

Neuroprotective Effect of α-Lipoic Acid against Aβ25–35-Induced Damage in BV2 Cells
 ,
,
Abstract
:1. Introduction
2. Results
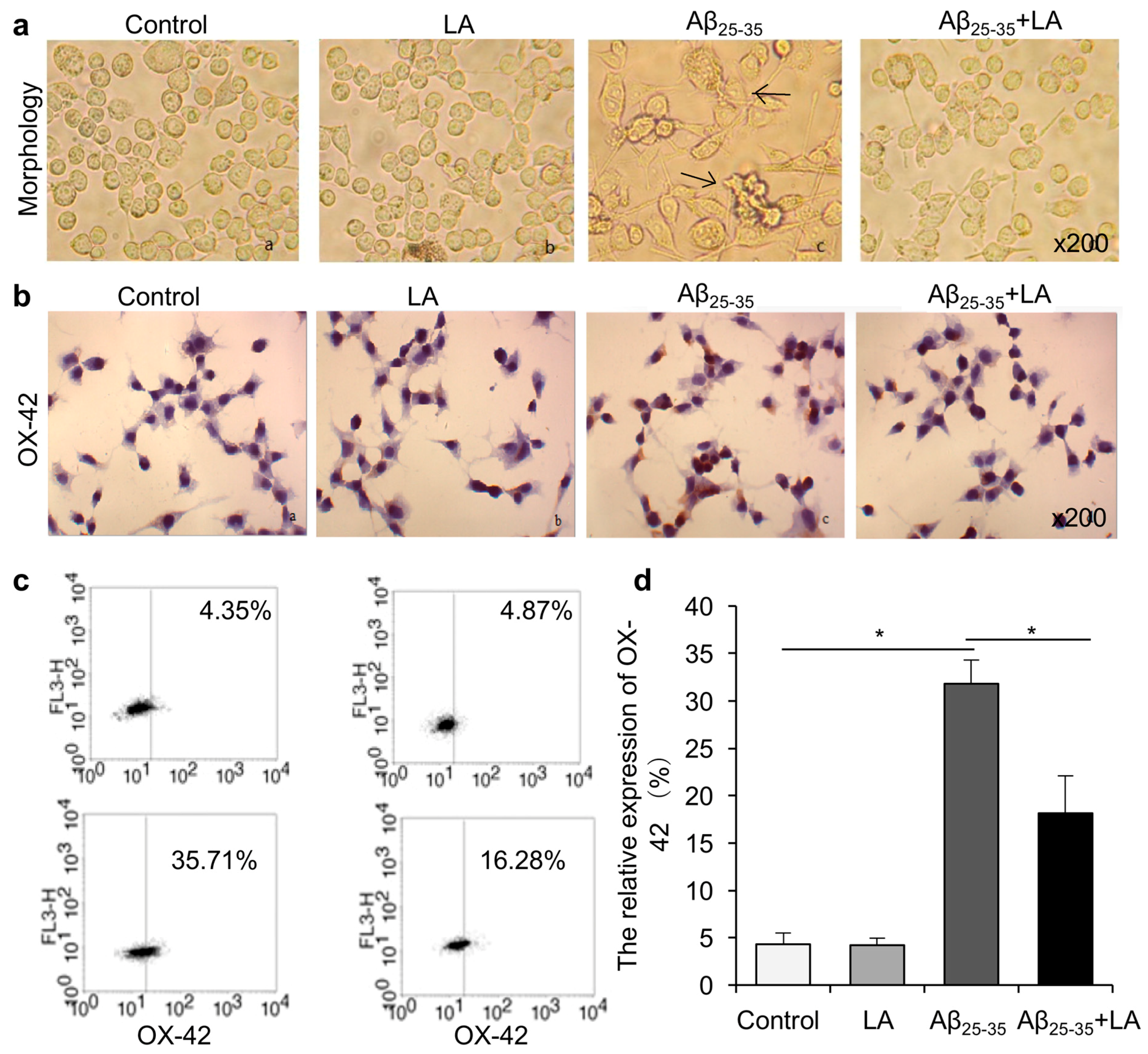
2.1. LA Improves Aβ25–35-Induced Morphology Changes and Activation in BV2 Cells
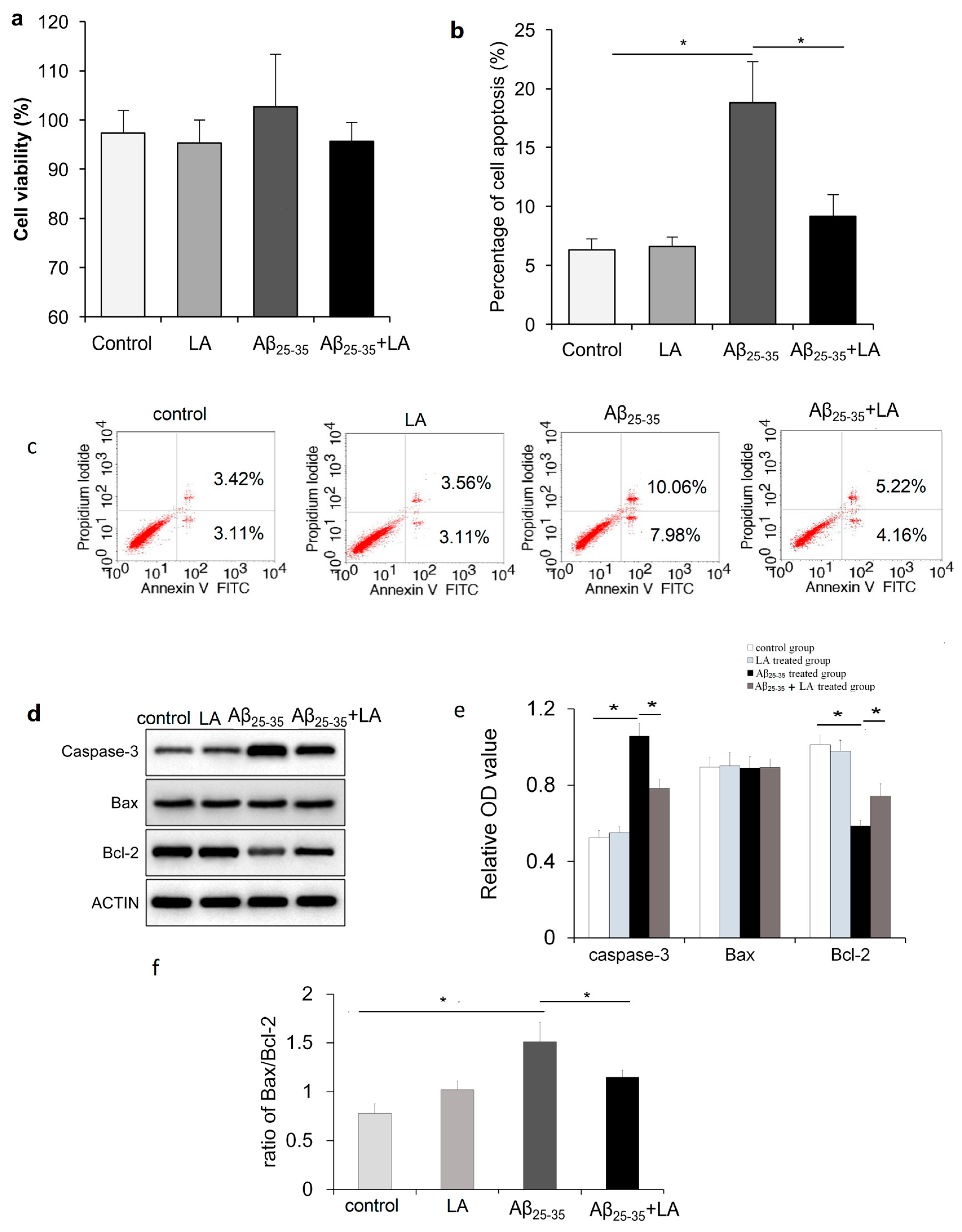
2.2. LA and Aβ25–35 Do Not Affect BV2 Cell Viability
2.3. LA rescues Cell Apoptosis Promoted by Aβ25–35
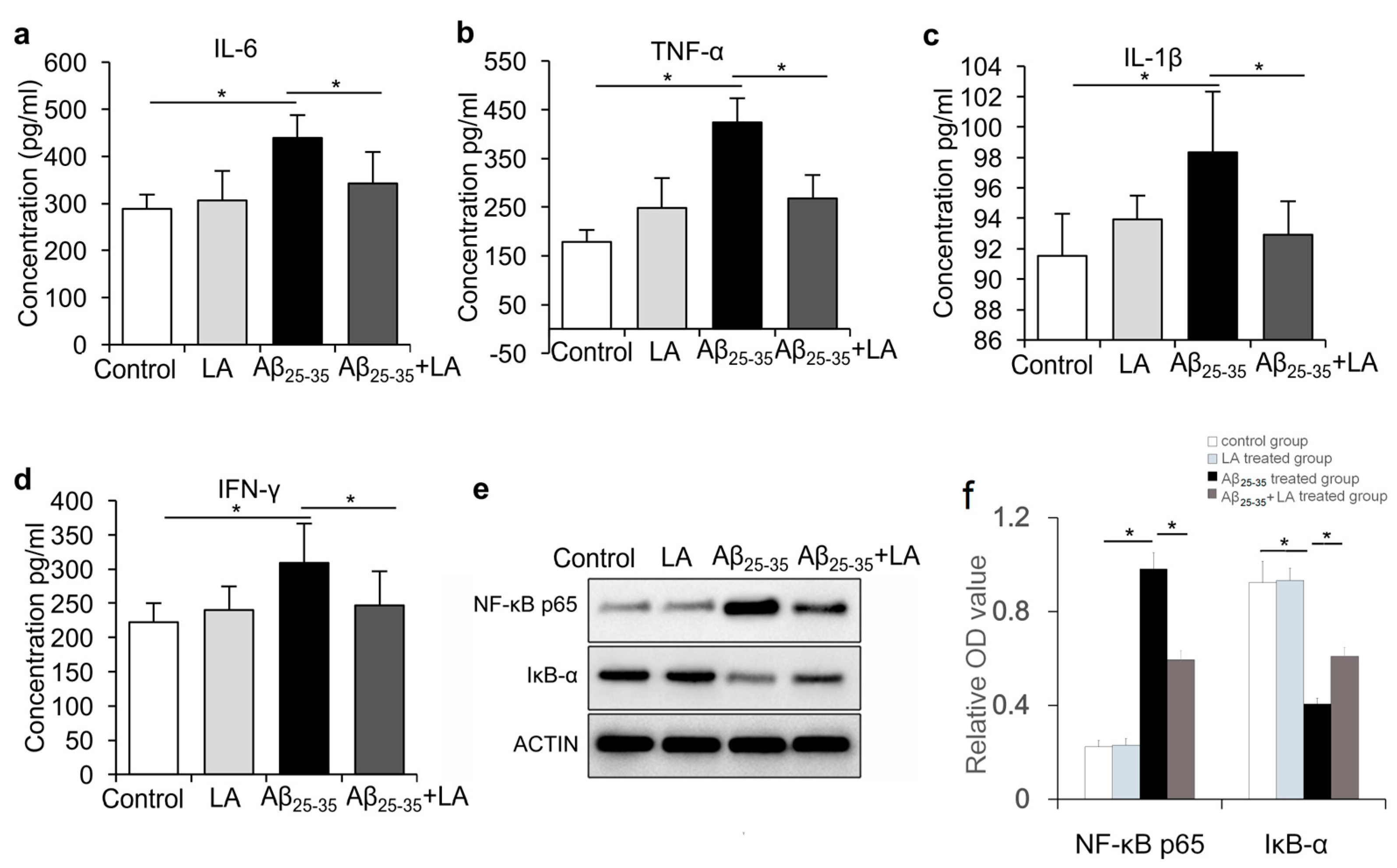
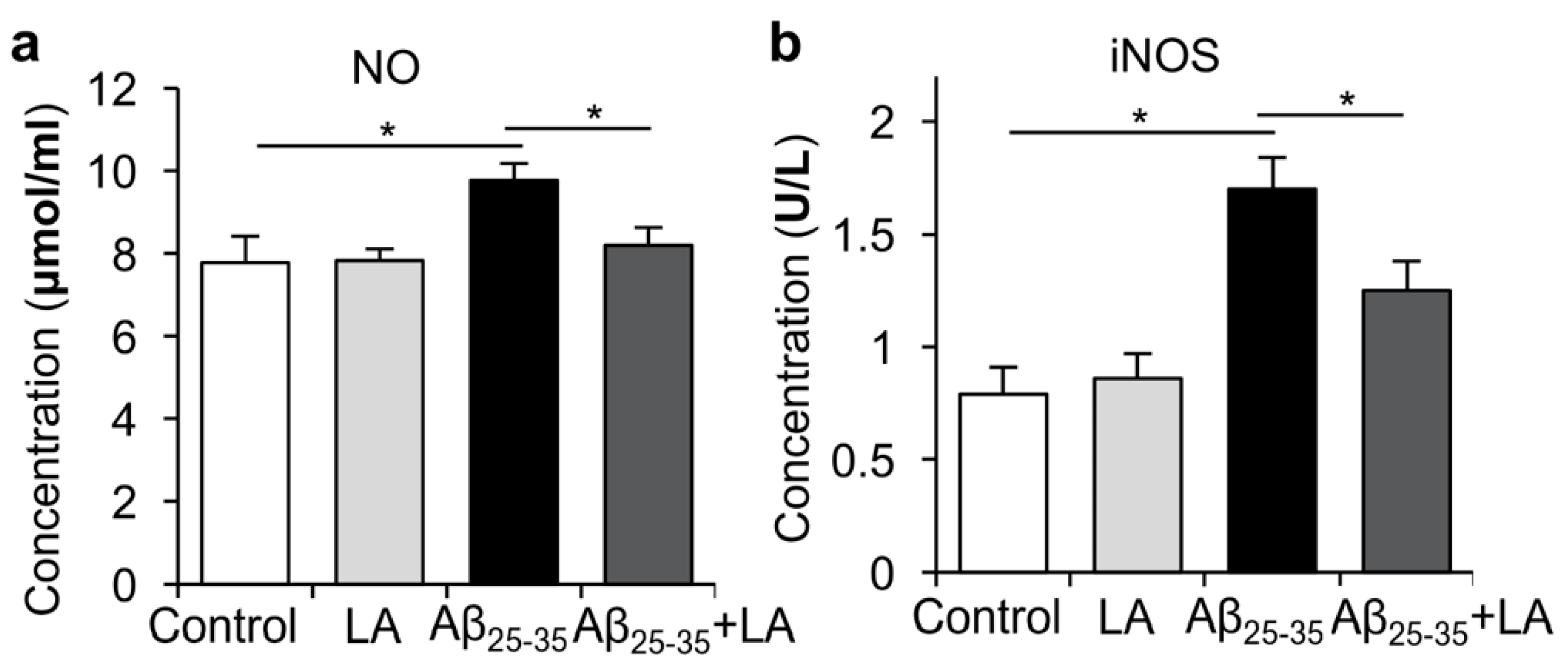
2.4. LA Is Involved in Mitigating the Inflammatory Response Induced by Aβ25–35
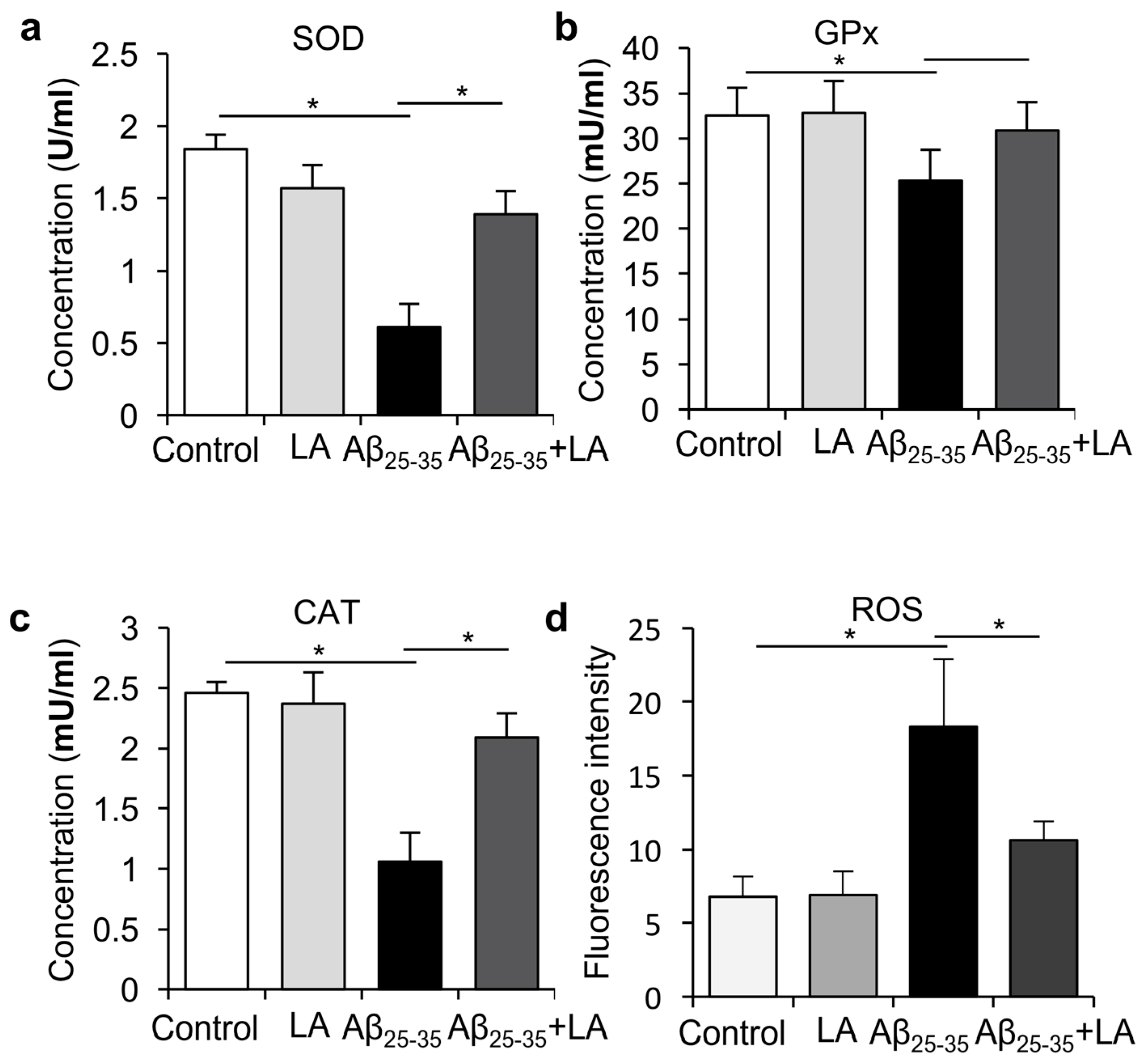
2.5. LA Downregulates ROS Levels Induced by Aβ25–35
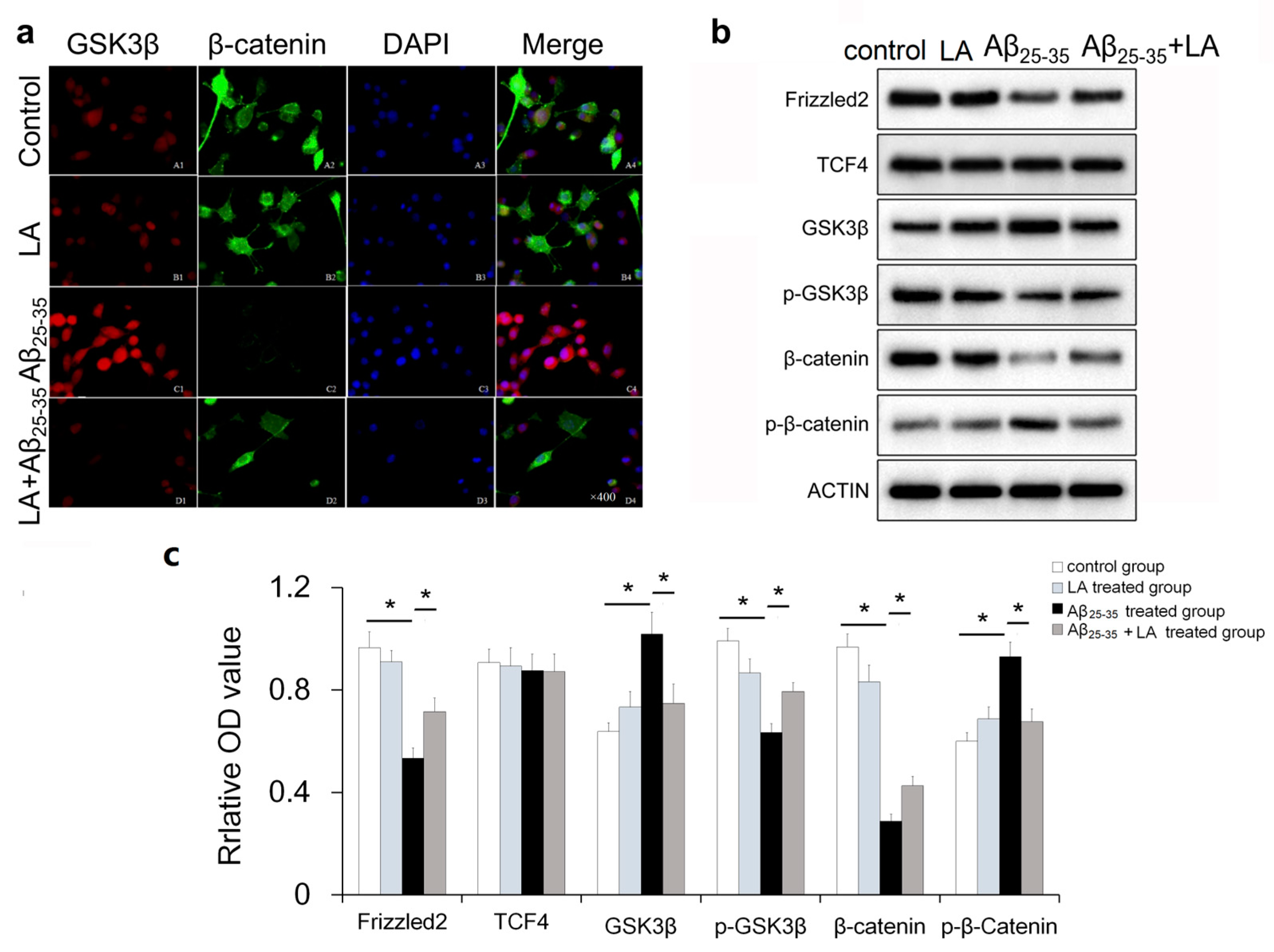
2.6. LA-regulated Wnt Pathway-Specific Protein Expression in Aβ25–35-Treated BV2 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Observation of Cell Morphology
4.3. Cell Growth Assays
4.4. ELISA Detection for IL-6, IL-1β, TNF-α, and IFN-γ
4.5. Detection of SOD, GPx, CAT, ROS, NO, and iNOS
4.6. Western Blot
4.7. Cell Apoptosis and Activation by Flow Cytometry
4.8. Immunofluorescence
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
References
- Hassan, A.A.; Mohammed, A. An update on the novel and approved drugs for Alzheimer disease. Saudi Pharm. J. 2022, 30, 1755–1764. [Google Scholar]
- The State of the Art of Dementia Research: New Frontiers. 2018. Available online: https://www.alzint.org/resource/world-alzheimer-report-2018/ (accessed on 12 February 2021).
- Yang, H.F.; Cong, J.Y.; Zang, X.Y.; Jiang, N.; Zhao, Y. A study on knowledge, attitudes and health behaviours regarding Alzheimer’s disease among community residents in Tianjin, China. J. Psychiatr. Ment. Health Nurs. 2015, 22, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Godoy, J.A.; Rios, J.A.; Zolezzi, J.M.; Braidy, N.; Inestrosa, N.C. Signaling pathway cross talk in Alzheimer’s disease. Cell Commun. Signal. 2014, 12, 23. [Google Scholar] [CrossRef] [Green Version]
- Ridge, P.G.; Ebbert, M.T.; Kauwe, J.S. Genetics of Alzheimer’s disease. BioMed Res. Int. 2013, 2013, 254954. [Google Scholar] [CrossRef] [Green Version]
- Delrieu, J.; Ousset, P.J.; Voisin, T.; Vellas, B. Amyloid beta peptide immunotherapy in Alzheimer disease. Rev. Neurol. 2014, 170, 739–748. [Google Scholar] [CrossRef]
- Waite, L.M. Treatment for Alzheimer’s disease: Has anything changed? Aust. Prescr. 2015, 38, 60–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, L.J.; DeBusk, B.G.; Gunsalus, I.C.; Hornberger, C.S., Jr. Crystalline alpha-lipoic acid: A catalytic agent associated with pyruvate dehydrogenase. Science 1951, 114, 93–94. [Google Scholar] [CrossRef]
- Gorąca, A.; Huk-Kolega, H.; Piechota, A.; Kleniewska, P.; Ciejka, E.; Skibska, B. Lipoic acid—Biological activity and therapeutic potential. Pharmacol. Rep. 2011, 63, 849–858. [Google Scholar] [CrossRef]
- Bingham, P.M.; Stuart, S.D.; Zachar, Z. Lipoic acid and lipoic acid analogs in cancer metabolism and chemotherapy. Expert Rev. Clin. Pharmacol. 2014, 7, 837–846. [Google Scholar] [CrossRef]
- Koufaki, M. Therapeutic applications of lipoic acid: A patent review (2011–2014). Expert Opin. Ther. Pat. 2014, 24, 993–1005. [Google Scholar] [CrossRef]
- Li, G.; Gao, L.; Jia, J.; Gong, X.; Zang, B.; Chen, W. α-Lipoic acid prolongs survival and attenuates acute kidney injury in a rat model of sepsis. Clin. Exp. Pharmacol. Physiol. 2014, 41, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Bergamini, C.; Fato, R.; Oiry, J.; Vasseur, J.J.; Smietana, M. Synthesis of new lipoic acid conjugates and evaluation of their free radical scavenging and neuroprotective activities. Chem. Biol. Drug Des. 2014, 83, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Rosini, M.; Simoni, E.; Bartolini, M.; Tarozzi, A.; Matera, R.; Milelli, A.; Hrelia, P.; Andrisano, V.; Bolognesi, M.L.; Melchiorre, C. Exploiting the lipoic acid structure in the search for novel multitarget ligands against Alzheimer’s disease. Eur. J. Med. Chem. 2011, 46, 5435–5442. [Google Scholar] [CrossRef] [PubMed]
- Baalman, K.; Marin, M.A.; Ho, T.S.; Godoy, M.; Cherian, L.; Robertson, C.; Rasband, M.N. Axon initial segment-associated microglia. J. Neurosci. 2015, 35, 2283–2292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, A.J.; Zelinka, C.; Milani-Nejad, N. Reactive retinal microglia, neuronal survival, and the formation of retinal folds and detachments. Glia 2015, 63, 313–327. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Cho, E.D.; Kim, H.K.; You, S.; Lee, H.J.; Hwang, D.; Lee, S.J. β1-integrin-dependent migration of microglia in response to neuron-released α-synuclein. Exp. Mol. Med. 2014, 46, e91. [Google Scholar] [CrossRef] [Green Version]
- Prokop, S.; Miller, K.R.; Heppner, F.L. Microglia actions in Alzheimer’s disease. Acta Neuropathol. 2013, 126, 461–477. [Google Scholar] [CrossRef]
- Biber, K.; Owens, T.; Boddeke, E. What is microglia neurotoxicity (Not)? Glia 2014, 62, 841–854. [Google Scholar] [CrossRef]
- Suzumura, A. Neuron-microglia interaction in neuroinflammation. Curr. Protein Pept. Sci. 2013, 14, 16–20. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Z. Microglia and Wnt Pathways: Prospects for Inflammation in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 110. [Google Scholar] [CrossRef]
- Halleskog, C.; Mulder, J.; Dahlström, J.; Mackie, K.; Hortobágyi, T.; Tanila, H.; Kumar Puli, L.; Färber, K.; Harkany, T.; Schulte, G. WNT signaling in activated microglia is proinflammatory. Glia 2011, 59, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguzzi, A.; Barres, B.A.; Bennett, M.L. Microglia: Scapegoat, saboteur, or something else? Science 2013, 339, 156–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasole, E.; Nicoletti, C.; Yang, Z.J.; Girelli, A.; Rubini, A.; Giuffreda, F.; Di Tano, A.; Camporesi, E.; Bosco, G. Effects of alpha lipoic acid and its R+ enantiomer supplemented to hyperbaric oxygen therapy on interleukin-6, TNF-α and EGF production in chronic leg wound healing. J. Enzym. Inhib. Med. Chem. 2014, 29, 297–302. [Google Scholar] [CrossRef] [PubMed]
- De Ferrari, G.V.; Avila, M.E.; Medina, M.A.; Perez-Palma, E.; Bustos, B.I.; Alarcon, M.A. Wnt/β-catenin signaling in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2014, 13, 745–754. [Google Scholar] [CrossRef]
- del Pino, J.; Ramos, E.; Aguilera, O.M.; Marco-Contelles, J.; Romero, A. Wnt signaling pathway, a potential target for Alzheimer’s disease treatment, is activated by a novel multitarget compound ASS234. CNS Neurosci. Ther. 2014, 20, 568–570. [Google Scholar] [CrossRef] [Green Version]
- Bettcher, B.M.; Kramer, J.H. Longitudinal inflammation, cognitive decline, and Alzheimer’s disease: A mini-review. Clin. Pharmacol. Ther. 2014, 96, 464–469. [Google Scholar] [CrossRef] [Green Version]
- Holmes, C. Review: Systemic inflammation and Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2013, 39, 51–68. [Google Scholar] [CrossRef]
- Lim, S.L.; Rodriguez-Ortiz, C.J.; Kitazawa, M. Infection, systemic inflammation, and Alzheimer’s disease. Microbes Infect. 2015, 17, 549–556. [Google Scholar] [CrossRef]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef]
- Togo, T.; Katsuse, O.; Iseki, E. Nitric oxide pathways in Alzheimer’s disease and other neurodegenerative dementias. Neurol. Res. 2004, 26, 563–566. [Google Scholar] [CrossRef]
- Balez, R.; Ooi, L. Getting to NO Alzheimer’s Disease: Neuroprotection versus Neurotoxicity Mediated by Nitric Oxide. Oxidative Med. Cell. Longev. 2016, 2016, 3806157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, A.R.; Godoy, J.A.; Mullendorff, K.; Olivares, G.H.; Bronfman, M.; Inestrosa, N.C. Wnt-3a overcomes beta-amyloid toxicity in rat hippocampal neurons. Exp. Cell Res. 2004, 297, 186–196. [Google Scholar] [CrossRef] [PubMed]
- He, P.; and Shen, Y. Interruption of beta-catenin signaling reduces neurogenesis in Alzheimer’s disease. J. Neurosci. 2009, 29, 6545–6557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.; Zhou, Q.; Xie, A.J.; Li, X.; Li, M.; Ye, J.; Li, S.; Ke, D.; Wang, Q.; Xu, Z.P.; et al. Tau acetylates and stabilizes beta-catenin thereby promoting cell survival. EMBO Rep. 2020, 21, e48328. [Google Scholar] [CrossRef] [PubMed]
- Hager, K.; Kenklies, M.; McAfoose, J.; Engel, J.; Münch, G. Alpha-lipoic acid as a new treatment option for Alzheimer’s disease—A 48 months follow-up analysis. J. Neural Transm. Suppl. 2007, 72, 189–193. [Google Scholar]
- Fava, A.; Pirritano, D.; Plastino, M.; Cristiano, D.; Puccio, G.; Colica, C.; Ermio, C.; De Bartolo, M.; Mauro, G.; Bosco, D. The Effect of Lipoic Acid Therapy on Cognitive Functioning in Patients with Alzheimer’s Disease. J. Neurodegener. Dis. 2013, 2013, 454253. [Google Scholar] [CrossRef] [Green Version]
- Maczurek, A.; Hager, K.; Kenklies, M.; Sharman, M.; Martins, R.; Engel, J.; Carlson, D.A.; Münch, G. Lipoic acid as an anti-inflammatory and neuroprotective treatment for Alzheimer’s disease. Adv. Drug Deliv. Rev. 2008, 60, 1463–1470. [Google Scholar] [CrossRef]
- Kandeil, M.A.; Amin, K.A.; Hassanin, K.A.; Ali, K.M.; Mohammed, E.T. Role of lipoic acid on insulin resistance and leptin in experimentally diabetic rats. J. Diabetes Complicat. 2011, 25, 31–38. [Google Scholar] [CrossRef]
- Christie, L.A.; Opii, W.O.; Head, E.; Araujo, J.A.; de Rivera, C.; Milgram, N.W.; Cotman, C.W. Short-term supplementation with acetyl-L-carnitine and lipoic acid alters plasma protein carbonyl levels but does not improve cognition in aged beagles. Exp. Gerontol. 2009, 44, 752–759. [Google Scholar] [CrossRef] [Green Version]
- Yuan, F.; Zhang, Y.; Ma, L.; Cheng, Q.; Li, G.; Tong, T. Enhanced NOLC1 promotes cell senescence and represses hepatocellular carcinoma cell proliferation by disturbing the organization of nucleolus. Aging Cell 2017, 16, 726–737. [Google Scholar] [CrossRef]
- Yuan, F.; Li, G.; Tong, T. Nucleolar and coiled-body phosphoprotein 1 (NOLC1) regulates the nucleolar retention of TRF2. Cell Death Discov. 2017, 3, 17043. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibodies. | Dilution | Manufacturer | Secondary Antibody Dilution |
|---|---|---|---|
| Frizzled 2 | 1:200 | Santa Cruz | 1:4000 |
| TCF4 | 1:1000 | Abcam | 1:4000 |
| GSK3β | 1:5000 | Abcam | 1:4000 |
| p-GSK3β | 1:10,000 | Abcam | 1:4000 |
| β-catenin | 1:5000 | Abcam | 1:4000 |
| p-β-catenin | 1:1000 | CST | 1:4000 |
| IκBα | 1:1000 | CST | 1:4000 |
| NF-kB p65 | 1:200 | CST | 1:4000 |
| Bcl-2 | 1:500 | Santa Cruz | 1:4000 |
| Bax | 1:300 | Santa Cruz | 1:4000 |
| p-NFκB p65 | 1:500 | CST | 1:4000 |
| Caspase-3 | 1:5000 | Abcam | 1:4000 |
| β-actin | 1:200 | Santa Cruz | 1:8000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pei, X.; Hu, F.; Hu, Z.; Luo, F.; Li, X.; Xing, S.; Sun, L.; Long, D. Neuroprotective Effect of α-Lipoic Acid against Aβ25–35-Induced Damage in BV2 Cells. Molecules 2023, 28, 1168. https://doi.org/10.3390/molecules28031168
Pei X, Hu F, Hu Z, Luo F, Li X, Xing S, Sun L, Long D. Neuroprotective Effect of α-Lipoic Acid against Aβ25–35-Induced Damage in BV2 Cells. Molecules. 2023; 28(3):1168. https://doi.org/10.3390/molecules28031168
Chicago/Turabian StylePei, Xinrong, Fangyan Hu, Zehui Hu, Feiya Luo, Xiaoling Li, Shuxia Xing, Lei Sun, and Dingxin Long. 2023. "Neuroprotective Effect of α-Lipoic Acid against Aβ25–35-Induced Damage in BV2 Cells" Molecules 28, no. 3: 1168. https://doi.org/10.3390/molecules28031168