
Computational and Experimental Evidence for Templated Macrocyclization: The Role of a Hydrogen Bond Network in the Quantitative Dimerization of 24-Atom Macrocycles

 ,
, 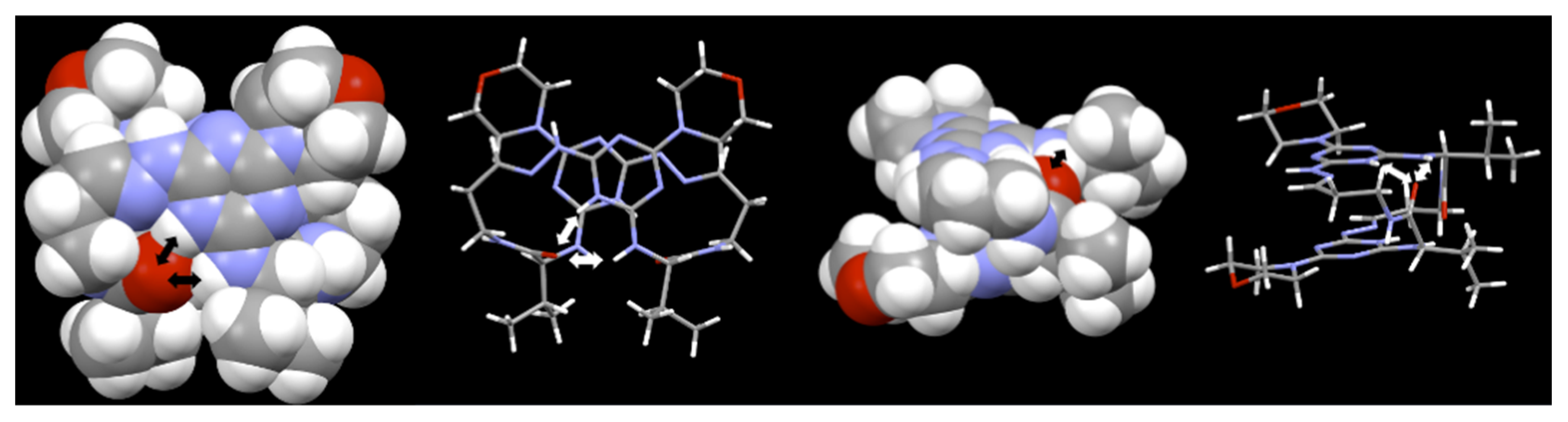{kind=link}
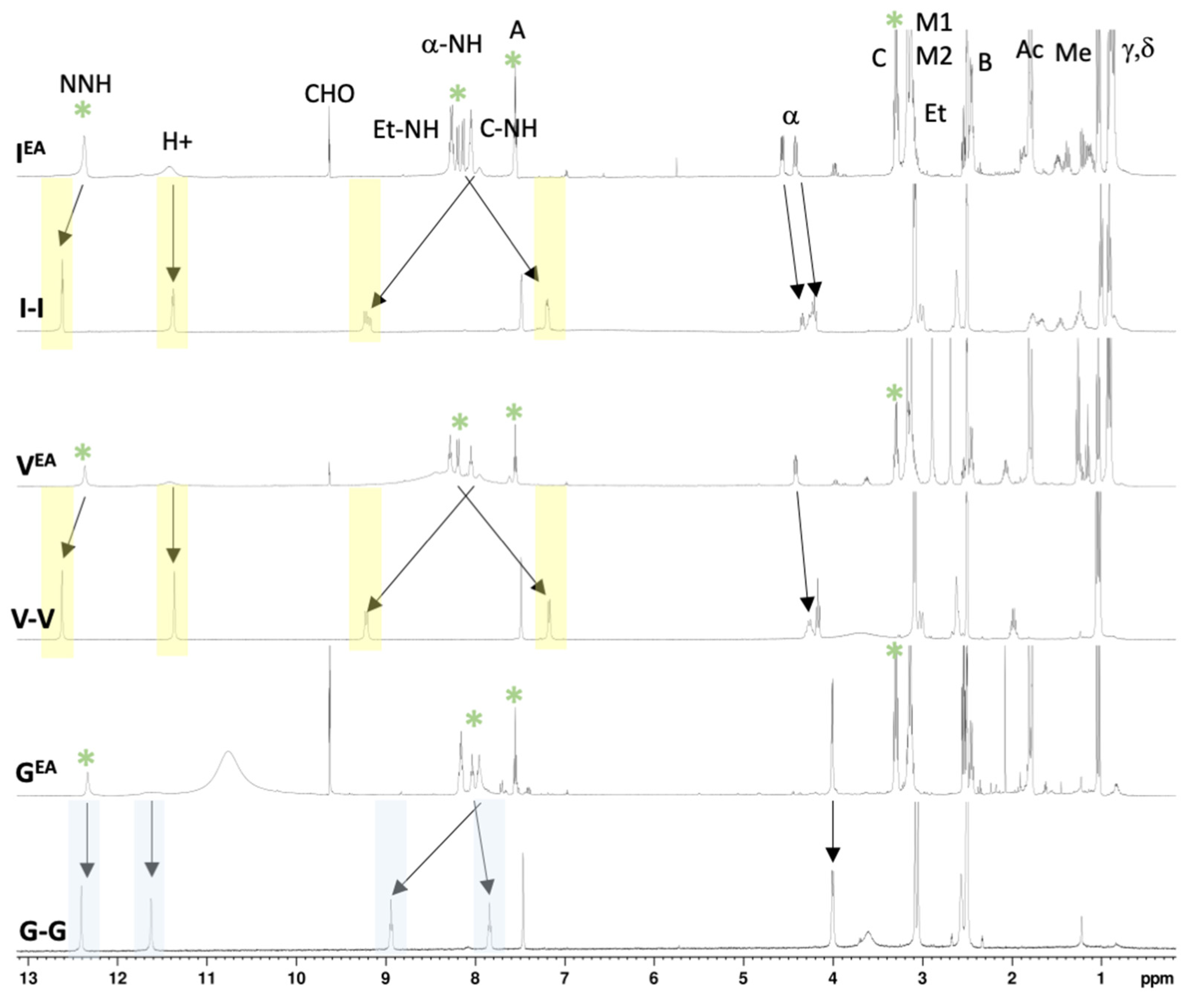{kind=link}
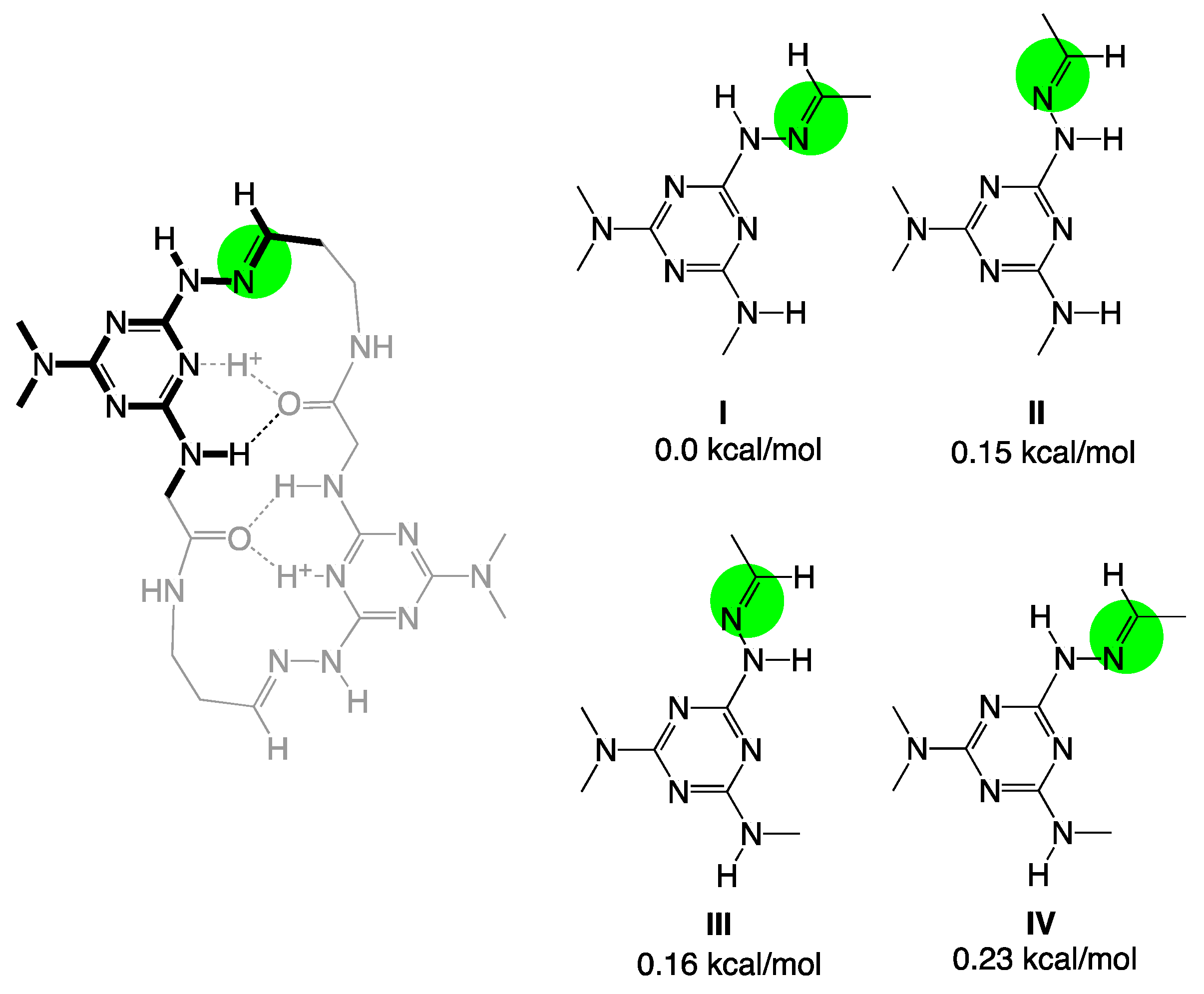{kind=link}
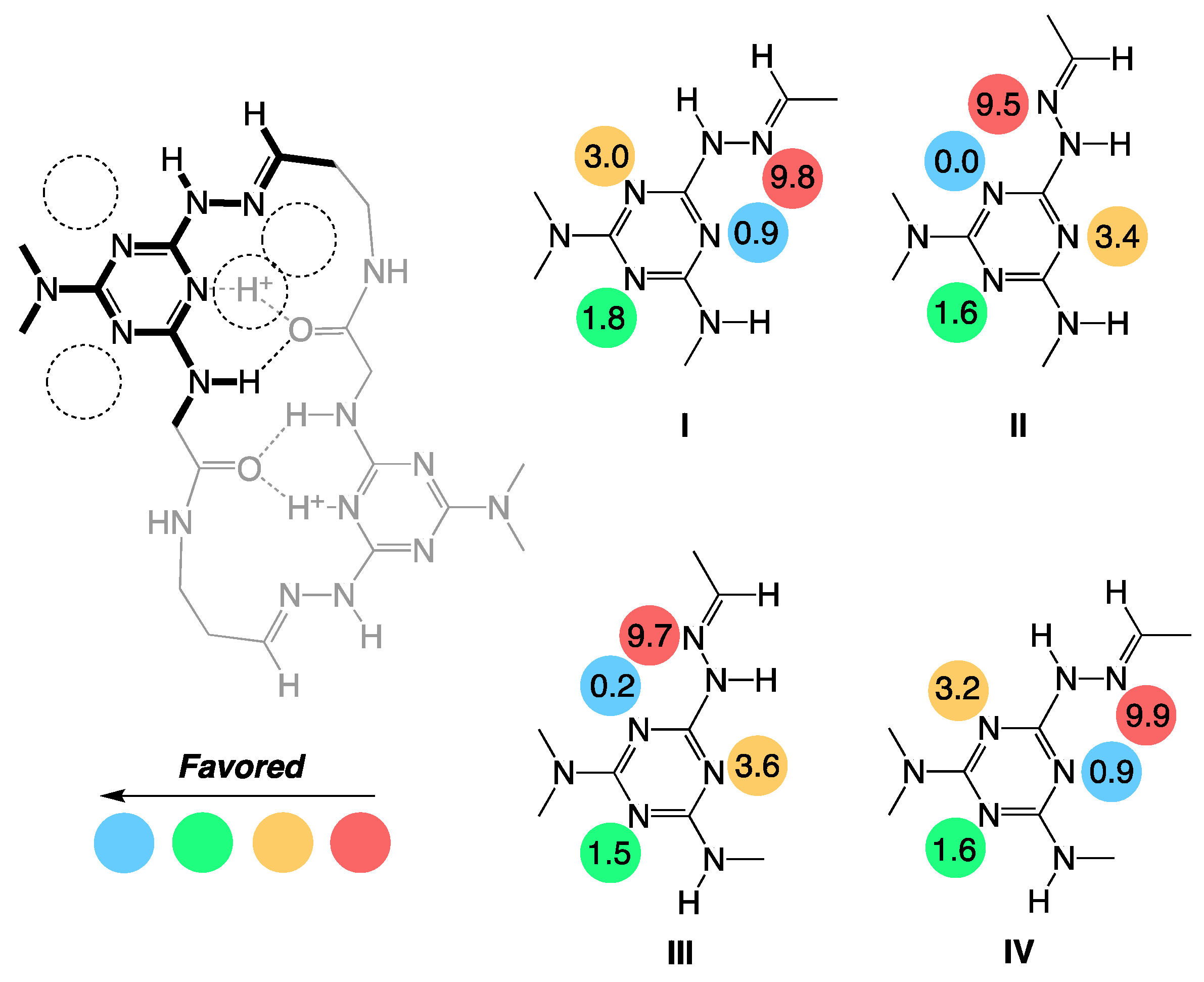{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
3. Experimental
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DeGoey, D.A.; Chen, H.J.; Cox, P.B.; Wendt, M.D. Beyond the Rule of 5: Lessons Learned from AbbVie’s Drugs and Compound Collection. J. Med. Chem. 2018, 61, 2636–2651. [Google Scholar] [CrossRef] [PubMed]
- Sebastiano, M.R.; Doak, B.C.; Backlund, M.; Poongavanam, V.; Over, B.; Ermondi, G.; Caron, G.; Matsson, P.; Kihlberg, J. Impact of Dynamically Exposed Polarity on Permeability and Solubility of Chameleonic Drugs Beyond the Rule of 5. J. Med. Chem. 2018, 61, 4189–4202. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.A.; Yin, Y.Z.; Suga, H. Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges. J. Am. Chem. Soc. 2019, 141, 4167–4181. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Danelius, E.; Poongavanam, V.; Peintner, S.; Wieske, L.H.E.; Erdelyi, M.; Kihlberg, J. Solution Conformations Explain the Chameleonic Behaviour of Macrocyclic Drugs. Chem. Eur. J. 2020, 26, 5231–5244. [Google Scholar] [CrossRef] [PubMed]
- Sedmera, P.; Jegorov, A.; Buchta, M.; Cvak, L. 11-Demethylcyclosporins exhibit a single conformation in methanol and dimethylsulfoxide. J. Pept. Res. 2001, 58, 229–236. [Google Scholar] [CrossRef]
- Naylor, M.R.; Ly, A.M.; Handford, M.J.; Ramos, D.P.; Pye, C.R.; Furukawa, A.; Klein, V.G.; Noland, R.P.; Edmondson, Q.; Turmon, A.C.; et al. Lipophilic Permeability Efficiency Reconciles the Opposing Roles of Lipophilicity in Membrane Permeability and Aqueous Solubility. J. Med. Chem. 2018, 61, 11169–11182. [Google Scholar] [CrossRef]
- Avdeef, A.; Kansy, M. “Flexible-Acceptor” General Solubility Equation for beyond Rule of 5 Drugs. Mol. Pharm. 2020, 17, 3930–3940. [Google Scholar] [CrossRef]
- Ono, S.; Naylor, M.R.; Townsend, C.E.; Okumura, C.; Okada, O.; Lee, H.W.; Lokey, R.S. Cyclosporin A: Conformational Complexity and Chameleonicity. J. Chem. Inf. Model. 2021, 61, 5601–5613. [Google Scholar] [CrossRef]
- Wenger, R.M. Synthesis of Cyclosporine. Total Syntheses of Cyclosporin-A and Cyclosporin-H, 2 Fungal Metabolites Isolaterd from the species Tolypocladium-inflatum gams. Helv. Chim. Acta 1984, 67, 502–525. [Google Scholar] [CrossRef]
- Marti-Centelles, V.; Pandey, M.D.; Burguete, M.I.; Luis, S.V. Macrocyclization Reactions: The Importance of Conformational, Configurational, and Template-Induced Preorganization. Chem. Rev. 2015, 115, 8736–8834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blankenstein, J.; Zhu, J.P. Conformation-directed macrocyclization reactions. Eur. J. Org. Chem. 2005, 10, 1949–1964. [Google Scholar] [CrossRef]
- Owston, P.G.; Peters, R.; Ramsammy, E.; Tasker, P.A.; Trotter, J. Non-template synthesis of N4 macrocyclic imine ligands with variable sizes—The importance of intramolecular hydrogen-bonding—x-ray crystal-structures of 3 macrocyclic and 2 open-chain ligands. J. Chem. Soc. Chem. Commun. 1980, 24, 1218–1220. [Google Scholar] [CrossRef]
- Clark, T.D.; Kobayashi, K.; Ghadiri, M.R. Covalent capture and stabilization of cylindrical beta-sheet peptide assemblies. Chem. Eur. J. 1999, 5, 782–792. [Google Scholar] [CrossRef]
- Lisowski, J. Imine- and Amine-Type Macrocycles Derived from Chiral Diamines and Aromatic Dialdehydes. Molecules 2022, 27, 4097. [Google Scholar] [CrossRef] [PubMed]
- Beeren, S.R.; Sanders, J.K.M. Ferrocene-amino acid macrocycles as hydrazone-based receptors for anions. Chem. Sci. 2011, 2, 1560–1567. [Google Scholar] [CrossRef]
- Taylor, C.M.; Kilah, N.L. Synthesis of 2 + 2 Schiff base macrocycles by a solvent templating strategy and halogen bonding directed assembly. J. Inclus. Phenom. Macro. Chem. Ournal Incl. Phenom. Macrocycl. Chem. 2022, 102, 543–555. [Google Scholar] [CrossRef]
- Jin, Y.H.; Wang, Q.; Taynton, P.; Zhang, W. Dynamic Covalent Chemistry Approaches toward Macrocycles, Molecular Cages, and Polymers. Acc. Chem. Res. 2014, 47, 1575–1586. [Google Scholar] [CrossRef]
- Jiao, T.Y.; Wu, G.C.; Zhang, Y.; Shen, L.B.; Lei, Y.; Wang, C.Y.; Fahrenbach, A.C.; Li, H. Self-Assembly in Water with N-Substituted Imines. Angew. Chem. Int. Ed. 2020, 59, 18350–18367. [Google Scholar] [CrossRef]
- Yepremyan, A.; Mehmood, A.; Asgari, P.; Janesko, B.G.; Simanek, E.E. Synthesis of Macrocycles Derived from Substituted Triazines. Chembiochem 2019, 20, 241–246. [Google Scholar] [CrossRef]
- Sharma, V.R.; Mehmood, A.; Janesko, B.G.; Simanek, E.E. Efficient syntheses of macrocycles ranging from 22–28 atoms through spontaneous dimerization to yield bis-hydrazones. RSC Adv. 2020, 10, 3217–3220. [Google Scholar] [CrossRef] [Green Version]
- Menke, A.J.; Gloor, C.J.; Claton, L.E.; Mekhail, M.A.; Pan, H.; Stewart, M.D.; Green, K.N.; Pavan, G.M.; Capelli, R.; Simanek, E.E. A Model for the Rapid Assessment of Solution-Structures for 24-Atom Macrocycles: The Impact of-Branched Amino Acids on Conformation. J. Org. Chem. 2022. under review. [Google Scholar]
- Katritzky, A.R.; Ghiviriga, I.; Steel, P.J.; Oniciu, D.C. Restricted rotations in 4,6-bis- and 2,4,6-tris-(N,N-dialkylamino)-s-triazines. JCS Perkins Trans. 2 1996, 3, 443–447. [Google Scholar] [CrossRef]
- Amm, M.; Platzer, N.; Guilhem, J.; Bouchet, J.P.; Volland, J.P. Structural and conformational study of substituted triazines by N-15 NMR and X-ray analysis. Magn. Res. Chem. 1998, 36, 587–596. [Google Scholar] [CrossRef]
- Birkett, H.E.; Harris, R.K.; Hodgkinson, P.; Carr, K.; Charlton, M.H.; Cherryman, J.C.; Chippendale, A.M.; Glover, R.P. NMR studies of exchange between triazine rotamers. Magn. Res. Chem. 2000, 38, 504–511. [Google Scholar] [CrossRef]
- Capelli, R.; Menke, A.J.; Pan, H.; Janesko, B.G.; Simanek, E.E.; Pavan, G.M. Well-Tempered Metadynamics Simulations Predict the Structural and Dynamic Properties of a Chiral 24-Atom Macrocycle in Solution. ACS Omega 2022, 7, 30291–30296. [Google Scholar] [CrossRef]
- List, M.; Puchinger, H.; Gabriel, H.; Monkowius, U.; Schwarzinger, C. N-Methylmelamines: Synthesis, Characterization, and Physical Properties. J. Org. Chem. 2016, 81, 4066–4075. [Google Scholar] [CrossRef]
- Jang, Y.H.; Hwang, S.; Chang, S.B.; Ku, J.; Chung, D.S. Acid Dissociation Constants of Melamine Derivatives from Density Functional Theory Calculations. J. Phys. Chem. A 2009, 113, 13036–13040. [Google Scholar] [CrossRef]
- Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019.
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford Univsrsity Press: Oxford, UK, 1989. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian-basis sets for molecular calculations. 1. 2nd row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Raghavachari, K.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. 20. Basis set for correlated wave-functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menke, A.J.; Henderson, N.C.; Kouretas, L.C.; Estenson, A.N.; Janesko, B.G.; Simanek, E.E. Computational and Experimental Evidence for Templated Macrocyclization: The Role of a Hydrogen Bond Network in the Quantitative Dimerization of 24-Atom Macrocycles. Molecules 2023, 28, 1144. https://doi.org/10.3390/molecules28031144
Menke AJ, Henderson NC, Kouretas LC, Estenson AN, Janesko BG, Simanek EE. Computational and Experimental Evidence for Templated Macrocyclization: The Role of a Hydrogen Bond Network in the Quantitative Dimerization of 24-Atom Macrocycles. Molecules. 2023; 28(3):1144. https://doi.org/10.3390/molecules28031144
Chicago/Turabian StyleMenke, Alexander J., Nicholas C. Henderson, Lola C. Kouretas, Anne N. Estenson, Benjamin G. Janesko, and Eric E. Simanek. 2023. "Computational and Experimental Evidence for Templated Macrocyclization: The Role of a Hydrogen Bond Network in the Quantitative Dimerization of 24-Atom Macrocycles" Molecules 28, no. 3: 1144. https://doi.org/10.3390/molecules28031144