (15H, m, Ar), 5.03, 4.78 (2 × 1H, 2 d, J = 11.4 Hz in both, PhCH2), 4.85, 4.64 (2 × 1H, 2 d, J = 10.8 Hz in both, PhCH2), 4.61, 4.56 (2 × 1H, 2 d, J = 12.3 Hz in both, PhCH2), 4.31 (1H, d, J = 9.7 Hz, H-1′), 3.81–3.73 (2H, m, H-6′a,b), 3.77 (1H, pt, J = 9.8, 8.5 Hz, H-3′ or H-4′), 3.71 (1H, m, H-5′), 3.60 (1H, pt, J = 9.2, 9.0 Hz, H-3′ or H-4′), 3.25 (1H, pt, J = 9.7, 9.6 Hz, H-2′), 1.68 (2H, br s, NH2); 13C NMR (100 MHz, CDCl3) δ (ppm): 154.2 (C-2), 145.0, 144.3, 143.5 (C-3, C-5, C-6), 138.6, 138.2, 138.1, 129.9–127.8 (Ar), 86.9, 81.4, 80.0, 78.8 (C-1′, C-3′–C-5′), 75.6, 75.0, 73.7 (3 × PhCH2), 69.2 (C-6′), 56.8 (C-2′). ESI-HRMS positive mode (m/z): calcd for C31H34N3O4+ [M+H]+ 512.2544; C31H33N3O4Na+ [M+Na]+ 534.2363. Found: [M+H]+ 512.2541, [M+Na]+ 534.2359.
2-(2′-Deoxy-2′-nitro-β-d-glucopyranosyl)pyridine (4a)
Prepared from compound 2a (0.30 g, 0.55 mmol) according to general procedure II. Reaction time: 0.5 h. Purification by column chromatography (9:1 CHCl3-MeOH) yielded 128 mg (85%) of a white, amorphous solid. Rf = 0.53 (4:1 CHCl3-MeOH). 1H NMR (400 MHz, CD3OD) δ (ppm): 8.53 (1H, d, J = 4.4 Hz, H-6), 7.85 (1H, t, J = 7.7 Hz, H-4), 7.54 (1H, d, J = 7.8 Hz, H-3), 7.40 (1H, m, H-5), 4.95 (1H, d, J = 9.7 Hz, H-1′), 4.74 (1H, pt, J = 10.1, 9.9 Hz, H-2′ or H-3′ or H-4′), 4.21 (1H, pt, J = 9.1, 8.7 Hz, H-2′ or H-3′ or H-4′), 3.91 (1H, m, H-6′a), 3.76 (1H, dd, J = 12.0, 4.2 Hz, H-6′b), 3.62–3.55 (2H, m, H-2′ or H-3′ or H-4′, H-5′); 13C NMR (90 MHz, CD3OD) δ (ppm): 156.4 (C-2), 150.2 (C-6), 138.9 (C-4), 125.6, 124.4 (C-3, C-5), 92.7, 82.7, 80.7, 76.4, 71.1 (C-1′–C-5′), 62.4 (C-6′). ESI-HRMS positive mode (m/z): calcd for C11H14N2O6Na+ [M+Na]+ 293.0744. Found: 293.0744.
3-(2′-Deoxy-2′-nitro-β-d-glucopyranosyl)pyridazine (4b)
Prepared from compound 2b (0.30 g, 0.55 mmol) according to general procedure II. Reaction time: 0.5 h. Purification by column chromatography (9:1 CHCl3-MeOH) yielded 133 mg (89%) of a white, amorphous solid. Rf = 0.39 (4:1 CHCl3-MeOH). 1H NMR (360 MHz, CD3OD) δ (ppm): 9.17 (1H, d, J = 4.9 Hz, H-6), 8.00 (1H, dd, J = 8.6, 1.4 Hz, H-4), 7.79 (1H, dd, J = 8.6, 4.9 Hz, H-5), 5.22 (1H, d, J = 10.0 Hz, H-1′), 4.82 (1H, pt, J = 10.1, 10.0 Hz, H-2′ or H-3′ or H-4′), 4.25 (1H, pt, J = 10.0, 9.9 Hz, H-2′ or H-3′ or H-4′), 3.94 (1H, dd, J = 12.2, 2.1 Hz, H-6′a), 3.77 (1H, dd, J = 12.2, 5.3 Hz, H-6′b), 3.67 (1H, ddd, J = 9.8, 5.3, 2.1 Hz, H-5′), 3.57 (1H, pt, J = 9.8, 9.5 Hz, H-2′ or H-3′ or H-4′); 13C NMR (90 MHz, CD3OD) δ (ppm): 160.3 (C-3), 153.0 (C-6), 129.7, 128.2 (C-4, C-5), 91.9, 82.9, 79.2, 76.3, 71.0 (C-1′–C-5′), 62.4 (C-6′). ESI-HRMS positive mode (m/z): calcd for C10H13N3O6Na+ [M+Na]+ 294.0697. Found: 294.0698.
2-(2′-Deoxy-2′-nitro-β-d-glucopyranosyl)pyrimidine (4c)
Prepared from compound 2c (0.10 g, 0.18 mmol) according to general procedure II. Reaction time: 0.5 h. Purification by column chromatography (9:1 CHCl3-MeOH) yielded 49 mg (98%) of a white, amorphous solid. Rf = 0.52 (2:1 CHCl3-MeOH). 1H NMR (360 MHz, CD3OD) δ (ppm): 8.99 (2H, d, J = 5.0 Hz, H-4, H-6), 7.70 (1H, t, J = 5.0 Hz, H-5), 5.24 (1H, d, J = 10.1 Hz, H-1′), 4.85 (1H, pt, J = 10.1, 10.0 Hz, H-2′ or H-3′ or H-4′), 4.20 (1H, pt, J = 10.0, 9.9 Hz, H-2′ or H-3′ or H-4′), 3.96 (1H, dd, J = 12.2, 2.1 Hz, H-6′a), 3.76 (1H, dd, J = 12.2, 5.5 Hz, H-6′b), 3.68 (1H, ddd, J = 9.5, 5.5, 2.1 Hz, H-5′), 3.52 (1H, pt, J = 9.9, 9.5 Hz, H-2′ or H-3′ or H-4′); 13C NMR (90 MHz, CD3OD) δ (ppm): 165.2 (C-2), 159.0 (C-4, C-6), 122.8 (C-5), 91.2, 82.9, 80.8, 76.3, 71.0 (C-1′–C-5′), 62.4 (C-6′). ESI-HRMS positive mode (m/z): calcd for C10H13N3O6Na+ [M+Na]+ 294.0697. Found: 294.0698.
2-(2′-Deoxy-2′-nitro-β-d-glucopyranosyl)pyrazine(4d)
Prepared from compound 2d (0.30 g, 0.55 mmol) according to general procedure II. Reaction time: 0.5 h. Purification by column chromatography (9:1 CHCl3-MeOH) yielded 117 mg (78%) of a white, amorphous solid. Rf = 0.48 (4:1 CHCl3-MeOH). 1H NMR (400 MHz, CD3OD) δ (ppm): 8.83 (1H, d, J = 1.5 Hz, H-3), 8.60 (1H, d, J = 2.6 Hz, H-6), 8.57 (1H, dd, J = 2.6, 1.5 Hz, H-5), 5.10 (1H, d, J = 9.9 Hz, H-1′), 4.81 (1H, pt, J = 10.0, 10.0 Hz, H-2′ or H-3′ or H-4′), 4.21 (1H, pt, J = 10.0, 9.7 Hz, H-2′ or H-3′ or H-4′), 3.93 (1H, dd, J = 12.2, 2.1 Hz, H-6′a), 3.76 (1H, dd, J = 12.2, 5.4 Hz, H-6′b), 3.64 (1H, ddd, J = 10.0, 5.4, 2.1 Hz, H-5′), 3.54 (1H, pt, J = 9.4, 9.3 Hz, H-2′ or H-3′ or H-4′); 13C NMR (90 MHz, CD3OD) δ (ppm): 152.9 (C-2), 146.2, 145.5, 145.2 (C-3, C-5, C-6), 91.6, 82.9, 78.6, 76.5, 71.1 (C-1′–C-5′), 62.4 (C-6′). ESI-HRMS positive mode (m/z): calcd for C10H13N3O6Na+ [M+Na]+ 294.0697. Found: 294.0698.
2-(2′-Deoxy-2′-nitro-β-d-glucopyranosyl)quinoline (4e)
Prepared from compound 2e (83 mg, 0.14 mmol) according to general procedure II. Reaction time: 1 h. Purification by column chromatography (9:1 CHCl3-MeOH) yielded 31 mg (76%) of a white, amorphous solid. Rf = 0.19 (9:1 CHCl3-MeOH). 1H NMR (400 MHz, CD3OD) δ (ppm): 8.36, 7.70 (2 × 1H, 2 d, J = 8.5 Hz in both, H-3, H-4), 8.00, 7.93 (2 ×1H, 2 d, J = 7.9 Hz in both, H-5, H-8), 7.77, 7.61 (2 × 1H, 2 t, J = 7.9 Hz in both, H-6, H-7), 5.14 (1H, d, J = 9.9 Hz, H-1′), 4.90 (1H, pt, J = 10.1, 10.0 Hz, H-2′ or H-3′ or H-4′), 4.27 (1H, pt, J = 9.7, 9.0 Hz, H-2′ or H-3′ or H-4′), 3.96 (1H, dd, J = 12.2, < 1Hz, H-6′a), 3.80 (1H, dd, J = 12.2, 4.7 Hz, H-6′b), 3.67 (1H, m, H-5′), 3.61 (1H, pt, J = 9.2, 9.1 Hz, H-2′ or H-3′ or H-4′); 13C NMR (90 MHz, CD3OD) δ (ppm): 157.2, 148.3 (C-2, C-8a), 138.9, 131.2, 129.7, 129.0, 128.4, 121.2 (C-3–C-8), 129.5 (C-4a), 92.3, 82.9, 81.1, 76.6, 71.2 (C-1′–C-5′), 62.5 (C-6′). ESI-HRMS positive mode (m/z): calcd for C15H16N2O6Na+ [M+Na]+ 343.0901. Found: 343.0900.
2-(2′-Amino-2′-deoxy-β-d-glucopyranosyl)pyridine (5a)
Method A: Prepared from compound 3a (65 mg, 0.13 mmol) according to general procedure II. Reaction time: 0.5 h. Purification by column chromatography (7:3 CHCl3-MeOH) yielded 15 mg (42%) of a white, amorphous solid.
Method B: Compound 3a (0.22 g, 0.43 mmol) and pentamethylbenzene (0.58 g, 3.88 mmol, 9 eq.) were dissolved in dry CH2Cl2 (22 mL), and the solution was cooled down to −78 °C. To this solution, a 1 M solution of BCl3 in CH2Cl2 (1.72 mL, 1.72 mmol, 4 eq.) was added dropwise over 5 min. The reaction mixture was stirred at this temperature until the TLC (9:1 CHCl3-MeOH) showed the completion of the reaction (0.5 h). Then, the reaction was quenched by the addition of MeOH (10 mL) and allowed to warm to rt. The solvents were then removed under reduced pressure. Purification of the residue by column chromatography (7:3 CHCl3-MeOH) yielded 113 mg (95%) of a white, amorphous solid. Rf = 0.23 (7:3 CHCl3-MeOH); [α]D = +84 (c 0.5, MeOH). 1H NMR (400 MHz, CD3OD) δ (ppm): 8.57 (1H, d, J = 4.5 Hz, H-6), 7.90 (1H, td, J = 7.8, 1.7 Hz, H-4), 7.72 (1H, d, J = 7.8 Hz, H-3), 7.41 (1H, dd, J = 7.5, 5.0 Hz, H-5), 4.57 (1H, d, J = 10.0 Hz, H-1′), 3.94 (1H, dd, J = 12.1, 1.8 Hz, H-6′a), 3.76 (1H, dd, J = 12.1, 4.9 Hz, H-6′b), 3.66 (1H, pt, J = 9.0, 8.9 Hz, H-3′), 3.54–3.45 (2H, m, H-4′, H-5′), 3.21 (1H, pt, J = 10.0, 9.9 Hz, H-2′); 13C NMR (90 MHz, CD3OD) δ (ppm): 158.7 (C-2), 149.6 (C-6), 139.1 (C-4), 125.1, 123.9 (C-3, C-5), 82.5, 78.7, 76.4, 71.6 (C-1′, C-3′–C-5′), 62.7 (C-6′), 57.6 (C-2′). ESI-HRMS positive mode (m/z): calcd for C11H17N2O4+ [M+H]+ 241.1183; C11H16N2O4Na+ [M+Na]+ 263.1001. Found: [M +H]+ 241.1183; [M+Na]+ 263.1002.
2-(2′-Amino-2′-deoxy-β-d-glucopyranosyl)pyrimidine (5c)
A degassed, vigorously stirred suspension of 10% Pd(C) (56 mg) in dry EtOH (11 mL) was saturated with H2, and compound 4c (0.11 g, 0.41 mmol) was added. The reaction mixture was heated at reflux temperature until the TLC (3:2 CHCl3-MeOH) indicated complete conversion of the starting material. After completion of the reaction (2 h), the catalyst was filtered off through a pad of celite and washed with MeOH. The resulting solution was then evaporated under reduced pressure. Purification of the remaining crude product by column chromatography (3:2 CHCl3-MeOH) yielded 37 mg (38%) of a white, amorphous solid. Rf = 0.10 (3:2 CHCl3-MeOH); [α]D = +22 (c 0.1, MeOH). 1H NMR (400 MHz, CD3OD) δ (ppm): 8.85 (2H, d, J = 4.9 Hz, H-4, H-6), 7.48 (1H, t, J = 4.9 Hz, H-5), 4.42 (1H, d, J = 9.7 Hz, H-1′), 3.87 (1H, dd, J = 12.1, 1.5 Hz, H-6′a), 3.73 (1H, dd, J = 12.1, 4.5 Hz, H-6′b), 3.50 (1H, pt, J = 9.5, 9.0 Hz, H-3′ or H-4′), 3.50–3.45 (1H, m, H-5′), 3.44 (1H, pt, J = 9.2, 9.0 Hz, H-3′ or H-4′), 3.08 (1H, pt, J = 9.5, 9.4 Hz, H-2′); 13C NMR (90 MHz, CD3OD) δ (ppm): 167.7 (C-2), 158.8 (C-4, C-6), 122.2 (C-5), 83.8, 82.6, 79.3, 71.4 (C-1′, C-3′–C-5′), 62.8 (C-6′), 57.6 (C-2′). ESI-HRMS positive mode (m/z): calcd for C10H15N3O4Na+ [M+Na]+ 264.0955. Found: 264.0957.
2-(2′-Amino-2′-deoxy-β-d-glucopyranosyl)pyrazine (5d)
A degassed, vigorously stirred suspension of 10% Pd(C) (60 mg) in dry EtOH (12 mL) was saturated with H2, and compound 4d (0.12 g, 0.43 mmol) was added. The reaction mixture was heated at reflux temperature until the TLC (3:2 CHCl3-MeOH) indicated complete conversion of the starting material. After completion of the reaction (6 h), the catalyst was filtered off through a pad of celite and washed with MeOH. The resulting solution was then evaporated under reduced pressure. Purification of the remaining crude product by column chromatography (7:3 CHCl3-MeOH) yielded 68 mg (66%) of a white, amorphous solid. Rf = 0.15 (3:2 CHCl3-MeOH); [α]D = +41 (c 0.5, MeOH). 1H NMR (400 MHz, CD3OD) δ (ppm): 8.79 (1H, d, J = 1.3 Hz, H-3), 8.62 (1H, dd, J = 2.4, 1.3 Hz, H-5), 8.57 (1H, d, J = 2.4 Hz, H-6), 4.38 (1H, d, J = 9.7 Hz, H-1′), 3.90 (1H, dd, J = 12.1, 1.7 Hz, H-6′a), 3.73 (1H, dd, J = 12.1, 5.0 Hz, H-6′b), 3.51–3.40 (3H, m, H-3′, H-4′, H-5′), 3.00 (1H, pt, J = 9.5, 9.4 Hz, H-2′); 13C NMR (90 MHz, CD3OD) δ (ppm): 155.4 (C-2), 146.0, 145.4, 145.1 (C-3, C-5, C-6), 82.6, 81.4, 79.2, 71.6 (C-1′, C-3′–C-5′), 62.8 (C-6′), 58.0 (C-2′). ESI-HRMS positive mode (m/z): calcd for C10H16N3O4Na+ [M+H]+ 242.1135. Found: 242.1133.
2-(2′-Amino-2′-deoxy-β-d-glucopyranosyl)quinoline (5e)
Compound 4e (0.10 g, 0.33 mmol) and tin powder (1.17 g, 9.82 mmol, 30 eq.) were suspended in a solvent mixture of THF (5 mL) and water (2.5 mL). This heterogenous mixture was cooled down in an ice bath, and ccHCl solution (0.85 mL, 9.88 mmol 30 eq.) was added. The reaction mixture was then stirred at rt. When the TLC (4:1 CHCl3-MeOH) showed complete conversion of 4e (1 d), a 2 M aq. solution of NaOH was added to the reaction mixture to obtain a slightly basic solution, which was then neutralized by the addition of sat. aq. NH4Cl solution. The solvents were removed under diminished pressure. The residue was treated with MeOH (20 mL), and the inseparable inorganic salts and the excess of the unreacted Sn were filtered off. The resulting solution was evaporated in vacuo. Column chromatographic purification of the residue (9:1 CHCl3-MeOH) yielded 28 mg (29%) of a pale yellow, amorphous solid. Rf = 0.11 (4:1 CHCl3-MeOH); [α]D = −11 (c 0.1, MeOH). 1H NMR (400 MHz, CD3OD) δ (ppm): 8.34 (1H, d, J = 8.5 Hz, H-4), 8.06 (1H, dd, J = 8.5, 1.2 Hz, H-8), 7.92 (1H, dd, J = 8.2, 1.4 Hz, H-5), 7.76 (1H, ddd, J = 8.5, 6.8, 1.4 Hz, H-7), 7.71 (1H, d, J = 8.5 Hz, H-3), 7.59 (1H, ddd, J = 8.2, 6.8, 1.2 Hz, H-6), 4.50 (1H, d, J = 9.7 Hz, H-1′), 3.94 (1H, dd, J = 12.2, 1.3 Hz, H-6′a), 3.79 (1H, ddd, J = 12.2, 3.2, 1.3 Hz, H-5′), 3.56–3.50 (3H, m, H-3′, H-4′, H-6′b), 3.12 (1H, pt, J = 9.5, 9.4 Hz, H-2′); 13C NMR (90 MHz, CD3OD) δ (ppm): 160.1, 148.3 (C-2, C-8a), 138.7, 131.0, 129.6, 129.0, 128.0, 121.7 (C-3–C-8), 129.4 (C-4a), 83.3, 82.6, 79.1, 71.7 (C-1′, C-3′–C-5′), 62.9 (C-6′) 58.3(C-2′). ESI-HRMS positive mode (m/z): calcd for C15H18N2O4Na+ [M+Na]+ 313.1159; C30H36N4O8Na+ [2M+Na]+ 603.2425. Found: [M+Na]+ 313.1158; [2M+Na]+ 603.2425.
2-(2′-Deoxy-2′-nitro-3′,4′,6′-tri-O-benzoyl-β-d-glucopyranosyl)pyridine (6a)
Prepared from compound 2a (95 mg, 0.18 mmol) and benzoyl chloride (0.13 mL, 1.12 mmol, 6 eq.) according to general procedure III. Reaction time: 5 d. Purified by column chromatography (1:4 EtOAc-hexane) yielded 90 mg (88%) of a white, amorphous solid. Rf = 0.38 (1:2 EtOAc-hexane). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.60 (1H, ddd, J = 4.9, 1.5, 0.9 Hz, H-6), 8.14–7.29 (18H, m, Ar, H-3, H-4, H-5), 6.39 (1H, pt, J = 10.0, 9.7 Hz, H-2′ or H-3′ or H-4′), 5.76 (1H, pt, J = 9.8, 9.8 Hz, H-2′ or H-3′ or H-4′), 5.46 (1H, pt, J = 10.1, 10.1 Hz, H-2′ or H-3′ or H-4′), 5.28 (1H, d, J = 9.9 Hz, H-1′), 4.66 (1H, dd, J = 12.4, 3.1 Hz, H-6′a), 4.53 (1H, dd, J = 12.4, 5.3 Hz, H-6′b), 4.41 (1H, ddd, J = 10.1, 5.3, 3.1 Hz, H-5′); 13C NMR (100 MHz, CDCl3) δ (ppm): 166.2, 165.4, 165.2 (3 × C=O), 153.8 (C-2), 149.7 (C-6), 137.4 (C-4), 133.8 (2), 133.3, 130.1–128.4 (Ar), 124.6, 123.1 (C-3, C-5), 87.2, 79.5, 76.9, 73.3, 69.4 (C-1′–C-5′), 63.3 (C-6′). ESI-HRMS positive mode (m/z): calcd for C32H27N2O9+ [M+H]+ 583.1711; C32H26N2O9Na+ [M+Na]+ 605.1531. Found: [M+H]+ 583.1713; [M+Na]+ 605.1532.
3-(2′-Deoxy-2′-nitro-3′,4′,6′-tri-O-benzoyl-β-d-glucopyranosyl)pyridazine (6b)
Prepared from compound 2b (50 mg, 0.092 mmol) and benzoyl chloride (64 μL, 0.55 mmol, 6 eq.) according to general procedure III. Reaction time: 10 d. Purification by column chromatography (1:1 EtOAc-hexane) yielded 52 mg (97%) of a white, amorphous solid. Rf = 0.32 (1:1 EtOAc-hexane). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.21 (1H, d, J = 5.0 Hz, H-6), 8.01–7.33 (17H, m, Ar, H-4, H-5), 6.45 (1H, pt, J = 9.8, 9.2 Hz, H-2′ or H-3′ or H-4′), 5.79 (1H, pt, J = 9.5, 9.5 Hz, H-2′ or H-3′ or H-4′), 5.60–5.49 (2H, m, H-1′, H-2′ or H-3′ or H-4′), 4.73–4.43 (3H, m, H-5′, H-6′a,b); 13C NMR (100 MHz, CDCl3) δ (ppm): 166.2, 165.3, 165.2 (3 × C=O), 157.0 (C-3), 152.1 (C-6), 133.9, 133.4, 130.1–129.8, 129.4, 128.6–128.5, 128.4, 128.2 (Ar), 127.5, 126.2 (C-4, C-5), 86.4, 78.0, 77.0, 73.0, 69.0 (C-1′–C-5′), 62.9 (C-6′). ESI-HRMS positive mode (m/z): calcd for C31H25N3O9Na+ [M+Na]+ 606.1483. Found: 606.1479.
2-(2′-Deoxy-2′-nitro-3′,4′,6′-tri-O-benzoyl-β-d-glucopyranosyl)pyrimidine (6c)
Prepared from compound 2c (0.50 g, 0.92 mmol) and benzoyl chloride (0.65 mL, 5.60 mmol, 6 eq.) according to general procedure III. Reaction time: 30 d. Purification by column chromatography (1:2 EtOAc-hexane) yielded 0.24 g (45%) of a white, amorphous solid. Rf = 0.34 (1:2 EtOAc-hexane). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.81 (2H, d, J = 4.8 Hz, H-4, H-6), 7.98–7.33 (16H, m, Ar, H-5), 6.39 (1H, pt, J = 10.0, 9.7 Hz, H-2′ or H-3′ or H-4′), 5.80 (1H, pt, J = 9.7, 9.6 Hz, H-2′ or H-3′ or H-4′), 5.68 (1H, pt, J = 10.2, 10.2 Hz, H-2′ or H-3′ or H-4′), 5.42 (1H, d, J = 10.0 Hz, H-1′), 4.65 (1H, dd, J = 12.3, 3.0 Hz, H-6′a), 4.52 (1H, dd, J = 12.3, 5.0 Hz, H-6′b), 4.44 (1H, ddd, J = 10.0, 5.0, 3.0 Hz, H-5′); 13C NMR (100 MHz, CDCl3) δ (ppm): 166.2, 165.3 (2), 162.8 (3 × C=O, C-2), 157.9 (C-4, C-6), 133.8, 133.2, 130.1–128.4 (Ar), 121.5 (C-5), 85.8, 80.4, 77.2, 73.0, 69.2 (C-1′–C-5′), 63.2 (C-6′). ESI-HRMS positive mode (m/z): calcd for C31H25N3O9Na+ [M+Na]+ 606.1483. Found: 606.1483.
2-(2′-Deoxy-2′-nitro-3′,4′,6′-tri-O-benzoyl-β-d-glucopyranosyl)pyrazine (6d)
Prepared from compound 2d (50 mg, 0.092 mmol) and benzoyl chloride (64 μL, 0.55 mmol, 6 eq.) according to general procedure III. Reaction time: 5 d. Purification by column chromatography (1:2 EtOAc-hexane) yielded 44 mg (82%) of a white, amorphous solid. Rf = 0.14 (1:2 EtOAc-hexane). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.83 (1H, d, J = 1.5 Hz, H-3), 8.62 (1H, d, J = 2.5 Hz, H-6), 8.55 (1H, dd, J = 2.5, 1.5 Hz, H-5), 8.01–7.35 (15H, m, Ar), 6.38 (1H, pt, J = 9.4, 9.3 Hz, H-2′ or H-3′ or H-4′), 5.74 (1H, pt, J = 9.7, 9.7 Hz, H-2′ or H-3′ or H-4′), 5.43 (1H, pt, J = 9.9, 9.5 Hz, H-2′ or H-3′ or H-4′), 5.39 (1H, d, J = 9.8 Hz, H-1′), 4.68 (1H, dd, J = 12.3, 2.9 Hz, H-6′a), 4.52 (1H, dd, J = 12.3, 5.4 Hz, H-6′b), 4.28 (1H, ddd, J = 10.0, 5.4, 2.9 Hz, H-5′); 13C NMR (100 MHz, CDCl3) δ (ppm): 166.2, 165.3, 165.2 (3 × C=O), 149.7 (C-2), 145.7, 144.7, 144.0 (C-3, C-5, C-6), 133.9, 133.4, 130.1–129.9, 129.5, 128.7–128.6, 128.5, 128.3 (Ar), 86.3, 77.4, 77.1, 73.1, 69.1 (C-1′–C-5′), 63.0 (C-6′). ESI-HRMS positive mode (m/z): calcd for C31H26N3O9+ [M+H]+ 584.1664; C31H25N3O9Na+ [M+Na]+ 606.1483. Found: [M+H]+ 584.1659; [M+Na]+ 606.1477.
2-(2′-Amino-2′-deoxy-3′,4′,6′-tri-O-benzoyl-β-d-glucopyranosyl)pyridine (7a)
Compound 6a (0.10 g, 0.17 mmol) and Zn powder (0.11 g, 1.71 mmol, 10 eq.) were suspended in a solvent mixture of THF (10 mL) and water (5 mL). To this stirred mixture, a 2 M aq. solution of HCl was added (2.6 mL, 5.14 mmol, 30 eq.). The reaction mixture was further stirred at rt until the TLC (95:5 CHCl3-MeOH) showed total consumption of the starting material (5 h). The reaction was quenched by the addition of sat. aq. NaHCO3 solution (50 mL). The insoluble inorganic salts and the rest of the Zn were filtered off, and the remaining solution was extracted with CH2Cl2 (2 × 50 mL). The combined organic phase was extracted with water (50 mL), then with brine (50 mL), dried over MgSO4 and filtered. The solvent was removed under reduced pressure. The residue was purified by column chromatography (100:1 CHCl3-MeOH) to yield 36 mg (38%) of a white, amorphous solid. Rf = 0.47 (50:1 CHCl3-MeOH); [α]D = −16 (c 0.5, CHCl3). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.60 (1H, ddd, J = 4.9, 1.9, 0.9 Hz, H-6), 8.01–7.25 (18H, m, Ar, H-3, H-4, H-5), 5.71 (1H, pt, J = 9.5, 9.5 Hz, H-4′), 5.66 (1H, pt, J = 9.5, 9.5 Hz, H-3′), 4.63 (1H, dd, J = 12.2, 3.1 Hz, H-6′a), 4.53 (1H, d, J = 9.5 Hz, H-1′), 4.52 (1H, dd, J = 12.2, 5.3 Hz, H-6′b), 4.25 (1H, ddd, J = 9.4, 5.3, 3.1 Hz, H-5′), 3.58 (1H, pt, J = 9.7, 9.6 Hz, H-2′), 1.71 (2H, s, NH2); 13C NMR (100 MHz, CDCl3) δ (ppm): 166.6, 166.3, 165.7 (3 × C=O), 157.7 (C-2), 149.1 (C-6), 137.2 (C-4), 133.4, 133.3, 133.1, 129.9–128.4 (Ar), 123.6, 122.9 (C-3, C-5), 83.9, 77.7, 76.6, 70.3 (C-1′, C-3′–C-5′), 64.0 (C-6′), 56.7 (C-2′). ESI-HRMS positive mode (m/z): calcd for C32H29N2O7+ [M+H]+ 553.1969; C32H28N2O7Na+ [M+Na]+ 575.1789. Found: [M+H]+ 553.1970; [M+Na]+ 575.1789.
3-(2′-Amino-2′-deoxy-3′,4′,6′-tri-O-benzoyl-β-d-glucopyranosyl)pyridazine (7b)
Prepared from compound 9b (105 mg, 0.16 mmol) according to general procedure VI. Purification by column chromatography (95:5 CHCl3-MeOH) yielded 78 mg (88%) of a white, amorphous solid. Rf = 0.31 (95:5 CHCl3-MeOH); [α]D = +7 (c 0.5, CHCl3). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.17 (1H, d, J = 5.0 Hz, H-6), 8.02–7.32 (17H, m, Ar, H-4, H-5), 5.70 (2H, m, H-3′, H-4′), 4.82 (1H, d, J = 9.7 Hz, H-1′), 4.65 (1H, dd, J = 12.2, 2.9 Hz, H-6′a), 4.52 (1H, dd, J = 12.2, 5.2 Hz, H-6′b), 4.32–4.27 (1H, m, H-5′), 3.55 (1H, pt, J = 9.7, 9.6 Hz, H-2′), 1.78 (2H, s, NH2); 13C NMR (100 MHz, CDCl3) δ (ppm): 166.6, 166.3, 165.6 (3 × C=O), 160.6 (C-3), 151.5 (C-6), 133.5, 133.4, 133.2, 129.9–129.8, 129.7, 129.3, 129.0, 128.5–128.4 (Ar), 127.5, 125.8 (C-4, C-5), 82.3, 77.4, 76.7, 69.9 (C-1′, C-3′–C-5′), 63.6 (C-6′), 56.7 (C-2′). ESI-HRMS positive mode (m/z): calcd for C31H28N3O7+ [M+H]+ 554.1922; C31H27N3O7Na+ [M+Na]+ 576.1741. Found: [M+H]+ 554.1922; [M+Na]+ 576.1740.
2-(2′-Amino-2′-deoxy-3′,4′,6′-tri-O-benzoyl-β-d-glucopyranosyl)pyrimidine (7c)
Prepared from compound 9c (90 mg, 0.14 mmol) according to general procedure VI. Purification by column chromatography (95:5 CHCl3-MeOH) yielded 73 mg (96%) of a white, amorphous solid. Rf = 0.33 (95:5 CHCl3-MeOH); [α]D = −15 (c 0.5, CHCl3). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.84 (2H, d, J = 4.9 Hz, H-4, H-6), 7.99–7.30 (16H, m, Ar, H-5), 5.74 (1H, pt, J = 9.7, 9.6 Hz, H-3′ or H-4′), 5.65 (1H, pt, J = 9.8, 9.6 Hz, H-3′ or H-4′), 4.67 (1H, d, J = 9.9 Hz, H-1′), 4.60 (1H, dd, J = 12.2, 3.2 Hz, H-6′a), 4.51 (1H, dd, J = 12.2, 5.4 Hz, H-6′b), 4.29 (1H, ddd, J = 9.2, 5.4, 3.2 Hz, H-5′), 3.88 (1H, pt, J = 10.0, 9.8 Hz, H-2′), 1.46 (2H, s, NH2); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.8, 166.3, 165.9, 165.6 (3 × C=O, C-2), 157.7 (C-4, C-6), 133.4, 133.3, 133.0, 130.0–129.8, 129.4, 129.2, 128.5–128.3 (Ar), 120.9 (C-5), 85.2, 77.8, 77.0, 70.4 (C-1′, C-3′–C-5′), 64.2 (C-6′), 55.3 (C-2′). ESI-HRMS positive mode (m/z): calcd for C31H28N3O7+ [M+H]+ 554.1922; C31H27N3O7Na+ [M+Na]+ 576.1741. Found: [M+H]+ 554.1916; [M+Na]+ 576.1734.
2-(2′-Amino-2′-deoxy-3′,4′,6′-tri-O-benzoyl-β-d-glucopyranosyl)pyrazine (7d)
Prepared from compound 9d (0.12 g, 0.18 mmol) according to general procedure VI. Purification by column chromatography (95:5 CHCl3-MeOH) yielded 98 mg (96%) of a white, amorphous solid. Rf = 0.32 (95:5 CHCl3-MeOH); [α]D = −7 (c 0.5, CHCl3). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.82 (1H, d, J = 1.2 Hz, H-3), 8.58–8.56 (2H, m, H-5, H-6), 8.01–7.31 (15H, m, Ar), 5.71 (1H, pt, J = 9.7, 9.4 Hz, H-3′ or H-4′), 5.68 (1H, pt, J = 9.7, 9.4 Hz, H-3′ or H-4′), 4.65 (1H, dd, J = 12.3, 3.0 Hz, H-6′a), 4.62 (1H, d, J = 9.8 Hz, H-1′), 4.51 (1H, dd, J = 12.3, 5.3 Hz, H-6′b), 4.28 (1H, ddd, J = 8.9, 5.3, 3.0 Hz, H-5′), 3.64 (1H, pt, J = 9.7, 9.7 Hz, H-2′), 1.82 (2H, s, NH2); 13C NMR (100 MHz, CDCl3) δ (ppm): 166.6, 166.2, 165.6 (3 × C=O), 153.1 (C-2), 145.1, 144.6, 143.6 (C-3, C-5, C-6), 133.5, 133.4, 133.1, 129.9–129.8, 129.7, 129.2, 129.0, 128.5–128.4 (Ar), 81.9, 77.4, 76.7, 70.0 (C-1′, C-3′–C-5′), 63.7 (C-6′), 56.1 (C-2′). ESI-HRMS positive mode (m/z): calcd for C31H28N3O7+ [M+H]+ 554.1922; C31H27N3O7Na+ [M+Na]+ 576.1741. Found: [M+H]+ 554.1916; [M+Na]+ 576.1735.
2-(2′-Amino-2′-deoxy-3′,4′,6′-tri-O-benzoyl-β-d-glucopyranosyl)quinoline (7e)
Prepared from compound 9e (0.12 g, 0.16 mmol) according to general procedure VI. Purification by column chromatography (1:1 EtOAc-hexane) yielded 83 mg (84%) of a white, amorphous solid. Rf = 0.18 (1:1 EtOAc-hexane); [α]D = +30 (c 0.5, CHCl3). 1H NMR (500 MHz, CDCl3) δ (ppm): 8.22–7.31 (21H, m, Ar, H-3–H-8), 5.77–5.71 (2H, m, H-3′, H-4′), 4.73 (1H, d, J = 9.5 Hz, H-1′), 4.66 (1H, dd, J = 12.2, 2.8 Hz, H-6′a), 4.54 (1H, dd, J = 12.2, 5.4 Hz, H-6′b), 4.31 (1H, ddd, J = 9.1, 5.4, 2.8 Hz, H-5′), 3.69 (1H, pt, J = 9.5, 9.1 Hz, H-2′), 1.83 (2H, br s, NH2); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.6, 166.3, 165.7 (3 × C=O), 157.9, 147.2 (C-2, C-8a), 137.4, 133.4, 133.3, 133.1, 130.0–129.6, 129.5, 129.2, 128.5–128.4, 128.0, 127.7, 127.0, 119.9 (Ar, C-3–C-8, C-4a), 84.2, 77.6, 76.7, 70.3 (C-1′, C-3′–C-5′), 63.9 (C-6′), 56.5 (C-2′). ESI-HRMS positive mode (m/z): calcd for C36H31N2O7+ [M+H]+ 603.2126. Found: 603.2123.
2-(2′-(tert-Butoxycarbonyl)amino-2′-deoxy-β-d-glucopyranosyl)pyrimidine (8c)
Prepared from compound 5c (60 mg, 0.25 mmol) and Boc2O (0.11 g, 0.50 mmol) according to general procedure IV. Purification by column chromatography (9:1 CHCl3-MeOH) yielded 57 mg (67%) of a white, amorphous solid. Rf = 0.37 (4:1 CHCl3-MeOH). 1H NMR (400 MHz, D2O) δ (ppm): 8.81 (2H, d, J = 5.0 Hz, H-4, H-6), 7.55 (1H, t, J = 5.0 Hz, H-5), 4.51 (1H, d, J = 9.7 Hz, H-1′), 3.94 (1H, dd, J = 12.4, 1.9 Hz, H-6′a), 3.86 (1H, dd, J = 12.4, 4.6 Hz, H-6′b), 3.74–3.61 (4H, m, H-2′–H-5′), 1.21 (6H, s, 2 × CH3), 1.10 (3H, s, CH3); 13C NMR (100 MHz, D2O + 2 drops of CD3OD) δ (ppm): 165.0 (C-2), 157.7 (C-4, C-6), 156.8 (C=O), 121.6 (C-5), 81.2, 79.7, 74.5, 69.8 (C-1′, C-3′–C-5′), 80.9 (C(CH3)3), 60.9 (C-6′), 56.8 (C-2′), 27.6 (3 × CH3). ESI-HRMS positive mode (m/z): calcd for C15H23N3O6Na+ [M+Na]+ 364.1479; C30H46N6O12Na+ [2M+Na]+ 705.3066. Found: [M+Na]+ 364.1474; [2M+Na]+ 705.3057.
2-(2′-(tert-Butoxycarbonyl)amino-2′-deoxy-β-d-glucopyranosyl)pyrazine (8d)
Prepared from compound 5d (0.10 g, 0.42 mmol) and Boc2O (0.18 g, 0.83 mmol) according to general procedure IV. Purification by column chromatography (9:1 CHCl3-MeOH) yielded 94 mg (67%) of a white, amorphous solid. Rf = 0.37 (4:1 CHCl3-MeOH). 1H NMR (400 MHz, CD3OD) δ (ppm): 8.72 (1H, d, J = 1.5 Hz, H-3), 8.56 (1H, dd, J = 2.7, 1.5 Hz, H-5), 8.53 (1H, d, J = 2.7 Hz, H-6), 4.41 (1H, d, J = 9.1 Hz, H-1′), 3.90 (1H, dd, J = 12.1, 2.2 Hz, H-6′a), 3.77 (1H, dd, J = 12.1, 5.1 Hz, H-6′b), 3.62–3.52 (3H, m, H-2′, H-3′, H-4′), 3.44 (1H, ddd, J = 9.0, 5.1, 2.2 Hz, H-5′), 1.22 (9H, s, 3 × CH3); 13C NMR (100 MHz, CD3OD) δ (ppm): 157.5, 155.3 (C=O, C-2), 145.0, 144.9, 144.8 (C-3, C-5, C-6), 82.3, 81.3, 76.6, 71.8 (C-1′, C-3′–C-5′), 79.9 (C(CH3)3), 62.7 (C-6′), 58.6 (C-2′), 28.6 (3 × CH3). ESI-HRMS positive mode (m/z): calcd for C15H23N3O6Na+ [M+Na]+ 364.1479. Found: 364.1471.
2-(2′-(tert-Butoxycarbonyl)amino-2′-deoxy-β-d-glucopyranosyl)quinoline (8e)
Prepared from compound 5e (25 mg, 0.086 mmol) and Boc2O (37.6 mg, 0.172 mmol) according to general procedure IV. Purification by column chromatography (9:1 CHCl3-MeOH) yielded 27 mg (80%) of a white, amorphous solid. Rf = 0.35 (4:1 CHCl3-MeOH). 1H NMR (360 MHz, CD3OD) δ (ppm): 8.29 (1H, d, J = 8.5 Hz, H-4), 8.05 (1H, dd, J = 8.5, 1.2 Hz, H-8), 7.90 (1H, dd, J = 8.1, 1.4 Hz, H-5), 7.75 (1H, ddd, J = 8.5, 6.9, 1.5 Hz, H-7), 7.69 (1H, d, J = 8.5 Hz, H-3), 7.58 (1H, ddd, J = 8.1, 6.9, 1.2 Hz, H-6), 4.49 (1H, d, J = 9.4 Hz, H-1′), 3.94 (1H, dd, J = 12.1, 2.3 Hz, H-6′a), 3.82 (1H, dd, J = 12.1, 5.2 Hz, H-6′b), 3.70–3.56 (3H, m, H-2′, H-3′, H-4′), 3.49 (1H, ddd, J = 9.5, 5.2, 2.3 Hz, H-5′), 1.00 (6H, s, 2 × CH3), 0.79 (3H, s, CH3); 13C NMR (90 MHz, CD3OD) δ (ppm): 160.2, 157.4, 148.0 (C=O, C-2, C-8a,), 138.2, 130.9, 129.2, 128.9, 127.8, 121.7 (C-3–C-8), 129.4 (C-4a), 83.6, 82.3, 77.0, 72.0 (C-1′, C-3′–C-5′), 79.8 (C(CH3)3), 62.9 (C-6′) 58.8 (C-2′), 28.4 (3 × CH3). ESI-HRMS positive mode (m/z): calcd for C20H26N2O6Na+ [M+Na]+ 413.1683. Found: 413.1683.
3-(2′-(tert-Butoxycarbonyl)amino-2′-deoxy-3′,4′,6′-tri-O-benzoyl-β-d-glucopyranosyl)pyridazine (9b)
A degassed, vigorously stirred suspension of 10% Pd(C) (65 mg) in dry EtOH (13 mL) was saturated with H2, and compound 4b (0.13 g, 0.48 mmol) was added. The reaction mixture was heated at reflux temperature until the TLC (3:2 CHCl3-MeOH) indicated complete conversion of the starting material. After completion of the reaction (3 h), the catalyst was filtered off through a pad of celite and washed with EtOH. The resulting solution was then evaporated under reduced pressure. Purification of the residue by column chromatography (3:2 CHCl3-MeOH) yielded 100 mg of a white, amorphous solid containing the desired 3-(2′-amino-2′-deoxy-β-d-glucopyranosyl)pyridazine 5b, along with unidentified impurities. This mixture was dissolved in a solvent mixture of water (5 mL) and 1,4-dioxane (5 mL), and Boc2O (0.21 g, 0.96 mmol) was added. The reaction mixture was stirred at rt until the TLC (9:1 CHCl3-MeOH) showed complete transformation of 5b (1 day). Then, the solvents were removed under reduced pressure. Column chromatographic purification of the residue (9:1 CHCl3-MeOH) resulted in 70 mg of 3-(2′-(tert-butoxycarbonyl)amino-2′-deoxy-β-d-glucopyranosyl)pyridazine (8b) contaminated with inseparable impurities. To a solution of the resulting 8b in dry pyridine (5 mL), benzoyl chloride (0.2 mL, 1.72 mmol) was added at rt. The reaction mixture was stirred at 60 °C for 1 h. Since the TLC (1:1 EtOAc-hexane) showed incompleteness of the reaction, an additional portion of benzoyl chloride (0.2 mL, 1.72 mmol) was added to the reaction mixture, and heating was continued for 1 h. The reaction mixture was allowed to cool to rt and further stirred overnight. The reaction mixture was then diluted with CH2Cl2 (50 mL) and extracted with sat. aq. solution of NaHCO3 (25 mL), then with water (25 mL). The separated organic phase was dried over MgSO4 and filtered, and the solvents were removed under diminished pressure. The residue was purified by column chromatography (1:1 EtOAc-hexane) to afford the title compound 9b (83 mg, 27% for 3 steps) as a white, amorphous solid. Rf = 0.28 (1:1 EtOAc-hexane. 1H NMR (400 MHz, CDCl3) δ (ppm): 9.12 (1H, dd, J = 4.9, 1.7 Hz, H-6), 8.04–7.32 (17H, m, Ar, H-4, H-5), 5.82 (1H, pt, J = 9.3, 9.3 Hz, H-3′ or H-4′), 5.79 (1H, pt, J = 9.7, 9.5 Hz, H-3′ or H-4′), 5.12 (1H, d, J = 9.9 Hz, NH), 5.02 (1H, d, J = 10.3 Hz, H-1′), 4.67 (1H, dd, J = 12.3, 2.8 Hz, H-6′a), 4.50 (1H, dd, J = 12.3, 4.8 Hz, H-6′b), 4.30 (1H, ddd, J = 9.5, 4.8, 2.8 Hz, H-5′), 4.28 (1H, pt, J = 10.1, 10.0 Hz, H-2′), 1.05 (9H, s, 3 × CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 166.7, 166.2, 165.4 (3 × C=O), 159.6, 155.0 (C=O, C-3), 151.4 (C-6), 133.6, 133.4, 133.2, 130.0–129.8, 129.7, 129.1, 128.9, 128.6–128.4 (Ar), 127.3, 125.7 (C-4, C-5), 81.3, 76.7, 74.3, 69.8 (C-1′, C-3′–C-5′), 80.0 (C(CH3)3), 63.3 (C-6′), 55.9 (C-2′), 27.9 (3 × CH3). ESI-HRMS positive mode (m/z): calcd for C36H35N3O9Na+ [M+Na]+ 676.2266; C72H70N6O18Na+ [2M+Na]+ 1329.4639. Found: [M+Na]+ 676.2256; [2M+Na]+ 1329.4641.
2-(2′-(tert-Butoxycarbonyl)amino-2′-deoxy-3′,4′,6′-tri-O-benzoyl-β-d-glucopyranosyl)pyrimidine (9c)
Prepared from compound 8c (60 mg, 0.18 mmol) and benzoyl chloride (0.15 mL, 1.29 mmol, 7.2 eq.) according to general procedure V. Purification by column chromatography (1:1 EtOAc-hexane) yielded 92 mg (80%) of a white, amorphous solid. Rf = 0.22 (1:1 EtOAc-hexane). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.82 (2H, d, J = 4.9 Hz, H-4, H-6), 7.98–7.27 (16H, m, Ar, H-5), 5.86 (1H, pt, J = 9.9, 9.6 Hz, H-3′ or H-4′), 5.80 (1H, pt, J = 9.6, 9.4 Hz, H-3′ or H-4′), 4.98 (1H, d, J = 9.5 Hz, NH), 4.90 (1H, d, J = 10.2 Hz, H-1′), 4.63 (1H, dd, J = 12.3, 3.5 Hz, H-6′a), 4.58 (1H, dd, J = 12.3, 5.4 Hz, H-6′b), 4.55 (1H, pt, J = 10.2, 9.7 Hz, H-2′), 4.30 (1H, ddd, J = 9.2, 5.4, 3.5 Hz, H-5′), 1.09 (9H, s, 3 × CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 166.7, 166.3, 165.4, 165.2 (3 × C=O, C-2), 154.6 (C=O), 157.4 (C-4, C-6), 133.4, 133.3, 133.0, 130.0–129.8, 129.7, 129.2, 129.0, 128.4–128.2 (Ar), 120.7 (C-5), 82.8, 76.7, 74.6, 70.2 (C-1′, C-3′–C-5′), 79.6 (C(CH3)3), 64.1 (C-6′), 55.2 (C-2′), 28.0 (3 × CH3). ESI-HRMS positive mode (m/z): calcd for C36H35N3O9Na+ [M+Na]+ 676.2266. Found: 676.2256.
2-(2′-(tert-Butoxycarbonyl)amino-2′-deoxy-3′,4′,6′-tri-O-benzoyl-β-d-glucopyranosyl)pyrazine (9d)
Prepared from compound 8d (90 mg, 0.26 mmol) and benzoyl chloride (0.22 mL, 1.89 mmol, 7.2 eq.) according to general procedure V. Purification by column chromatography (1:1 EtOAc-hexane) yielded 122 mg (71%) of a white, amorphous solid. Rf = 0.36 (1:1 EtOAc-hexane). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.83 (1H, s, H-3), 8.55–8.53 (2H, m, H-5, H-6), 8.02–7.30 (15H, m, Ar), 5.88 (1H, d, J = 9.8, 9.6 Hz, H-3′ or H-4′), 5.83 (1H, d, J = 9.5, 9.3 Hz, H-3′ or H-4′), 5.15 (1H, d, J = 9.3 Hz, NH), 4.83 (1H, d, J = 10.2 Hz, H-1′), 4.68 (1H, dd, J = 12.3, 2.9 Hz, H-6′a), 4.53 (1H, dd, J = 12.3, 4.9 Hz, H-6′b), 4.35 (1H, pt, J = 9.8, 9.7 Hz, H-2′), 4.34–4.29 (H, m, H-5′), 1.09 (9H, s, 3 × CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.8, 166.3, 165.4 (3 × C=O), 154.8, 152.4 (C=O, C-2), 144.6 (2), 143.4 (C-3, C-5, C-6), 133.5, 133.4, 133.2, 130.0–129.8, 129.7, 129.1, 129.0, 128.5–128.4 (Ar), 80.7, 76.8, 74.3, 70.0 (C-1′, C-3′–C-5′), 79.9 (C(CH3)3), 63.5 (C-6′), 55.9 (C-2′), 28.0 (3 × CH3). ESI-HRMS positive mode (m/z): calcd for C36H35N3O9Na+ [M+Na]+ 676.2266. Found: 676.2260.
2-(2′-(tert-Butoxycarbonyl)amino-2′-deoxy-3′,4′,6′-tri-O-benzoyl-β-d-glucopyranosyl)quinoline (9e)
Prepared from compound 8e (50 mg, 0.13 mmol) and benzoyl chloride (0.11 mL, 0.95 mmol, 7.2 eq.) according to general procedure V. Purification by column chromatography (1:2 EtOAc-hexane) yielded 73 mg (81%) of a white, amorphous solid. Rf = 0.21 (1:2 EtOAc-hexane). 1H NMR (500 MHz, CDCl3) δ (ppm): 8.23–7.32 (21H, m, Ar, H-3–H-8), 5.84 (1H, pt, J = 9.6, 9.5 Hz, H-3′ or H-4′), 5.77 (1H, dd, J = 10.0, 9.8 Hz, H-3′ or H-4′), 4.98 (1H, d, J = 9.8 Hz, NH), 4.89 (1H, d, J = 10.2 Hz, H-1′), 4.67 (1H, dd, J = 12.2, 2.9 Hz, H-6′a), 4.52 (1H, dd, J = 12.2, 4.8 Hz, H-6′b), 4.36 (1H, q, J = 10.2 Hz, H-2′), 4.28 (1H, ddd, J = 9.7, 4.8, 2.9 Hz, H-5′), 0.86 (9H, s, 3 × CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 170.8, 166.4, 165.5 (3 × C=O), 157.4, 155.1, 146.5 (C=O, C-2, C-8a), 137.9, 133.5, 133.3, 133.2, 130.3–129.8, 129.4, 129.1, 128.5–128.3, 128.2, 127.9, 127.0, 119.5 (Ar, C-3–C-8, C-4a), 82.5, 76.6, 74.7, 70.1 (C-1′, C-3′–C-5′), 79.5 (C(CH3)3), 63.5 (C-6′), 56.1 (C-2′), 27.7 (3 × CH3). ESI-HRMS positive mode (m/z): calcd for C41H38N2O9Na+ [M+Na]+ 725.2470. Found: 725.2473.
Complex Ru-3a
Prepared from compound 3a (47 mg, 0.092 mmol, 1.9 eq.), Ru-dimer (30 mg, 0.049 mmol) and TlPF6 (34 mg, 0.097 mmol) according to general procedure VII. Purification by column chromatography (95:5 CHCl3-MeOH) yielded 63 mg (74%) of a yellow solid. Rf = 0.50 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.02 (1H, d, J = 5.5 Hz, H-6), 7.86 (1H, dd, J = 8.1, 1.8 Hz, H-3), 7.82 (1H, td, J = 8.1, 1.5 Hz, H-4), 7.40–7.26 (16H, m, Ar, H-5), 5.84, 5.80, 5.59, 5.13 (4 × 1H, 4 d, J = 5.9 Hz in each, 4 × p-cym-CHAr), 5.34 (1H, pt, J = 10.8 Hz, NH2), 4.89, 4.59 (2 × 1H, 2 d, J = 12.1 Hz in both, PhCH2), 4.78, 4.56 (2 × 1H, 2 d, J = 12.3 Hz in both, PhCH2), 4.75, 4.65 (2 × 1H, 2 d, J = 11.3 Hz in both, PhCH2), 4.54 (1H, d, J = 10.2 Hz, H-1′), 4.23 (1H, pt, J = 8.9, 8.8 Hz, H-3′), 3.99 (1H, ddd, J = 9.4, 5.6, 2.6, H-5′), 3.84–3.77 (2H, m, H-6′a,b), 3.53 (1H, pt, J = 9.1, 9.0 Hz, H-4′), 3.20 (1H, dd, J = 10.8, 5.3 Hz, NH2), 2.58 (1H, hept, J = 6.9 Hz, i-Pr-CH), 2.03–1.94 (1H, m, H-2′), 1.70 (3H, s, C6H4-CH3), 1.16, 1.02 (2 × 3H, 2 d, J = 6.9 Hz in both, 2 × i-Pr-CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 162.0 (C-2), 156.8 (C-6), 139.4 (C-4), 138.4, 138.1 (2), 129.1–127.8 (Ar), 124.5, 123.2 (C-3, C-5), 104.9, 98.7 (2 × p-cym-CqAr), 86.6, 84.8, 83.8, 83.4, 82.9, 77.7, 76.8, 76.0 (4 × p-cym-CHAr, C-1′, C-3′–C-5′), 75.1, 74.2, 73.5 (3 × PhCH2), 68.7 (C-6′), 53.6 (C-2′), 31.0 (i-Pr-CH), 23.2, 21.5 (2 × i-Pr-CH3), 17.7 (C6H4-CH3). ESI-HRMS positive mode (m/z): calcd for C42H48ClN2O4Ru+ [M-PF6]+ 781.2349. Found: 781.2346.
Complex Os-3a
Prepared from compound 3a (12.3 mg, 0.024 mmol, 1.9 eq.), Os-dimer (10.0 mg, 0.013 mmol) and TlPF6 (8.7 mg, 0.025 mmol) according to general procedure VII. After purification by column chromatography (95:5 CHCl3-MeOH), the complex was dissolved in CHCl3 (1 mL), and diisopropyl ether (8 mL) was added. The precipitated product was filtered off, then washed with CHCl3-diisopropyl ether (1:8, 1 mL) to yield 15.6 mg (64%) of a dark purple solid. Rf = 0.58 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.87 (1H, dd, J = 6.0, 1.5 Hz, H-6), 7.90 (1H, d, J = 7.8 Hz, H-3), 7.81 (1H, td, J = 7.8, 1.5 Hz, H-4), 7.41–7.26 (16H, m, Ar, H-5), 6.09, 6.08, 5.82, 5.27 (4 × 1H, 4 d, J = 5.7 Hz in each, 4 × p-cym-CHAr), 5.90–5.76 (1H, broad signal, NH2), 4.91, 4.77 (2 × 1H, 2 d, J = 12.3 Hz in both, PhCH2), 4.76, 4.67 (2 × 1H, 2 d, J = 11.5 Hz in both, PhCH2), 4.59, 4.55 (2 × 1H, 2 d, J = 12.2 Hz in both, PhCH2), 4.50 (1H, d, J = 10.2 Hz, H-1′), 4.22 (1H, pt, J = 9.0, 8.8 Hz, H-3′ or H-4′), 4.00–3.98 (1H, m, H-5′), 3.84–3.77 (2H, m, H-6′a,b), 3.67 (1H, dd, J = 11.4, 4.6 Hz, NH2), 3.57 (1H, pt, J = 9.2, 9.1 Hz, H-3′ or H-4′), 2.47 (1H, hept, J = 7.0 Hz, i-Pr-CH), 2.32–2.23 (1H, m, H-2′), 1.75 (3H, s, C6H4-CH3), 1.19, 0.98 (2 × 3H, 2 d, J = 6.9 Hz in both, 2 × i-Pr-CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 161.3 (C-2), 157.7 (C-6), 139.5 (C-4), 138.4, 138.1 (2), 129.9–127.8 (Ar), 124.9, 122.5 (C-4, C-5), 95.0, 89.9 (2 × p-cym-CqAr), 82.7, 78.3, 77.7, 76.8, 76.3, 75.6, 75.4, 73.6 (4 × p-cym-CHAr, C-1′, C-3′–C-5′), 75.2, 74.2, 73.5 (3 × PhCH2), 68.7 (C-6′), 53.6 (C-2′), 31.1 (i-Pr-CH), 23.6, 21.6 (2 × i-Pr-CH3), 17.6 (C6H4-CH3). ESI-HRMS positive mode (m/z): calcd for C42H47N2O4Os+ [M-HCl-PF6]+ 835.3148. Found: 835.3143.
Complex Ir-3a
Prepared from compound 3a (12.2 mg, 0.024 mmol, 1.9 eq.), Ir-dimer (10.0 mg, 0.013 mmol) and TlPF6 (8.8 mg, 0.025 mmol) according to general procedure VII. Purification by column chromatography (95:5 CHCl3-MeOH) yielded 22.7 mg (93%) of a yellow solid. Rf = 0.51 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.58 (1H, d, J = 5.8 Hz, H-6), 7.92–7.87 (2H, m, H-3, H-4), 7.42–7.27 (16H, m, Ar, H-5), 4.83, 4.70 (2 × 1H, 2 d, J = 12.6 Hz in both, PhCH2), 4.78, 4.69 (2 × 1H, 2 d, J = 11.4 Hz in both, PhCH2), 4.60, 4.56 (2 × 1H, 2 d, J = 12.1 Hz in both, PhCH2), 4.25 (1H, d, J = 10.0 Hz, H-1′), 4.12–4.05 (2H, m, H-5′, NH2), 4.08 (1H, pt, J = 8.1, 8,0 Hz, H-3′ or H-4′), 3.95 (1H, dd, J = 11.5, 5.5 Hz, NH2), 3.85 (1H, dd, J = 10.8, 4.1 Hz, H-6′a), 3.79 (1H, dd, J = 10.8, 3.0 Hz, H-6′b), 3.69 (1H, pt, J = 8.0, 7.9 Hz, H-3′ or H-4′), 2.55–2.47 (1H, m, H-2′), 1.41 (15H, s, Cp*-CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 160.8 (C-2), 155.3 (C-6), 139.9 (C-4), 138.3, 138.1, 137.8, 129.9, 129.3–127.9 (Ar), 126.0, 123.0 (C-4, C-5), 88.1 (Cp*), 81.5, 77.5, 77.4, 77.1 (C-1′, C-3′–C-5′), 74.3, 74.1, 73.5 (3 × PhCH2), 69.0 (C-6′), 54.1 (C-2′), 8.5 (Cp*-CH3). ESI-HRMS positive mode (m/z): calcd for C42H49ClN2O4Ir+ [M-PF6]+ 873.2999. Found: 873.2997.
Complex Rh-3a
Prepared from compound 3a (15.7 mg, 0.031 mmol, 1.9 eq.), Rh-dimer (10.0 mg, 0.016 mmol) and TlPF6 (11.3 mg, 0.032 mmol) according to general procedure VII. After purification by column chromatography (95:5 CHCl3-MeOH), the complex was dissolved in CHCl3 (1 mL), and diisopropyl ether (8 mL) was added. The precipitated product was filtered off, then washed with CHCl3-diisopropyl ether (1:8, 1 mL) to yield 23.6 mg (83%) of an orange solid. Rf = 0.30 (95:5 CHCl3-MeOH).1H NMR (400 MHz, CDCl3) δ (ppm): 8.61 (1H, dd, J = 5.7, 1.7 Hz, H-6), 7.94–7.85 (2H, m, H-3, H-4), 7.46–7.28 (16H, m, Ar, H-5), 4.81, 4.68 (2 × 1H, 2 d, J = 12.5 Hz in both, PhCH2), 4.77, 4.70 (2 × 1H, 2 d, J = 11.2 Hz in both, PhCH2), 4.61, 4.58 (2 × 1H, 2 d, J = 12.8 Hz in both, PhCH2), 4.26 (1H, d, J = 9.9 Hz, H-1′), 4.03 (1H, ddd, J = 7.6, 4.1, 3.2 Hz, H-5′), 3.96 (1H, pt, J = 7.7, 7.6 Hz, H-3′ or H-4′), 3.85 (1H, dd, J = 10.8, 4.1 Hz, H-6′a), 3.79 (1H, dd, J = 10.8, 3.2 Hz, H-6′b), 3.72 (1H, pt, J = 7.7, 7.7 Hz, H-3′ or H-4′), 3.47 (1H, dd, J = 11.1, 5.9 Hz, NH2), 3.24 (1H, pt, J = 10.2 Hz, NH2), 2.40–2.32 (1H, m, H-2′), 1.43 (15H, s, Cp*-CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 160.8 (C-2), 154.2 (C-6), 139.8 (C-4), 138.2, 138.1, 137.7, 129.9, 129.2–127.9 (Ar), 125.6, 123.2 (C-3, C-5), 96.5, 96.4 (Cp*), 82.3, 77.7, 77.6, 76.9 (C-1′, C-3′–C-5′), 74.3, 74.0, 73.6 (3 × PhCH2), 69.0 (C-6′), 54.4 (C-2′), 8.9 (Cp*-CH3). ESI-HRMS positive mode (m/z): calcd for C42H49ClN2O4Rh+ [M-PF6]+ 783.2430. Found: 783.2430.
Complex Ru-3d
Prepared from compound 3d (16.7 mg, 0.033 mmol, 2 eq.), Ru-dimer (10.0 mg, 0.016 mmol) and TlPF6 (11.4 mg, 0.033 mmol) according to general procedure VII. After purification by column chromatography (100:1 CHCl3-MeOH), the complex was dissolved in CHCl3 (1.5 mL), and diisopropyl ether (12 mL) was added. The precipitated product was filtered off, then washed with diisopropyl ether (1 mL) to yield 10.0 mg (33%) of a brown solid. Rf = 0.31 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.09 (1H, s, H-3), 8.94 (1H, dd, J = 3.2, 1.1 Hz, H-5), 8.65 (1H, d, J = 3.2 Hz, H-6), 7.43–7.26 (15H, m, Ar), 5.89, 5.85, 5.54, 5.15 (4 × 1H, 4 d, J = 6.0 Hz in each, 4 × p-cym-CHAr), 5.22–5.16 (1H, broad signal, NH2), 4.92, 4.77 (2 × 1H, 2 d, J = 12.3 Hz in both, PhCH2), 4.76, 4.66 (2 × 1H, 2 d, J = 11.3 Hz in both, PhCH2), 4.67 (1H, d, J = 10.3 Hz, H-1′), 4.56 (2H, s, PhCH2), 4.20 (1H, pt, J = 9.1, 8.9 Hz, H-3′ or H-4′), 3.99–3.94 (1H, m, H-5′), 3.81–3.77 (2H, m, H-6′a,b), 3.56 (1H, pt, J = 9.2, 9.1 Hz, H-3′ or H-4′), 3.16 (1H, dd, J = 11.1, 5.1 Hz, NH2), 2.56 (1H, hept, J = 6.9 Hz, i-Pr-CH), 2.04–1.95 (1H, m, H-2′), 1.76 (3H, s, C6H4-CH3), 1.16, 1.03 (2 × 3H, 2 d, J = 6.9 Hz in both, 2 × i-Pr-CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 156.5 (C-2), 149.9, 145.3, 145.2 (C-3, C-5, C-6), 138.4, 138.0, 137.9, 129.1–127.9 (Ar), 105.7, 98.9 (2 × p-cym-CqAr), 86.8, 85.2, 84.1, 84.0, 83.3, 77.5, 77.2, 75.1 (4 × p-cym-CHAr, C-1′, C-3′–C-5′), 75.3, 74.4, 73.5 (3 × PhCH2), 68.5 (C-6′), 53.2 (C-2′), 31.1 (i-Pr-CH), 23.0, 21.6 (2 × i-Pr-CH3), 17.8 (C6H4-CH3). ESI-HRMS positive mode (m/z): calcd for C41H47ClN3O4Ru+ [M-PF6]+ 782.2307. Found: 782.2315.
Complex Os-3d
Prepared from compound 3d (13.0 mg, 0.025 mmol, 2 eq.), Os-dimer (10.0 mg, 0.013 mmol) and TlPF6 (8.7 mg, 0.025 mmol) according to general procedure VII. After purification by column chromatography (100:1 CHCl3-MeOH), the complex was dissolved in CHCl3 (1.5 mL), and diisopropyl ether (12 mL) was added. The precipitated product was filtered off, then washed with diisopropyl ether (1 mL) to yield 12.0 mg (47%) of a greenish–brown solid. Rf = 0.27 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.10 (1H, s, H-3), 8.76 (1H, dd, J = 3.3, 1.1 Hz, H-5), 8.56 (1H, d, J = 3.3 Hz, H-6), 7.42–7.27 (15H, m, Ar), 6.12, 6.11, 5.80, 5.29 (4 × 1H, 4 d, J = 5.8 Hz in each, 4 × p-cym-CHAr), 5.90–5.77 (1H, broad signal, NH2), 4.92, 4.75 (2 × 1H, 2 d, J = 12.3 Hz in both, PhCH2), 4.77, 4.68 (2 × 1H, 2 d, J = 11.4 Hz in both, PhCH2), 4.70–4.65 (1H, broad signal, NH2), 4.62 (1H, d, J = 10.4 Hz, H-1′), 4.56 (2H, s, PhCH2), 4.23 (1H, pt, J = 9.1, 9.0 Hz, H-3′ or H-4′), 3.99–3.95 (1H, m, H-5′), 3.83–3.76 (2H, m, H-6′a,b), 3.59 (1H, pt, J = 9.3, 9.2 Hz, H-3′ or H-4′), 2.46 (1H, hept, J = 6.9 Hz, i-Pr-CH), 2.33–2.24 (1H, m, H-2′), 1.80 (3H, s, C6H4-CH3), 1.18, 0.99 (2 × 3H, 2 d, J = 6.9 Hz in both, 2 × i-Pr-CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 155.4 (C-2), 150.4, 146.0, 144.8 (C-3, C-5, C-6), 138.4, 138.0, 137.9, 129.1–127.9 (Ar), 96.2, 90.3 (2 × p-cym-CqAr), 82.5, 78.5, 77.5, 77.1, 76.2, 76.0, 75.3, 74.7 (4 × p-cym-CHAr, C-1′, C-3′–C-5′), 75.3, 74.4, 73.5 (3 × PhCH2), 68.4 (C-6′), 53.0 (C-2′), 31.1 (i-Pr-CH), 23.4, 21.7 (2 × i-Pr-CH3), 17.6 (C6H4-CH3). ESI-HRMS positive mode (m/z): calcd for C41H47ClN3O4Os+ [M-PF6]+ 872.2861. Found: 872.2867.
Complex Ru-5a
Prepared from compound 5a (26 mg, 0.094 mmol, 2.3 eq.), Ru-dimer (25 mg, 0.041 mmol) and TlPF6 (62 mg, 0.176 mmol, 4.3 eq.) according to general procedure VII. Column chromatographic purification (95:5 CHCl3-MeOH) yielded 23 mg (43%) of a yellow solid. Rf = 0.50 (7:3 CHCl3-MeOH). 1H NMR (400 MHz, CD3OD) δ (ppm); 9.09 (1H, ddd, J = 5.8, 1.4, 0.8 Hz, H-6), 8.07–8.00 (2H, m, H-3, H-4), 7.53–4.49 (1H, ddd, J = 7.8, 5.8, 2.0 Hz, H-5), 5.96, 5.94, 5.81, 5.64 (4 × 1H, 4 d, J = 6.0 Hz in each, 4 × p-cym-CHAr), 5.70 (1H, dd, J = 11.8, 5.6 Hz, NH2), 4.41 (1H, d, J = 10.1 Hz, H-1′), 4.01 (1H, dd, J = 12.2, 2.1 Hz, H-6′a), 3.87–3.79 (1H, broad signal, NH2), 3.84 (1H, pt, J = 9.6, 8.9 Hz, H-3′), 3.82 (1H, dd, J = 12.2, 5.6 Hz, H-6′b), 3.67 (1H, ddd, J = 9.5, 5.6, 2.1 Hz, H-5′), 3.39 (1H, pt, J = 9.3, 9.2 Hz, H-4′), 2.83 (1H, hept, J = 6.9 Hz, i-Pr-CH), 2.22–2.18 (1H, m, H-2), 1.91 (3H, s, CH3), 1.28, 1.23 (2 × 3H, 2 d, J = 6.9 Hz in both, 2 × i-Pr-CH3); 13C NMR (100 MHz, CD3OD) δ (ppm): 161.8 (C-2), 158.1 (C-6), 140.9 (C-4), 125.8, 123.6 (C-3, C-5), 106.0, 100.8 (2 × p-cym-CqAr), 87.8, 85.2, 84.1, 83.4, 81.8, 80.2, 77.5, 71.3 (4 × p-cym-CHAr, C-1′, C-3′–C-5′), 62.6 (C-6′), 56.1 (C-2′), 32.3 (i-Pr-CH), 23.3, 21.9 (2 × i-Pr-CH3), 18.0 (C6H4-CH3). ESI-HRMS positive mode (m/z): calcd for C21H30ClN2O4Ru+ [M-PF6]+ 511.0935. Found: 511.0934.
Complex Ru-7a
Prepared from compound 7a (18.0 mg, 0.033 mmol, 2 eq.), Ru-dimer (10.0 mg, 0.016 mmol) and TlPF6 (11.4 mg, 0.033 mmol) according to general procedure VII. The crude product was dissolved in CHCl3 (3 mL), and diisopropyl ether (12 mL) was added. The precipitation was filtered off, then washed with CHCl3-diisopropyl ether (1:2, 1 mL) to yield 25.5 mg (81%) of a yellow solid. Rf = 0.49 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.02 (1H, d, J = 6.1 Hz, H-6), 8.12–7.31 (18H, m, Ar, H-3, H-4, H-5), 6.68–6.59 (1H, broad signal, NH2), 6.15 (1H, d, J = 5.9 Hz, p-cym-CHAr), 6.10 (1H, pt, J = 9.4, 9.3 Hz, H-3′), 6.03, 6.00 (2 × 1H, 2 d, J = 6.0 Hz in both, 2 × p-cym-CHAr), 5.75 (1H, pt, J = 9.8, 9.6 Hz, H-4′), 5.47 (1H, d, J = 6.0 Hz, p-cym-CHAr), 5.01–4.97 (2H, m, H-1′, H-6′a), 4.86 (1H, ddd, J = 10.0, 3.4, 2.8 Hz, H-5′), 4.49 (1H, dd, J = 12.7, 3.4 Hz, H-6′b), 3.39 (1H, dd, J = 11.2, 7.3 Hz, NH2), 2.89 (1H, hept, J = 6.9 Hz, i-Pr-CH), 2.65–2.57 (1H, m, H-2′), 1.81 (3H, s, C6H4-CH3), 1.25, 1.23 (2 × 3H, 2 d, J = 6.9 Hz in both, 2 × i-Pr-CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 168.7, 166.3, 165.6 (3 × C=O), 160.2 (C-2), 156.7 (C-6), 140.0 (C-4), 134.4, 133.8, 133.3, 130.4–130.0, 129.9, 128.7–128.6, 127.8 (Ar), 125.3, 122.9 (C-3, C-5), 104.3, 100.7 (2 × p-cym-CqAr), 88.0, 84.5, 82.6, 82.3, 77.9, 77.2, 74.4, 67.6 (4 × p-cym-CHAr, C-1′, C-3′–C-5′), 62.1 (C-6′), 54.3 (C-2′), 31.0 (i-Pr-CH), 22.8, 22.2 (2 × i-Pr-CH3), 17.9 (C6H4-CH3). ESI-HRMS positive mode (m/z): calcd for C42H42ClN2O7Ru+ [M-PF6]+ 823.1727. Found: 823.1727.
Complex Os-7a
Prepared from compound 7a (14.0 mg, 0.025 mmol, 2 eq.), Os-dimer (10.0 mg, 0.013 mmol) and TlPF6 (8.7 mg, 0.025 mmol) according to general procedure VII. After purification by column chromatography (95:5 CHCl3-MeOH), the complex was dissolved in CHCl3 (2 mL), and diisopropyl ether (16 mL) was added. The precipitated product was filtered off, then washed with CHCl3-diisopropyl ether (1:4, 1 mL) to yield 11.5 mg (43%) of a dark purple solid. Rf = 0.45 (95:5 CHCl3-MeOH). 1H NMR (500 MHz, CDCl3) δ (ppm): 8.86 (1H, dd, J = 5.9, 1.6 Hz, H-6), 8.15–7.34 (18H, m, Ar, H-3, H-4, H-5), 7.24–7.16 (1H, broad signal, NH2), 6.52, 6.38, 6.20, 5.66 (4 × 1H, 4 d, J = 5.6 Hz in each, 4 × p-cym-CHAr), 5.96 (1H, pt, J = 9.3, 9.1 Hz, H-3′ or H-4′), 5.78 (1H, pt, J = 9.7, 9.6 Hz, H-3′ or H-4′), 4.99 (1H, dd, J = 12.7, 2.1 Hz, H-6′a), 4.88 (1H, d, J = 10.1 Hz, H-1′), 4.84 (1H, ddd, J = 10.2, 3.3, 2.1 Hz, H-5′), 4.49 (1H, dd, J = 12.7, 3.3 Hz, H-6′b), 4.24 (1H, dd, J = 11.6, 7.2 Hz, NH2), 2.87–2.78 (2H, m, H-2′, i-Pr-CH), 1.85 (3H, s, C6H4-CH3), 1.27, 1.26 (2 × 3H, 2 d, J = 6.9 Hz in both, 2 × i-Pr-CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 168.8, 166.3, 165.6 (3 × C=O), 159.6 (C-2), 156.8 (C-6), 140.2 (C-4), 134.5, 133.8, 133.3, 130.4–130.0, 129.9, 128.8–128.6, 127.7 (Ar), 125.5, 122.4 (C-3, C-5), 94.3, 92.2 (2 × p-cym-CqAr), 80.6, 79.0, 77.1, 76.2, 74.3, 73.2, 73.1, 67.3 (4 × p-cym-CHAr, C-1′, C-3′–C-5′), 62.0 (C-6′), 54.2 (C-2′), 31.2 (i-Pr-CH), 23.1, 22.6 (2 × i-Pr-CH3), 17.8 (C6H4-CH3). ESI-HRMS positive mode (m/z): calcd for C42H42ClN2O7Os+ [M-PF6]+ 913.2292. Found: 913.2288.
Complex Ir-7a
Prepared from compound 7a (13.9 mg, 0.025 mmol, 2 eq.), Ir-dimer (10.0 mg, 0.013 mmol) and TlPF6 (8.8 mg, 0.025 mmol) according to general procedure VII. The crude product was dissolved in CHCl3 (3 mL), and diisopropyl ether (12 mL) was added. The precipitation was filtered off, then washed with CHCl3-diisopropyl ether (1:1, 1 mL) to yield 20.1 mg (76%) of a yellow solid. Rf = 0.36 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.67 (1H, ddd, J = 5.8, 1.6, 0.7 Hz, H-6), 8.14–7.36 (18H, m, Ar, H-3, H-4, H-5), 6.08 (1H, dd, J = 11.9, 6.0 Hz, NH2), 5.82 (1H, pt, J = 9.9, 9.7 Hz, H-4′), 5.69 (1H, pt, J = 9.6, 9.5 Hz, H-3′), 5.08 (1H, dd, J = 12.8, 2.2 Hz, H-6′a), 4.78 (1H, ddd, J = 10.0, 3.2, 2.2 Hz, H-5′), 4.53 (1H, dd, J = 12.8, 3.2 Hz, H-6′b), 4.49 (1H, d, J = 10.5 Hz, H-1′), 4.22 (1H, dd, J = 11.9, 8.1 Hz, NH2), 3.09–3.00 (1H, m, H-2′), 1.76 (15H, s, Cp*-CH3); 13C NMR (100 MHz, acetone-d6) δ (ppm): 167.9, 166.5, 166.0 (3 × C=O), 158.9 (C-2), 156.3 (C-6), 141.4 (C-4), 134.7, 134.6, 134.2, 130.8, 130.7–130.4, 130.0, 129.9, 129.5–129.4 (Ar), 127.2, 123.4 (C-3, C-5), 89.0 (Cp*), 81.8, 76.4, 76.3, 70.1 (C-1′, C-3′–C-5′), 63.3 (C-6′), 54.8 (C-2′), 9.2 (Cp*-CH3). ESI-HRMS positive mode (m/z): calcd for C42H43ClN2O7Ir+ [M-PF6]+ 915.2377. Found: 915.2381.
Complex Rh-7a
Prepared from compound 7a (17.9 mg, 0.032 mmol, 2 eq.), Rh-dimer (10.0 mg, 0.016 mmol) and TlPF6 (11.3 mg, 0.032 mmol) according to general procedure VII. The crude product was dissolved in CHCl3 (3 mL), and diisopropyl ether (12 mL) was added. The precipitation was filtered off, then washed with CHCl3-diisopropyl ether (1:1, 0.5 mL) to yield 27.1 mg (86%) of an orange solid. Rf = 0.32 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.68 (1H, d, J = 5.3 Hz, H-6), 8.13–7.43 (18H, m, Ar, H-3, H-4, H-5), 5.81 (1H, pt, J = 9.8, 9.7 Hz, H-3′ or H-4′), 5.66 (1H, pt, J = 9.8, 9.6 Hz, H-3′ or H-4′), 5.15 (1H, dd, J = 11.0, 5.0 Hz, NH2), 5.06 (1H, dd, J = 12.9, 2.2 Hz, H-6′a), 4.72 (1H, ddd, J = 10.2, 3.1, 2.2 Hz, H-5′), 4.53 (1H, d, J = 10.0 Hz, H-1′), 4.51 (1H, dd, J = 12.9, 3.1 Hz, H-6′b), 3.51 (1H, pt, J = 10.2, 9.8 Hz, NH2), 2.88–2.80 (1H, m, H-2′), 1.76 (15H, s, Cp*-CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 169.3, 166.2, 165.6 (3 × C=O), 158.0 (C-2), 153.8 (C-6), 140.4 (C-4), 134.6, 133.8, 133.3, 130.5–130.0, 129.9, 128.8–128.7, 128.6, 127.6 (Ar), 126.0, 123.4 (C-3, C-5), 97.4, 97.3 (Cp*), 79.1, 78.3, 75.3, 67.2 (C-1′, C-3′–C-5′), 62.0 (C-6′), 55.0 (C-2′), 9.3 (Cp*-CH3). ESI-HRMS positive mode (m/z): calcd for C42H43ClN2O7Rh+ [M-PF6]+ 825.1808. Found: 825.1807.
Complex Ru-7b
Prepared from compound 7b (18.5 mg, 0.033 mmol, 2.05 eq.), Ru-dimer (10.0 mg, 0.016 mmol) and TlPF6 (11.4 mg, 0.033 mmol) according to general procedure VII. The crude product was dissolved in CHCl3 (3 mL), and diisopropyl ether (12 mL) was added. The precipitation was filtered off, then washed with CHCl3-diisopropyl ether (1:2, 2 mL) to yield 28.0 mg (88%) of a brownish–orange solid. Rf = 0.51 (95:5 CHCl3-MeOH). Diastereomeric ratio = 2:1. 1H NMR (400 MHz, CDCl3) δ (ppm): 9.18 (dd, J = 4.9, 2.0 Hz, major H-6), 9.08 (dd, J = 4.8, 2.0 Hz, minor H-6), 8.21 (dd, J = 8.5, 2.0 Hz, major H-4), 8.19 (dd, J = 8.4, 2.0 Hz, minor H-4), 8.08–7.25 (m, minor and major Ar), 7.79 (dd, J = 8.5, 4.9 Hz, minor H-5), 7.77 (dd, J = 8.5, 4.9 Hz, major H-5), 6.33 (pt, J = 10.6 Hz, major NH2), 6.09 (pt, J = 9.2, 9.0 Hz, major H-3′), 6.06 (pt, J = 9.5, 9.1 Hz, minor H-3′), 6.03–5.96 (broad signal, minor NH2), 6.00, 5.94, 5.76, 5.42 (4 d, J = 6.0 Hz in each, 4 × major p-cym-CHAr), 5.82, 5.80, 5.77, 5.69 (4 d, J = 6.0 Hz in each, 4 × minor p-cym-CHAr), 5.65 (pt, J = 9.9, 9.6 Hz, major H-4′), 5.57 (pt, J = 9.2, 9.1 Hz, minor H-4′), 5.23 (d, J = 10.3 Hz, major H-1′), 5.18 (d, J = 10.5 Hz, minor H-1′), 4.97 (dd, J = 12.7, 2.2 Hz, major H-6′a), 4.72–4.68 (m, minor H-6′a or H-6′b), 4.68 (ddd, J = 10.1, 3.4, 2.2 Hz, major H-5′), 4.49–4.44 (m, minor H-6′a or H-6′b), 4.46 (dd, J = 12.7, 3.4 Hz, major H-6′b), 4.44–4.39 (m, minor H-5′), 4.13 (dd, J = 11.3, 4.9 Hz, major NH2), 3.79 (pt, J = 11.9 Hz, minor NH2), 3.22–3.15 (m, minor H-2′), 3.01 (hept, J = 6.9 Hz, minor i-Pr-CH), 2.93–2.85 (m, major H-2′), 2.80 (hept, J = 6.9 Hz, major i-Pr-CH), 2.28 (s, minor C6H4-CH3), 1.85 (s, major C6H4-CH3), 1.28, 1.27 (2 d, J = 6.9 Hz in both, 2 × minor i-Pr-CH3), 1.22, 1.17 (2 d, J = 6.9 Hz in both, 2 × major i-Pr-CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 167.7, 166.4, 165.4, 165.3 (3 × major C=O, major C-3), 166.9, 166.3, 165.3, 164.1 (3 × minor C=O, minor C-3), 152.3 (major C-6), 151.1 (minor C-6), 134.1, 133.9, 133.8, 133.7, 133.5 (2), 130.3–127.8 (minor and major Ar, C-4, C-5), 105.7, 100.5 (2 × minor p-cym-CqAr), 104.9, 100.5 (2 × major p-cym-CqAr), 88.5, 85.7, 85.5, 83.2 (4 × major p-cym-CHAr), 86.6, 86.5, 86.0, 83.3 (4 × minor p-cym-CHAr), 77.4, 75.0, 74.4, 67.7 (major C-1′, C-3′–C-5′), 76.3, 75.7, 73.3, 69.2 (minor C-1′, C-3′–C-5′), 62.7 (minor C-6′), 61.9 (major C-6′), 53.6 (minor C-2′), 53.6 (major C-2′), 30.9 (minor i-Pr-CH), 30.8 (major i-Pr-CH), 22.9, 21.8 (2 × major i-Pr-CH3), 22.5, 22.3 (2 × minor i-Pr-CH3), 18.2 (minor C6H4-CH3), 17.9 (major C6H4-CH3); ESI-HRMS positive mode (m/z): calcd for C41H41ClN3O7Ru+ [M-PF6]+ 824.1679. Found: 824.1674.
Complex Os-7b
Prepared from compound 7b (14.2 mg, 0.026 mmol, 2.05 eq.), Os-dimer (10.0 mg, 0.0126 mmol) and TlPF6 (8.7 mg, 0.025 mmol) according to general procedure VII. The crude product was dissolved in CHCl3 (3 mL), and diisopropyl ether (6 mL) was added. The precipitation was filtered off, then washed with CHCl3-diisopropyl ether (1:2, 3 mL) to yield 19.6 mg (73%) of a dark green solid. Rf = 0.38 (95:5 CHCl3-MeOH). Diastereomeric ratio = 5:4. 1H NMR (400 MHz, CDCl3) δ (ppm): 9.08 (dd, J = 4.9, 2.0 Hz, major H-6), 8.93 (dd, J = 4.8, 2.0 Hz, minor H-6), 8.30 (dd, J = 8.7, 2.0 Hz, minor H-4), 8.24 (dd, J = 8.6, 2.0 Hz, major H-4), 8.12–7.31 (m, minor and major Ar), 7.85 (dd, J = 8.7, 4.8 Hz, minor H-5), 7.69 (dd, J = 8.6, 4.9 Hz, major H-5), 6.89 (pt, J = 11.0 Hz, major NH2), 6.72–6.65 (broad signal, minor NH2), 6.27, 6.22, 6.19, 5.70 (4 d, J = 5.8 Hz in each, 4 × major p-cym-CHAr), 6.08, 6.04, 6.03, 5.92 (4 d, J = 5.7 Hz in each, 4 × minor p-cym-CHAr), 5.94 (pt, J = 9.0, 9.0 Hz, major H-3′), 5.88 (pt, J = 9.2, 8.9 Hz, minor H-3′), 5.73 (pt, J = 9.9, 9.8 Hz, major H-4′), 5.63 (pt, J = 9.4, 9.3 Hz, minor H-4′), 5.49 (d, J = 10.5 Hz, minor H-1′), 5.12 (d, J = 10.3 Hz, major H-1′), 4.98 (dd, J = 12.7, 2.2 Hz, major H-6′a), 4.73 (dd, J = 12.5, 2.5 Hz, minor H-6′a), 4.70 (ddd, J = 10.1, 3.4, 2.2 Hz, major H-5′), 4.64 (dd, J = 11.7, 4.9 Hz, major NH2), 4.52–4.44 (m, minor and major H-6′b), 4.44–4.34 (m, minor H-5′, NH2), 3.38–3.27 (m, minor H-2′), 3.13–3.05 (m, major H-2′), 2.88 (hept, J = 6.9 Hz, minor i-Pr-CH), 2.77 (hept, J = 6.9 Hz, major i-Pr-CH), 2.26 (s, minor C6H4-CH3), 1.93 (s, major C6H4-CH3), 1.26–1.20 (m, 2 × minor and major i-Pr-CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 167.9, 167.0, 166.4, 166.3, 165.4, 165.3, 164.4, 163.6 (3 × minor and major C=O, minor and major C-3), 152.7 (major C-6), 152.0 (minor C-6), 134.2, 133.9, 133.8, 133.7, 133.5, 133.4, 130.9–127.9 (minor and major Ar, C-4, C-5), 96.5, 94.2 (2 × minor p-cym-CqAr), 94.9, 92.5 (2 × major p-cym-CqAr), 80.3, 79.8 (2), 78.3 (2), 77.0, 76.5, 75.2 (2), 75.9, 75.8, 74.3, 73.6, 73.3, 69.0, 67.4 (4 × minor and major p-cym-CHAr, minor and major C-1′, C-3′–C-5′), 62.6 (minor C-6′), 61.8 (major C-6′), 54.5 (minor C-2′), 53.5 (major C-2′), 31.0 (minor i-Pr-CH), 30.9 (major i-Pr-CH), 22.4, 21.9 (2 × major i-Pr-CH3), 22.7, 22.6 (2 × minor i-Pr-CH3), 18.2 (minor C6H4-CH3), 17.9 (major C6H4-CH3). ESI-HRMS positive mode (m/z): calcd for C41H41ClN3O7Os+ [M-PF6]+ 914.2234. Found: 914.2239.
Complex Ir-7b
Prepared from compound 7b (14.2 mg, 0.026 mmol, 2.05 eq.), Ir-dimer (10.0 mg, 0.013 mmol) and TlPF6 (8.8 mg, 0.025 mmol) according to general procedure VII. The crude product was dissolved in CHCl3 (3 mL), and diisopropyl ether (6 mL) was added. The precipitation was filtered off, then washed with CHCl3-diisopropyl ether (1:1, 4 mL) to yield 18.5 mg (69%) of a yellow solid. Rf = 0.33 (95:5 CHCl3-MeOH). Diastereomeric ratio = 9:1. 1H NMR (400 MHz, CDCl3) δ (ppm): 9.09 (dd, J = 4.9, 2.0 Hz, major H-6), 8.96 (dd, J = 4.8, 2.0 Hz, minor H-6), 8.34 (dd, J = 8.7, 2.0 Hz, major H-4), 8.29 (dd, J = 8.7, 2.0 Hz, minor H-4), 8.12–7.34 (m, minor and major Ar), 7.86 (dd, J = 8.7, 4.8 Hz, minor H-5), 7.78 (dd, J = 8.7, 4.9 Hz, major H-5), 5.96 (d, J = 10.2 Hz, minor H-1′), 5.87 (pt, J = 9.1, 9.0 Hz, major H-3′ or H-4′), 5.81 (pt, J = 9.8, 9.3 Hz, minor H-3′ or H-4′), 5.78 (pt, J = 9.9, 9.5 Hz, major H-3′ or H-4′), 5.66 (pt, J = 9.2, 9.1 Hz, minor H-3′ or H-4′), 5.59 (pt, J = 10.6 Hz, major NH2), 5.36–5.29 (m, minor NH2), 5.12–5.05 (m, minor NH2), 4.99 (dd, J = 12.7, 2.2 Hz, major H-6′a), 4.89 (d, J = 10.3 Hz, major H-1′), 4.75 (ddd, J = 10.3, 3.6, 2.2 Hz, major H-5′), 4.77–4.66 (m, major NH2, minor H-6′a), 4.54 (dd, J = 12.5, 5.2 Hz, minor H-6′b), 4.48 (dd, J = 12.7, 3.6 Hz, major H-6′b), 4.29 (ddd, J = 10.0, 5.2, 2.7 Hz, minor H-5′), 3.75–3.67 (m, minor H-2′), 3.44–3.35 (m, major H-2′), 1.69 (s, major Cp*-CH3), 1.57 (s, minor Cp*-CH3); 13C NMR (100 MHz, CDCl3) δ (ppm); only the major isomer can be clearly assigned: 168.4, 166.4, 165.5, 163.7 (3 × C=O, C-3), 153.5 (C-6), 134.3, 133.8, 133.4, 130.5–130.0, 129.8, 129.6–127.6, 127.9 (Ar, C-4, C-5), 89.3 (Cp*), 78.0, 76.4, 74.6, 67.3 (C-1′, C-3′–C-5′), 61.9 (C-6′), 54.3 (C-2′), 8.8 (Cp*-CH3). ESI-HRMS positive mode (m/z): calcd for C41H42ClN3O7Ir+ [M-PF6]+ 916.2329. Found: 916.2322.
Complex Rh-7b
Prepared from compound 7b (9.2 mg, 0.017 mmol, 2.05 eq.), Rh-dimer (5.0 mg, 0.008 mmol) and TlPF6 (5.6 mg, 0.016 mmol) according to general procedure VII. The crude product was dissolved in CHCl3 (1.5 mL), and diisopropyl ether (3 mL) was added. The precipitation was filtered off, then washed with CHCl3-diisopropyl ether (1:1, 2 mL) to yield 13.5 mg (86%) of an orange solid. Rf = 0.38 (95:5 CHCl3-MeOH). Diastereomeric ratio = 5:1. 1H NMR (400 MHz, CDCl3) δ (ppm): 9.17 (dd, J = 4.9, 1.9 Hz, major H-6), 9.10 (dd, J = 4.8, 1.9 Hz, minor H-6), 8.28 (dd, J = 8.7, 1.9 Hz, major H-4), 8.17 (dd, J = 8.7, 1.9 Hz, minor H-4), 8.12–7.32 (m, minor and major Ar), 7.80 (dd, J = 8.7, 4.8 Hz, minor H-5), 7.77 (dd, J = 8.7, 4.9 Hz, major H-5), 6.01 (d, J = 10.0 Hz, minor H-1′), 5.89 (pt, J = 9.2, 9.1 Hz, major H-3′ or H-4′), 5.79 (pt, J = 9.7, 9.6 Hz, minor H-3′ or H-4′), 5.74 (pt, J = 9.8, 9.6 Hz, major H-3′ or H-4′), 5.61 (pt, J = 9.6, 9.4 Hz, minor H-3′ or H-4′), 5.07 (d, J = 10.3 Hz, major H-1′), 4.95 (dd, J = 12.7, 2.2 Hz, major H-6′a), 4.92–4.87 (broad signal, minor NH2), 4.81 (pt, J = 10.6 Hz, major NH2), 4.73 (ddd, J = 10.2, 3.7, 2.2 Hz, major H-5′), 4.63 (dd, J = 12.4, 2.6 Hz, minor H-6′a), 4.50 (dd, J = 12.4, 5.4 Hz, minor H-6′b), 4.48 (dd, J = 12.7, 3.7 Hz, major H-6′b), 4.37 (pt, J = 9.1 Hz, minor NH2), 4.25 (dd, J = 10.6, 4.4 Hz, major NH2), 4.21 (ddd, J = 10.1, 5.4, 2.6 Hz, minor H-5′), 3.57–3.47 (m, minor H-2′), 3.30–3.21 (m, major H-2′), 1.73 (s, major Cp*-CH3); 1.65 (s, minor Cp*-CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 170.0, 165.1, 164.5, 162.7 (3 × minor C=O, minor C-3), 168.3, 166.4, 165.5, 164.5 (3 × major C=O, major C-3), 152.8 (major C-6), 151.4 (minor C-6), 134.5, 134.2, 133.7 (2), 133.4, 133.3, 130.5–128.1 (minor and major C-4, C-5, Ar), 98.0, 97.9 (minor Cp*), 97.5, 97.4 (major Cp*), 79.1, 76.4, 74.3, 68.9 (minor C-1′, C-3′–C-5′), 78.9, 75.5, 74.6, 67.5 (major C-1′, C-3′–C-5′), 63.0 (minor C-6′), 62.0 (major C-6′), 54.8 (minor C-2′), 52.6 (major C-2′), 9.0 (major Cp*-CH3), 8.7 (minor Cp*-CH3). ESI-HRMS positive mode (m/z): calcd for C41H42ClN3O7Rh+ [M-PF6]+ 826.1761. Found: 826.1757.
Complex Ru-7c
Prepared from compound 7c (18.5 mg, 0.033 mmol, 2.05 eq.), Ru-dimer (10.0 mg, 0.016 mmol) and TlPF6 (11.4 mg, 0.033 mmol) according to general procedure VII. The crude product was dissolved in CHCl3 (3 mL), and diisopropyl ether (12 mL) was added. The precipitation was filtered off, then washed with CHCl3-diisopropyl ether (1:2, 2 mL) to yield 31.2 mg (99%) of a brownish–orange solid. Rf = 0.44 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.33, 8.93 (2 ×1H, 2 dd, J = 5.8, 2.2 Hz and 4.7, 2.2 Hz, respectively, H-4, H-6), 8.02–7.21 (16H, m, Ar, H-5), 6.19 (1H, dd, J = 11.6, 8.3 Hz, NH2), 6.11 (1H, pt, J = 9.2, 9.2 Hz, H-3′ or H-4′), 6.01, 5.91, 5.89, 5.40 (4 × 1H, 4 d, J = 6.0 Hz in each, 4 × p-cym-CHAr), 5.71 (1H, pt, J = 9.7, 9.6 Hz, H-3′ or H-4′), 5.11 (1H, d, J = 10.3 Hz, H-1′), 4.82 (1H, dd, J = 12.7, 2.4 Hz, H-6′a), 4.73 (1H, m, H-5′), 4.60 (1H, dd, J = 12.7, 3.6 Hz, H-6′b), 3.67 (1H, dd, J = 11.6, 6.2 Hz, NH2), 2.94–2.86 (1H, m, H-2′), 2.75 (1H, hept, J = 6.9 Hz, i-Pr-CH), 1.72 (3H, s, C6H4-CH3), 1.23, 1.09 (2 × 3H, 2 d, J = 6.9 Hz in both, 2 × i-Pr-CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 168.5, 166.4, 166.2, 165.2 (3 × C=O, C-2), 165.1 (C-6), 159.4 (C-4), 134.4, 133.8, 133.2, 130.3–128.5, 127.4 (Ar), 122.1 (C-5), 105.4, 100.8 (2 × p-cym-CqAr), 86.2, 84.2, 83.5, 82.6 (4 × p-cym-CHAr), 78.0, 77.4, 74.3, 68.2 (C-1′, C-3′–C-5′), 62.7 (C-6′), 54.2 (C-2′), 31.0 (i-Pr-CH), 23.0, 21.8 (2 × i-Pr-CH3), 18.0 (C6H4-CH3). ESI-HRMS positive mode (m/z): calcd for C41H41ClN3O7Ru+ [M-PF6]+ 824.1679. Found: 824.1681.
Complex Os-7c
Prepared from compound 7c (14.2 mg, 0.026 mmol, 2.05 eq.), Os-dimer (10.0 mg, 0.013 mmol) and TlPF6 (8.7 mg, 0.025 mmol) according to general procedure VII. The crude product was dissolved in CHCl3 (3 mL), and diisopropyl ether (6 mL) was added. The precipitation was filtered off, then washed with CHCl3-diisopropyl ether (1:1, 2 mL) to yield 26.3 mg (98%) of a dark green solid. Rf = 0.27 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.21, 8.86 (2 ×1H, 2 dd, J = 5.9, 2.2 Hz and 4.7, 2.2 Hz, respectively, H-4, H-6), 8.03–7.21 (16H, m, Ar, H-5), 6.86 (1H, dd, J = 12.0, 8.5 Hz, NH2), 6.23, 6.18, 6.14, 5.57 (4 × 1H, 4 d, J = 5.7 Hz in each, 4 × p-cym-CHAr), 6.07 (1H, pt, J = 9.2, 9.2 Hz, H-3′ or H-4′), 5.74 (1H, pt, J = 9.8, 9.6 Hz, H-3′ or H-4′), 5.03 (1H, d, J = 10.3 Hz, H-1′), 4.82 (1H, dd, J = 12.7, 2.4 Hz, H-6′a), 4.71 (1H, ddd, J = 10.2, 3.5, 2.4 Hz, H-5′), 4.60 (1H, dd, J = 12.7, 3.5 Hz, H-6′b), 4.43 (1H, dd, J = 12.0, 6.0 Hz, NH2), 3.19–3.10 (1H, m, H-2′), 2.68 (1H, hept, J = 6.9 Hz, i-Pr-CH), 1.75 (3H, s, C6H4-CH3), 1.23, 1.06 (2 × 3H, 2 d, J = 6.9 Hz in both, 2 × i-Pr-CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 168.4, 166.2, 165.6, 165.2 (3 × C=O, C-2), 165.5 (C-6), 159.2 (C-4), 134.5, 133.9, 133.2, 130.3–130.0, 129.8, 128.8–128.5, 127.3 (Ar), 122.4 (C-5), 96.0, 92.5 (2 × p-cym-CqAr), 78.3, 77.9, 77.0, 75.8, 74.5, 74.2, 73.5, 68.0 (4 × p-cym-CHAr, C-1′, C-3′–C-5′), 62.6 (C-6′), 53.9 (C-2′), 31.1 (i-Pr-CH), 23.4, 21.9 (2 × i-Pr-CH3), 17.9 (C6H4-CH3). ESI-HRMS positive mode (m/z): calcd for C41H41ClN3O7Os+ [M-PF6]+ 914.2234. Found: 914.2234.
Complex Ir-7c
Prepared from compound 7c (14.2 mg, 0.026 mmol, 2.05 eq.), Ir-dimer (10.0 mg, 0.013 mmol) and TlPF6 (8.8 mg, 0.025 mmol) according to general procedure VII. The crude product was dissolved in CHCl3 (3 mL), and diisopropyl ether (6 mL) was added. The precipitation was filtered off, then washed with CHCl3-diisopropyl ether (1:1, 4 mL) to yield 26.4 mg (99%) of a yellow solid. Rf = 0.30 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.98, 8.84 (2 ×1H, 2 dd, J = 4.8, 2.2 Hz and 5.9, 2.2 Hz, respectively, H-4, H-6), 8.01–7.29 (16H, m, Ar, H-5), 5.94 (1H, dd, J = 11.8, 5.2 Hz, NH2), 5.83 (1H, pt, J = 9.6, 9.6 Hz, H-3′ or H-4′), 5.66 (1H, pt, J = 9.5, 9.6 Hz, H-3′ or H-4′), 4.81–4.56 (4H, m, H-1′, H-5′, H-6′a,b), 4.29 (1H, dd, J = 11.8, 8.2 Hz, NH2), 3.37–3.29 (1H, m, H-2′), 1.74 (15H, s, Cp*-CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 169.3, 166.3, 165.5, 164.1 (3 × C=O, C-2), 162.3, 160.5 (C-4, C-6), 134.8, 133.8, 133.2, 130.6–130.0, 129.8, 128.9–128.5, 127.5 (Ar), 123.2 (C-5), 89.4 (Cp*), 80.5, 77.9, 75.2, 67.7 (C-1′, C-3′–C-5′), 62.8 (C-6′), 54.9 (C-2′), 9.1 (Cp*-CH3). ESI-HRMS positive mode (m/z): calcd for C41H42ClN3O7Ir+ [M-PF6]+ 916.2329. Found: 916.2324.
Complex Rh-7c
Prepared from compound 7c (9.2 mg, 0.017 mmol, 2.05 eq.), Rh-dimer (5.0 mg, 0.008 mmol) and TlPF6 (5.6 mg, 0.016 mmol) according to general procedure VII. The crude product was dissolved in CHCl3 (1.5 mL), and diisopropyl ether (3 mL) was added. The precipitation was filtered off, then washed with CHCl3-diisopropyl ether (1:1, 4 mL) to yield 15.5 mg (99%) of an orange solid. Rf = 0.24 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.02, 8.92 (2 ×1H, 2 dd, J = 4.9, 2.3 Hz and 5.7, 2.3 Hz, respectively, H-4, H-6), 8.03–7.29 (16H, m, Ar, H-5), 5.84 (1H, pt, J = 9.6, 9.6 Hz, H-3′ or H-4′), 5.67 (1H, pt, J = 9.7, 9.6 Hz, H-3′ or H-4′), 5.15 (1H, dd, J = 12.1, 3.7 Hz, NH2), 4.85–4.58 (3H, m, H-5′, H-6′a,b), 4.72 (1H, d, J = 10.2 Hz, H-1′), 3.46 (1H, pt, J = 10.0 Hz, NH2), 3.21–3.13 (1H, m, H-2′), 1.81 (15H, s, Cp*-CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 169.4, 166.3, 165.5, 164.7 (3 × C=O, C-2), 161.7, 160.4 (C-4, C-6), 134.8, 133.8, 133.2, 130.6–130.0, 129.9, 128.9–128.5, 127.5 (Ar), 122.8 (C-5), 97.9, 97.8 (Cp*), 80.0, 78.4, 75.4, 67.8 (C-1′, C-3′–C-5′), 62.8 (C-6′), 55.3 (C-2′), 9.5 (Cp*-CH3). ESI-HRMS positive mode (m/z): calcd for C41H42ClN3O7Rh+ [M-PF6]+ 826.1761. Found: 826.1757.
Complex Ru-7d
Prepared from compound 7d (18.5 mg, 0.033 mmol, 2.05 eq.), Ru-dimer (10.0 mg, 0.016 mmol) and TlPF6 (11.4 mg, 0.033 mmol) according to general procedure VII. The crude product was dissolved in CHCl3 (3 mL), and diisopropyl ether (6 mL) was added. The precipitation was filtered off, then washed with CHCl3-diisopropyl ether (1:2, 6 mL) to yield 28.8 mg (91%) of a brown solid. Rf = 0.35 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.09 (1H, s, H-3), 9.02 (1H, dd, J = 3.2, 1.1 Hz, H-5), 8.76 (1H, d, J = 3.2 Hz, H-6), 8.08–7.26 (15H, m, Ar), 6.26 (1H, dd, J = 11.5, 8.3 Hz, NH2), 6.07 (1H, pt, J = 9.3, 9.2 Hz, H-3′ or H-4′), 6.01–5.96 (3H, m, 3 × p-cym-CHAr), 5.72 (1H, pt, J = 9.7, 9.7 Hz, H-3′ or H-4′), 5.44 (1H, d, J = 6.0 Hz, p-cym-CHAr), 5.03 (1H, d, J = 10.2 Hz, H-1′), 4.98 (1H, dd, J = 12.8, 2.2 Hz, H-6′a), 4.76 (1H, ddd, J = 10.2, 3.4, 2.2 Hz, H-5′), 4.48 (1H, dd, J = 12.8, 3.4 Hz, H-6′b), 3.54 (1H, dd, J = 11.5, 6.7 Hz, NH2), 2.81 (1H, hept, J = 6.9 Hz, i-Pr-CH), 2.73–2.64 (1H, m, H-2′), 1.77 (3H, s, C6H4-CH3), 1.23, 1.16 (2 × 3H, 2 d, J = 6.9 Hz in both, 2 × i-Pr-CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 168.4, 166.3, 165.4 (3 × C=O), 154.7 (C-2), 150.1, 146.3, 144.6 (C-3, C-5, C-6), 134.4, 133.9, 133.4, 130.3–130.0, 129.7, 128.8–128.6, 127.6 (Ar), 105.4, 101.0 (2 × p-cym-CqAr), 87.4, 84.9, 83.4, 83.3 (4 × p-cym-CHAr), 77.0, 76.9, 74.6, 67.7 (C-1′, C-3′–C-5′), 61.9 (C-6′), 53.8 (C-2′), 31.0 (i-Pr-CH), 22.9, 21.9 (2 × i-Pr-CH3), 17.9 (C6H4-CH3). ESI-HRMS positive mode (m/z): calcd for C41H41ClN3O7Ru+ [M-PF6]+ 824.1679. Found: 824.1673.
Complex Os-7d
Prepared from compound 7d (14.2 mg, 0.026 mmol, 2.05 eq.), Os-dimer (10.0 mg, 0.013 mmol) and TlPF6 (8.7 mg, 0.025 mmol) according to general procedure VII. The crude product was dissolved in CHCl3 (3 mL), and diisopropyl ether (6 mL) was added. The precipitation was filtered off, then washed with CHCl3-diisopropyl ether (1:2, 3 mL) to yield 24.4 mg (91%) of a brown solid. Rf = 0.29 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.09 (1H, s, H-3), 8.87 (1H, dd, J = 3.2, 1.1 Hz, H-5), 8.69 (1H, d, J = 3.2 Hz, H-6), 8.09–7.28 (15H, m, Ar), 6.96 (1H, dd, J = 11.9, 8.4 Hz, NH2), 6.27–6.24 (2H, m, 2 × p-cym-CHAr), 6.20 (1H, d, J = 5.7 Hz, p-cym-CHAr), 6.03 (1H, pt, J = 9.2, 9.2 Hz, H-3′ or H-4′), 5.75 (1H, pt, J = 9.7, 9.6 Hz, H-3′ or H-4′), 5.62 (1H, d, J = 5.7 Hz, p-cym-CHAr), 4.99 (1H, dd, J = 12.8, 2.2 Hz, H-6′a), 4.94 (1H, d, J = 10.2 Hz, H-1′), 4.75 (1H, ddd, J = 10.2, 3.4, 2.2 Hz, H-5′), 4.49 (1H, dd, J = 12.8, 3.4 Hz, H-6′b), 4.28 (1H, dd, J = 11.9, 6.4 Hz, NH2), 2.97–2.88 (1H, m, H-2′), 2.74 (1H, hept, J = 6.9 Hz, i-Pr-CH), 1.80 (3H, s, C6H4-CH3), 1.23, 1.14 (2 × 3H, 2 d, J = 6.9 Hz in both, 2 × i-Pr-CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 168.5, 166.3, 165.3 (3 × C=O), 153.6 (C-2), 150.4, 147.1, 144.3 (C-3, C-5, C-6), 134.5, 133.9, 133.4, 130.3–130.0, 129.7, 128.8–128.6, 127.5 (Ar), 95.9, 92.7 (2 × p-cym-CqAr), 79.5, 77.6, 76.7, 76.6, 74.6, 74.3, 74.1, 67.5 (4 × p-cym-CHAr, C-1′, C-3′–C-5′), 61.8 (C-6′), 53.6 (C-2′), 31.1 (i-Pr-CH), 23.3, 22.1 (2 × i-Pr-CH3), 17.7 (C6H4-CH3). ESI-HRMS positive mode (m/z): calcd for C41H41ClN3O7Os+ [M-PF6]+ 914.2234. Found: 914.2233.
Complex Ir-7d
Prepared from compound 7d (14.2 mg, 0.026 mmol, 2.05 eq.), Ir-dimer (10.0 mg, 0.013 mmol) and TlPF6 (8.8 mg, 0.025 mmol) according to general procedure VII. The crude product was dissolved in CHCl3 (3 mL), and diisopropyl ether (6 mL) was added. The precipitation was filtered off, then washed with CHCl3-diisopropyl ether (1:1, 4 mL) to yield 26.5 mg (99%) of a yellow solid. Rf = 0.24 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.18 (1H, s, H-3), 8.76 (1H, d, J = 3.2 Hz, H-6), 8.54 (1H, dd, J = 3.2, 1.2 Hz, H-5), 8.10–7.32 (15H, m, Ar), 5.94 (1H, dd, J = 11.8, 6.4 Hz, NH2), 5.84 (1H, pt, J = 9.8, 9.7 Hz, H-3′ or H-4′), 5.68 (1H, pt, J = 9.6, 9.5 Hz, H-3′ or H-4′), 5.03 (1H, dd, J = 12.9, 2.2 Hz, H-6′a), 4.74 (1H, ddd, J = 10.1, 3.0, 2.2 Hz, H-5′), 4.56 (1H, d, J = 10.1 Hz, H-1′), 4.50 (1H, dd, J = 12.9, 3.0 Hz, H-6′b), 4.27 (1H, dd, J = 11.8, 7.9 Hz, NH2), 3.17–3.09 (1H, m, H-2′), 1.73 (15H, s, Cp*-CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 169.3, 166.2, 165.5 (3 × C=O), 152.1 (C-2), 147.8, 147.5, 145.5 (C-3, C-5, C-6), 134.8, 133.8, 133.3, 130.6–130.0, 129.8, 128.8–128.6, 128.5, 127.5 (Ar), 89.7 (Cp*), 78.9, 77.7, 75.4, 67.1 (C-1′, C-3′–C-5′), 61.8 (C-6′), 54.5 (C-2′), 8.9 (Cp*-CH3). ESI-HRMS positive mode (m/z): calcd for C41H42ClN3O7Ir+ [M-PF6]+ 916.2329. Found: 916.2331.
Complex Rh-7d
Prepared from compound 7d (9.2 mg, 0.017 mmol, 2.05 eq.), Rh-dimer (5.0 mg, 0.008 mmol) and TlPF6 (5.6 mg, 0.016 mmol) according to general procedure VII. The crude product was dissolved in CHCl3 (1.5 mL), and diisopropyl ether (3 mL) was added. The precipitation was filtered off, then washed with CHCl3-diisopropyl ether (1:1, 4 mL) to yield 15.1 mg (96%) of an orange solid. Rf = 0.21 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.19 (1H, s, H-3), 8.83 (1H, d, J = 3.1 Hz, H-6), 8.61 (1H, dd, J = 3.1, 1.2 Hz, H-5), 8.11–7.32 (15H, m, Ar), 5.83 (1H, pt, J = 9.8, 9.7 Hz, H-3′ or H-4′), 5.69 (1H, pt, J = 9.7, 9.6 Hz, H-3′ or H-4′), 5.13 (1H, dd, J = 11.4, 6.1 Hz, NH2), 5.05 (1H, dd, J = 12.9, 2.2 Hz, H-6′a), 4.75 (1H, ddd, J = 10.2, 2.9, 2.2 Hz, H-5′), 4.69 (1H, d, J = 10.1 Hz, H-1′), 4.51 (1H, dd, J = 12.9, 2.9 Hz, H-6′b), 3.54 (1H, pt, J = 9.9 Hz, NH2), 3.02–2.93 (1H, m, H-2′), 1.78 (15H, s, Cp*-CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 169.4, 166.2, 165.5 (3 × C=O), 152.6 (C-2), 147.1, 146.8, 145.6 (C-3, C-5, C-6), 134.8, 133.8, 133.3, 130.6–130.0, 129.8–128.7, 128.6, 127.5 (Ar), 98.1, 98.0 (Cp*), 78.3, 78.2, 75.5, 67.1 (C-1′, C-3′–C-5′), 61.7 (C-6′), 54.9 (C-2′), 9.3 (Cp*-CH3). ESI-HRMS positive mode (m/z): calcd for C41H42ClN3O7Rh+ [M-PF6]+ 826.1761. Found: 826.1754.
Complex Ru-7e
Prepared from compound 7e (21.2 mg, 0.035 mmol, 2.15 eq.), Ru-dimer (10.0 mg, 0.016 mmol) and TlPF6 (11.4 mg, 0.033 mmol) according to general procedure VII. The crude product was dissolved in CHCl3 (3 mL), and diisopropyl ether (12 mL) was added. The precipitation was filtered off, then washed with CHCl3-diisopropyl ether (1:2, 2 mL) to yield 27.2 mg (82%) of an orange solid. Rf = 0.66 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.38 (1H, d, J = 9.3 Hz, H-3 or H-4 or H-5 or H-8), 8.37 (1H, d, J = 8.7 Hz, H-3 or H-4 or H-5 or H-8), 8.16–7.34 (19H, m, Ar, H-3 and/or H-4 and/or H-5 and/or H-8, H-6, H-7), 6.58 (1H, dd, J = 11.4, 6.3 Hz, NH2), 6.22–6.18 (3H, m, 3 × p-cym-CHAr), 6.04 (1H, pt, J = 9.5, 9.2 Hz, H-3′ or H-4′), 5.83 (1H, pt, J = 9.7, 9.7 Hz, H-3′ or H-4′), 5.51 (1H, d, J = 6.1 Hz, p-cym-CHAr), 5.14 (1H, d, J = 10.1 Hz, H-1′), 5.05 (1H, dd, J = 12.7, 2.1 Hz, H-6′a), 4.93 (1H, ddd, J = 9.8, 3.3, 2.1 Hz, H-5′), 4.53 (1H, dd, J = 12.7, 3.3 Hz, H-6′b), 3.61 (1H, dd, J = 11.4, 7.6 Hz, NH2), 2.89 (1H, hept, J = 6.9 Hz, i-Pr-CH), 2.82–2.74 (1H, m, H-2), 1.59 (3H, s, C6H4-CH3), 1.26, 1.19 (2 × 3H, 2 d, J = 6.9 Hz in both, 2 × i-Pr-CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 168.9, 166.3, 165.7, 163.5, 149.3 (3 × C=O, C-2, C-8a), 141.3, 134.4, 133.8, 133.3, 131.4, 131.3, 130.4–130.0, 129.9, 129.3, 129.0–128.6, 127.8, 120.0 (Ar, C-3–C-8, C-4a), 104.4, 100.6 (2 × p-cym-CqAr), 87.3, 85.2, 83.3, 82.3, 79.9, 77.4, 74.6, 67.3 (4 × p-cym-CHAr, C-1′, C-3′–C-5′), 62.1 (C-6′), 54.6 (C-2′), 31.0 (i-Pr-CH), 22.8, 21.7 (2 × i-Pr-CH3), 17.7 (C6H4-CH3). ESI-HRMS positive mode (m/z): calcd for C46H44ClN2O7Ru+ [M-PF6]+ 873.1885. Found: [M-PF6]+ 873.1876.
Complex Os-7e
Prepared from compound 7e (15.6 mg, 0.026 mmol, 2.05 eq.), Os-dimer (10.0 mg, 0.013 mmol) and TlPF6 (8.7 mg, 0.025 mmol) according to general procedure VII. The crude product was dissolved in CHCl3 (1 mL), and diisopropyl ether (8 mL) was added. The precipitation was filtered off, then washed with CHCl3-diisopropyl ether (1:4, 2 mL) to yield 26.9 mg (96%) of a dark green solid. Rf = 0.58 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.18 (1H, d, J = 9.1 Hz, H-3 or H-4 or H-5 or H-8), 8.29 (1H, d, J = 8.7 Hz, H-3 or H-4 or H-5 or H-8), 8.16–7.33 (19H, m, Ar, H-3 and/or H-4 and/or H-5 and/or H-8, H-6, H-7), 7.25 (1H, dd, J = 11.7, 6.9 Hz, NH2), 6.54–6.51 (2H, m, 2 × p-cym-CHAr), 6.48 (1H, d, J = 5.7 Hz, p-cym-CHAr), 5.96 (1H, pt, J = 9.2, 9.1 Hz, H-3′ or H-4′), 5.83 (1H, pt, J = 9.8, 9.6 Hz, H-3′ or H-4′), 5.71 (1H, d, J = 5.7 Hz, p-cym-CHAr), 5.04 (1H, dd, J = 12.7, 2.3 Hz, H-6′a), 4.99 (1H, d, J = 10.2 Hz, H-1′), 4.90 (1H, ddd, J = 10.0, 3.4, 2.3 Hz, H-5′), 4.58 (1H, dd, J = 11.9, 7.3 Hz, NH2), 4.52 (1H, dd, J = 12.7, 3.4 Hz, H-6′b), 3.03–2.95 (1H, m, H-2′), 2.82 (1H, hept, J = 6.9 Hz, i-Pr-CH), 1.63 (3H, s, C6H4-CH3), 1.28, 1.18 (2 × 3H, 2 d, J = 6.9 Hz in both, 2 × i-Pr-CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 168.9, 166.3, 165.7, 163.2, 149.4 (3 × C=O, C-2, C-8a), 141.6, 134.4, 133.8, 133.3, 132.5, 131.4, 130.5–130.0, 129.9, 129.4, 129.0–128.6, 127.8, 119.5 (C-3–C-8, C-4a, Ar), 95.0, 92.4 (2 × p-cym-CqAr), 81.0, 79.3, 77.4, 76.8, 75.2, 74.5, 73.5, 67.2 (4 × p-cym-CHAr, C-1′, C-3′–C-5′), 62.1 (C-6′), 54.8 (C-2′), 31.1 (i-Pr-CH), 23.1, 21.9 (2 × i-Pr-CH3), 17.6 (C6H4-CH3). ESI-HRMS positive mode (m/z): calcd for C46H44ClN2O7Os+ [M-PF6]+ 963.2439. Found: 963.2432.
Complex Ir-7e
Prepared from compound 7e (15.5 mg, 0.026 mmol, 2.05 eq.), Ir-dimer (10.0 mg, 0.013 mmol) and TlPF6 (8.8 mg, 0.025 mmol) according to general procedure VII, with a slight modification. During the reaction, the desired product Ir-7e also precipitated from the reaction mixture. Therefore, after completion of the reaction, the solvents were removed under reduced pressure. Then, the residue was treated with CH3CN (10 mL), and the insoluble TlCl was filtered off. The resulting solution was evaporated in vacuo. The residue was dissolved in a mixture of CH3CN (0.1 mL) and CHCl3 (2 mL), and diisopropyl ether (8 mL) was added. The precipitated product was filtered off, then washed with CHCl3-diisopropyl ether (1:2, 2 mL) to yield 24.6 mg (88%) of a dark yellow solid. Rf = 0.41 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CD3CN) δ (ppm): 8.72 (1H, d, J = 9.0 Hz, H-3 or H-4 or H-5 or H-8), 8.60 (1H, d, J = 8.7 Hz, H-3 or H-4 or H-5 or H-8), 8.16–7.43 (19H, Ar, H-3 and/or H-4 and/or H-5 and/or H-8, H-6, H-7), 5.80 (1H, pt, J = 9.8, 9.4 Hz, H-3′ or H-4′), 5.73 (1H, pt, J = 9.6, 9.3 Hz, H-3′ or H-4′), 5.27 (1H, d, J = 12.9, NH2), 4.80 (1H, dd, J = 12.4, 2.2 Hz, H-6′a), 4.82–4.73 (1H, broad signal, NH2), 4.63 (1H, dd, J = 12.4, 3.9 Hz, H-6′b), 4.62–4.56 (1H, m, H-5′), 4.55 (1H, d, J = 10.5 Hz, H-1′), 3.50–3.52 (1H, m, H-2′), 1.70 (15H, s, Cp*-CH3); 13C NMR (90 MHz, CD3CN) δ (ppm): 168.1, 166.8, 166.2, 162.6, 147.1 (3 × C=O, C-2, C-8a), 142.9, 134.9, 134.8, 134.4, 132.6, 131.9, 130.6–130.0, 129.9, 129.6–129.4, 120.4 (Ar, C-3–C-8, C-4a), 89.7 (Cp*), 84.3, 76.4 (2), 70.0 (C-1′, C-3′–C-5′), 63.3 (C-6′), 54.4 (C-2′), 9.9 (Cp*-CH3). ESI-HRMS positive mode (m/z): calcd for C46H46N2O7Ir+ [M+H-Cl-PF6]+ 931.2929. Found: 9631.2920.
Complex Rh-7e
Prepared from compound 7e (20.0 mg, 0.033 mmol, 2.05 eq.), Rh-dimer (10.0 mg, 0.016 mmol) and TlPF6 (11.3 mg, 0.032 mmol) according to general procedure VII, with a slight modification. During the reaction, the desired product, Rh-7e, also precipitated from the reaction mixture. Therefore, after completion of the reaction, the solvents were removed under reduced pressure. Then, the residue was treated with CH3CN (10 mL), and the insoluble TlCl was filtered off. The resulting solution was evaporated in vacuo. The residue was dissolved in a mixture of CH3CN (0.1 mL) and CHCl3 (2 mL), and diisopropyl ether (8 mL) was added. The precipitated product was filtered off, then washed with CHCl3-diisopropyl ether (1:4, 2 mL) to yield 29.9 mg (90%) of a red solid. Rf = 0.58 (95:5 CHCl3-MeOH). 1H NMR (400 MHz, CD3CN) δ (ppm): 8.86 (1H, d, J = 8.9 Hz, H-3 or H-4 or H-5 or H-8), 8.61 (1H, d, J = 8.7 Hz, H-3 or H-4 or H-5 or H-8), 8.12–7.43 (19H, m, Ar, H-3 and/or H-4 and/or H-5 and/or H-8, H-6, H-7), 5.83 (1H, dd, J = 10.2, 9.3 Hz, H-3′ or H-4′), 5.68 (1H, pt, J = 9.8, 9.4 Hz, H-3′ or H-4′), 4.80 (1H, dd, J = 12.6, 2.6 Hz, H-6′a), 4.71 (1H, d, J = 10.3 Hz, H-1′), 4.64 (1H, dd, J = 12.6, 4.0 Hz, H-6′b), 4.63–4.58 (1H, m, H-5′), 4.49 (1H, d, J = 12.6 Hz, NH2), 3.95 (1H, pt, J = 10.4 Hz, NH2), 3.24–3.16 (1H, m, H-2′), 1.68 (15H, s, Cp*-CH3); 13C NMR (90 MHz, CD3CN) δ (ppm): 168.2, 166.9, 166.3, 162.7, 147.2 (3 × C=O, C-2, C-8a), 142.3, 134.9, 134.8, 134.4, 132.0, 131.6, 130.8, 130.7–130.5, 130.0, 129.9, 129.8–129.3, 120.8 (Ar, C-3–C-8, C-4a), 98.1, 98.0 (Cp*), 82.1, 77.3, 76.3, 70.0 (C-1′, C-3′–C-5′), 63.4 (C-6′), 54.4 (C-2′), 10.0 (Cp*-CH3). ESI-HRMS positive mode (m/z): calcd for C46H44ClN2O7Rh+ [M-PF6]+ 875.1965. Found: 875.1953.

 ,
, 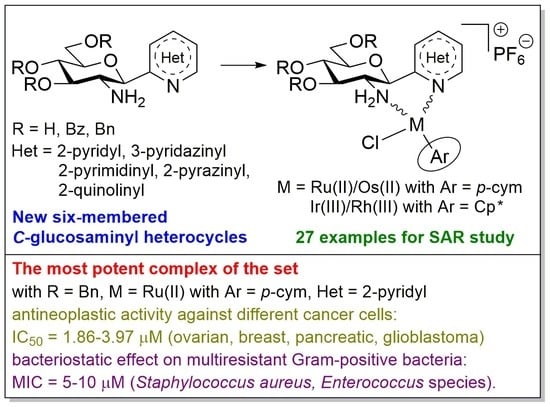











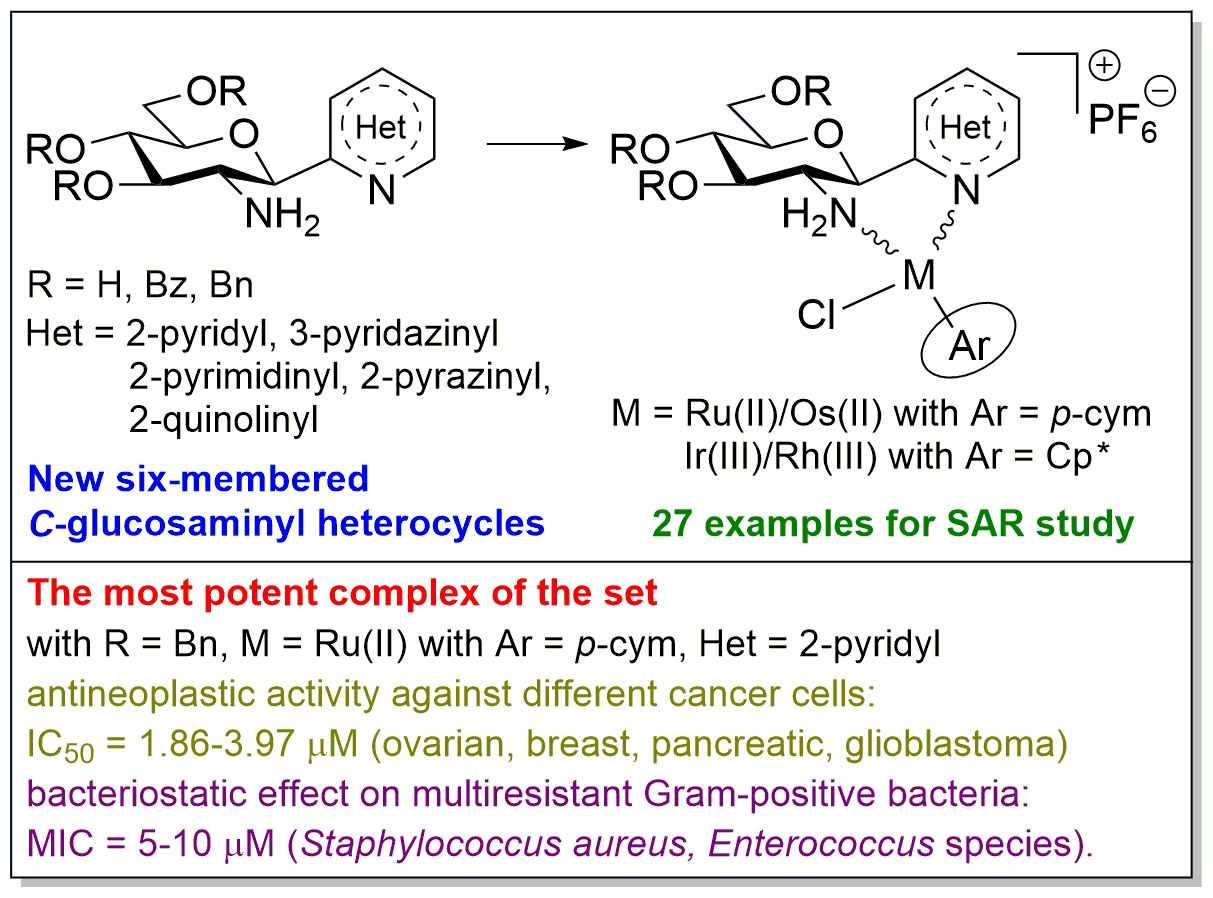{kind=link}
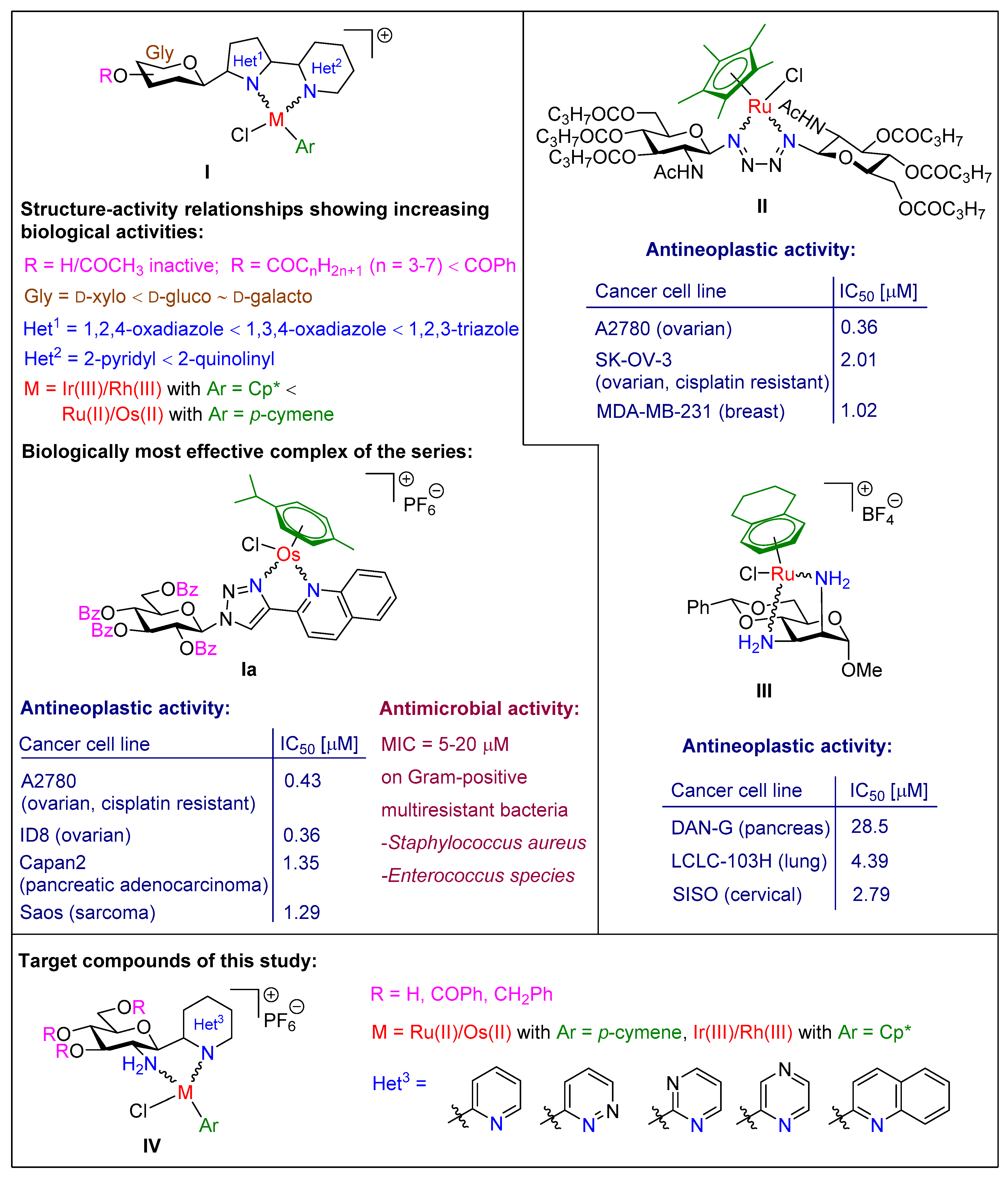{kind=link}
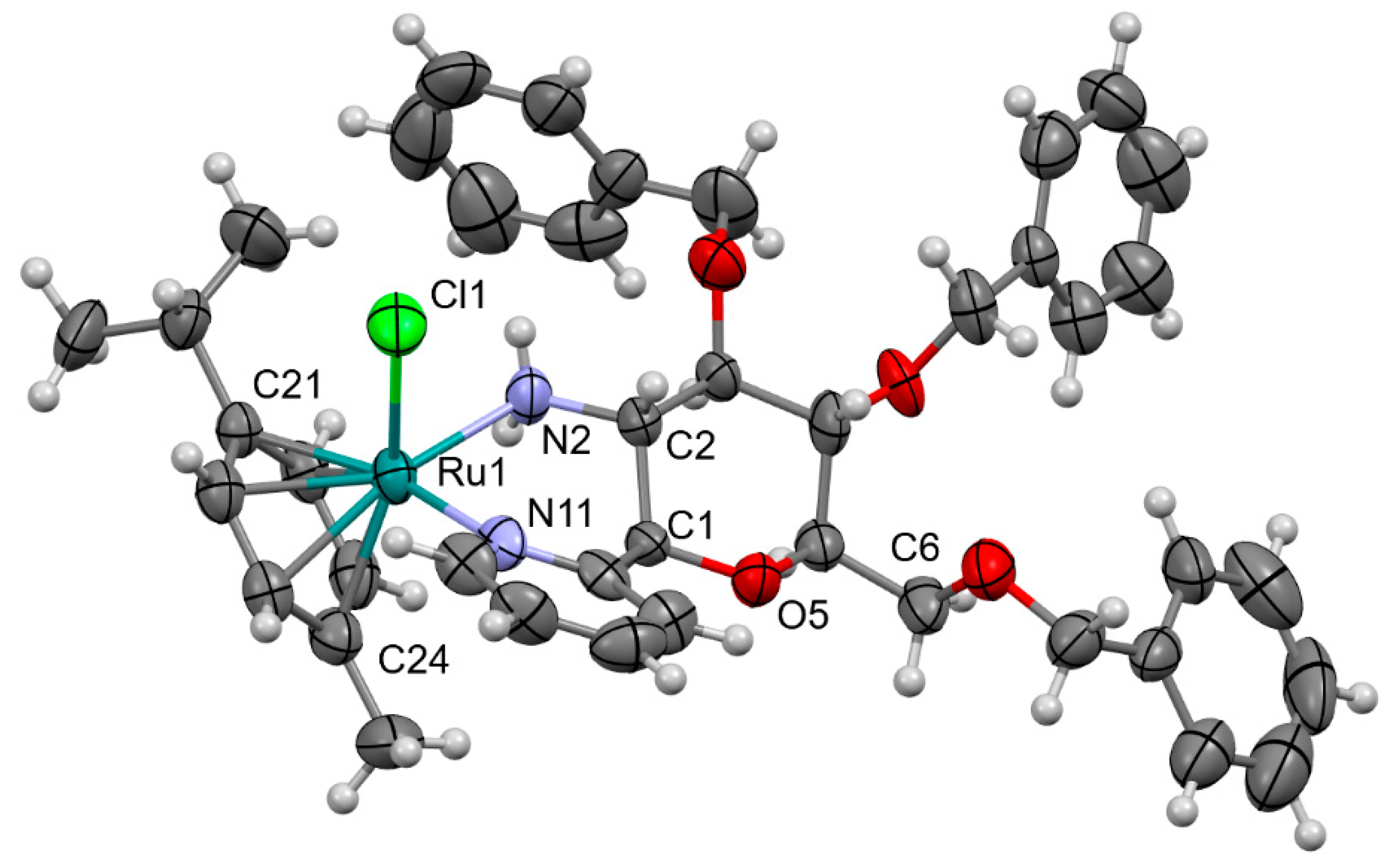{kind=link}
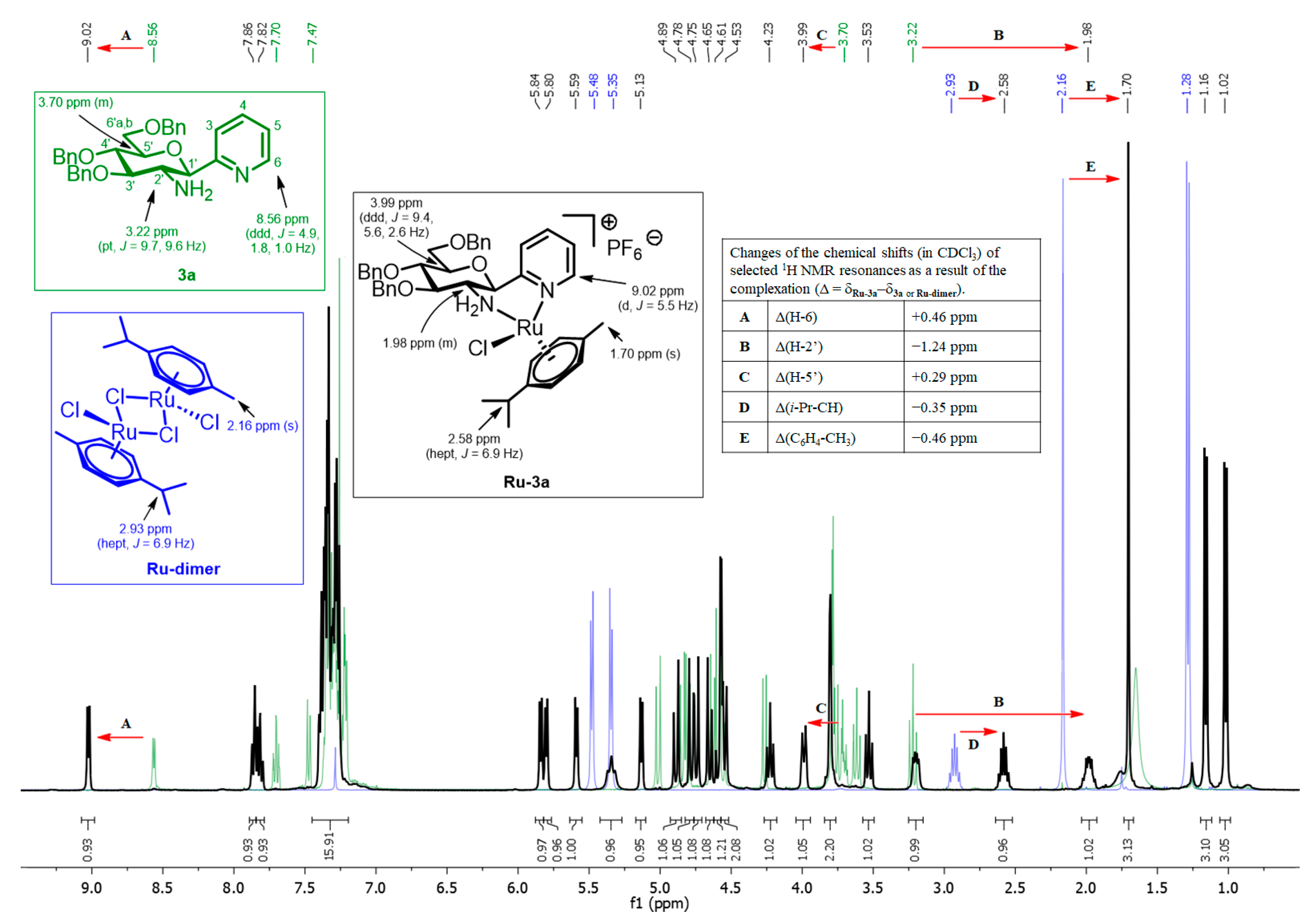{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
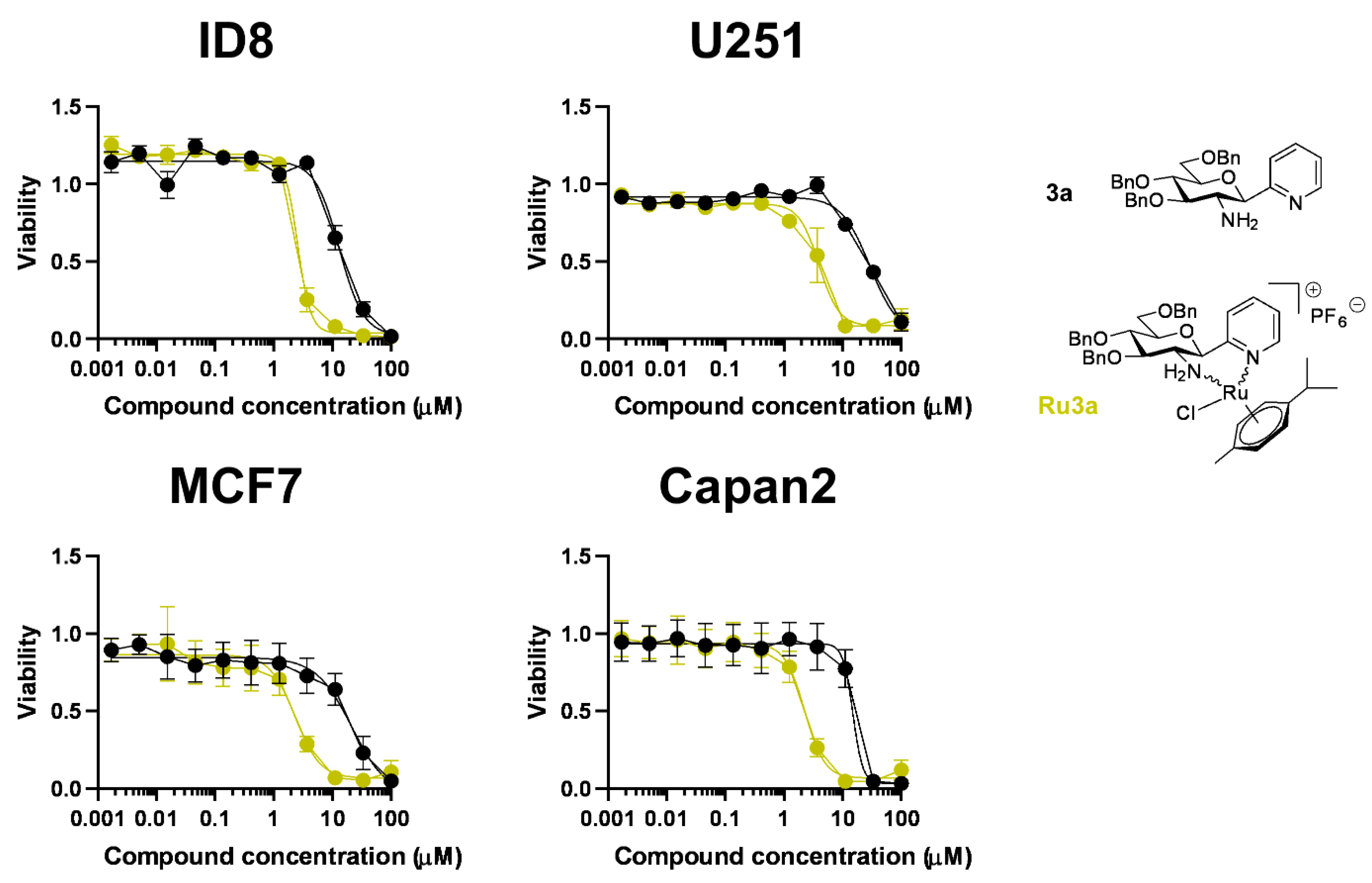{kind=link}
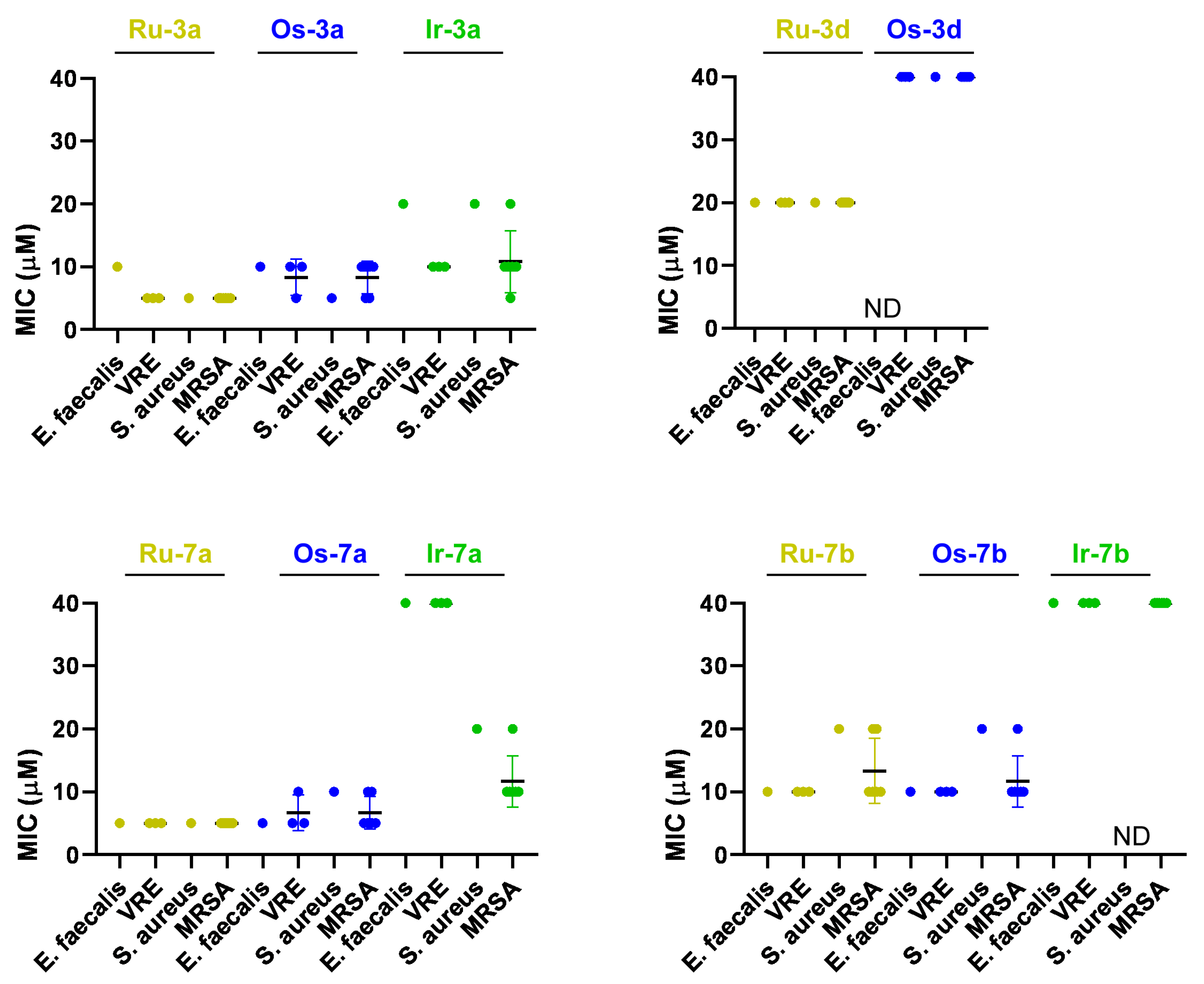{kind=link}
{kind=link}
 Conditions: (i) 1.2–2.0 equiv. of Het-Hlg (Hlg = Br, I), 2.5 M solution of n-Bu-Li in hexane (1.2–2.0 equiv.), dry THF,
Conditions: (i) 1.2–2.0 equiv. of Het-Hlg (Hlg = Br, I), 2.5 M solution of n-Bu-Li in hexane (1.2–2.0 equiv.), dry THF, 




 Conditions: (i) 1M solution of BCl3 in CH2Cl2 (5 equiv.), dry CH2Cl2, −78 °C; (ii) 1M solution of BCl3 in CH2Cl2 (4 equiv.), pentamethylbenzene (9 equiv.), dry CH2Cl2, −78 °C; (iii) H2, Pd(C), dry EtOH, reflux; (iv) Sn powder, ccHCl, THF-H2O (2:1), r.t.
Conditions: (i) 1M solution of BCl3 in CH2Cl2 (5 equiv.), dry CH2Cl2, −78 °C; (ii) 1M solution of BCl3 in CH2Cl2 (4 equiv.), pentamethylbenzene (9 equiv.), dry CH2Cl2, −78 °C; (iii) H2, Pd(C), dry EtOH, reflux; (iv) Sn powder, ccHCl, THF-H2O (2:1), r.t.




 Conditions: (i) 6 equiv. of benzoyl chloride, 2 equiv. of Zn(OTf)2, dry ClCH2CH2Cl, r.t.; (ii) Zn powder, ccHCl or 2M aq. HCl, THF-H2O (2:1), r.t.; (iii) H2, Pd(C), dry EtOH, reflux; (iv) 2 equiv. of Boc2O, 1,4-dioxane-H2O (1:1), r.t.; (v) 7.2 equiv. of benzoyl chloride, dry pyridine, 60 °C; (vi) 2 equiv. of CF3COOH, dry CH2Cl2, r.t.
Conditions: (i) 6 equiv. of benzoyl chloride, 2 equiv. of Zn(OTf)2, dry ClCH2CH2Cl, r.t.; (ii) Zn powder, ccHCl or 2M aq. HCl, THF-H2O (2:1), r.t.; (iii) H2, Pd(C), dry EtOH, reflux; (iv) 2 equiv. of Boc2O, 1,4-dioxane-H2O (1:1), r.t.; (v) 7.2 equiv. of benzoyl chloride, dry pyridine, 60 °C; (vi) 2 equiv. of CF3COOH, dry CH2Cl2, r.t.










