
Molecular and Electronic Structures of Macrocyclic Compounds Formed at Template Synthesis in the M(II)—Thiocarbohydrazide—Diacetyl Triple Systems: A Quantum-Chemical Analysis by DFT Methods


Abstract
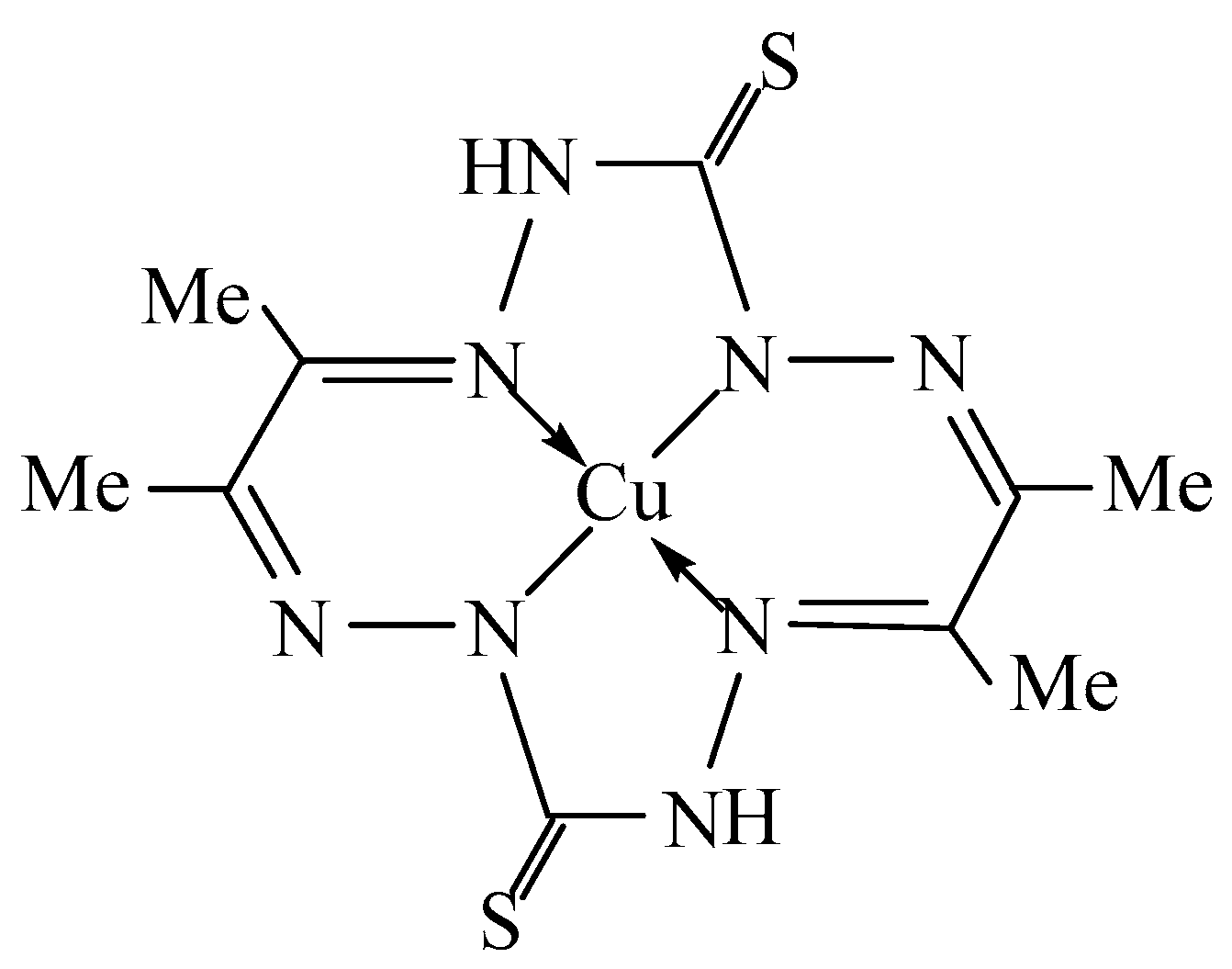
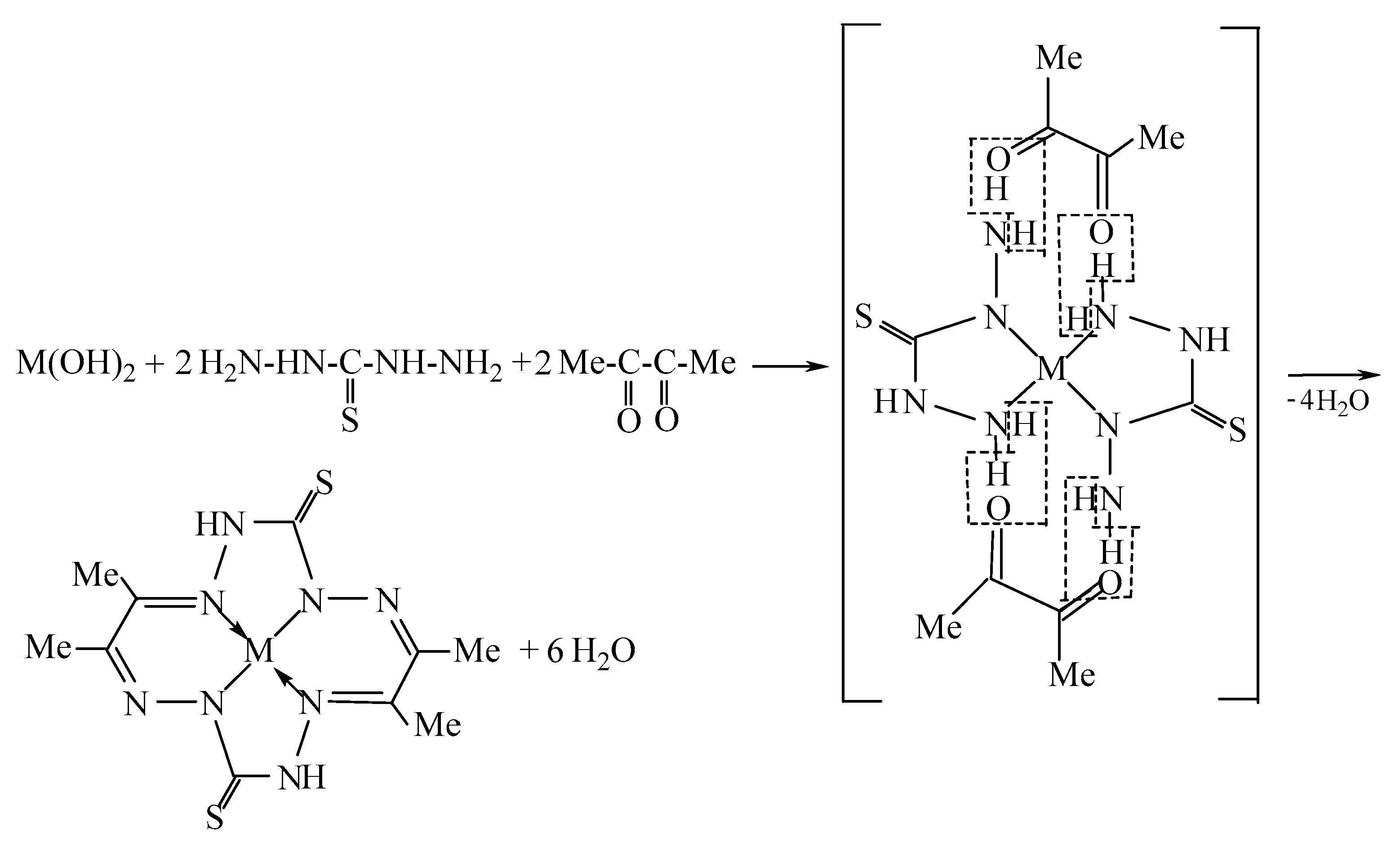
:1. Introduction
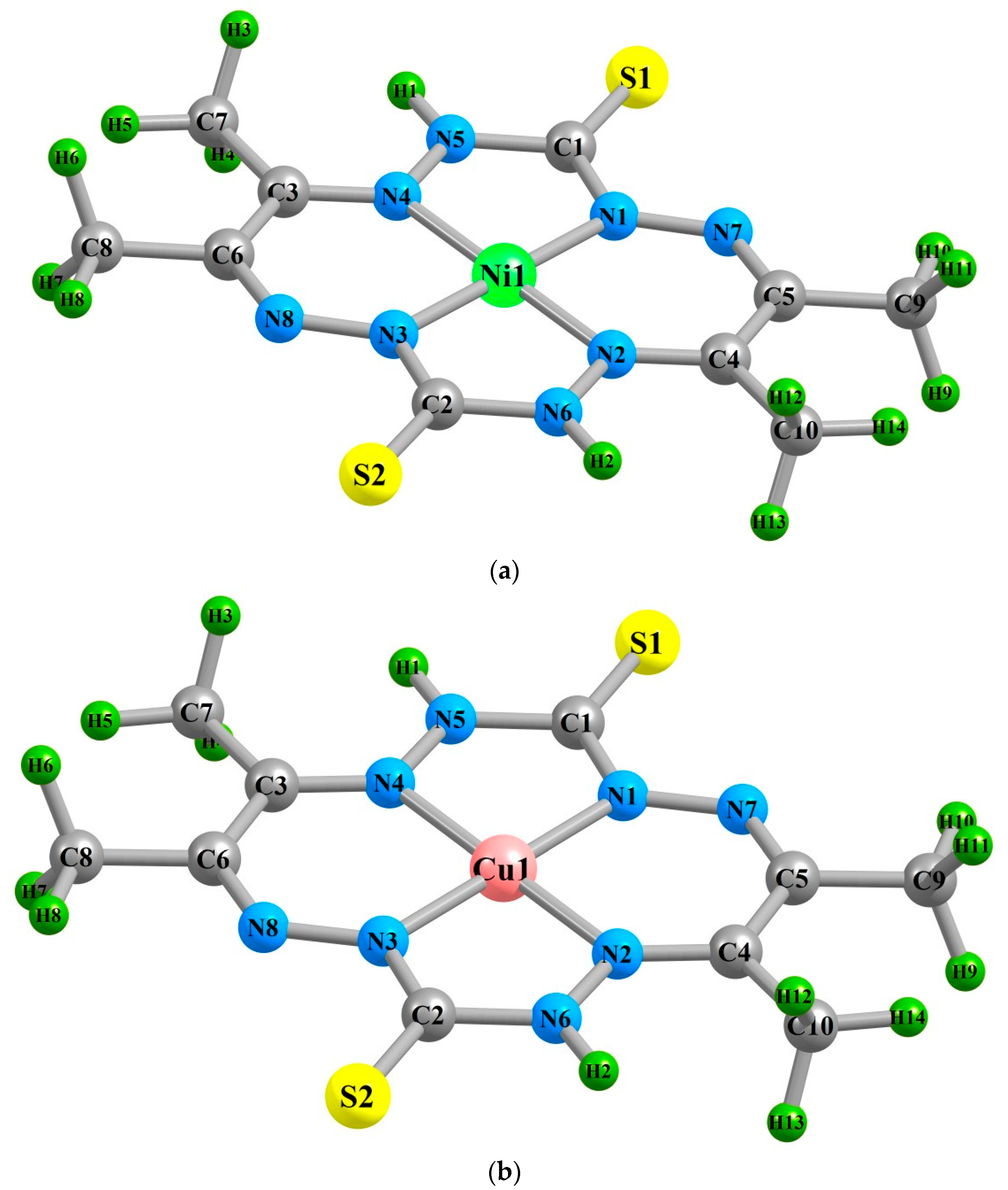
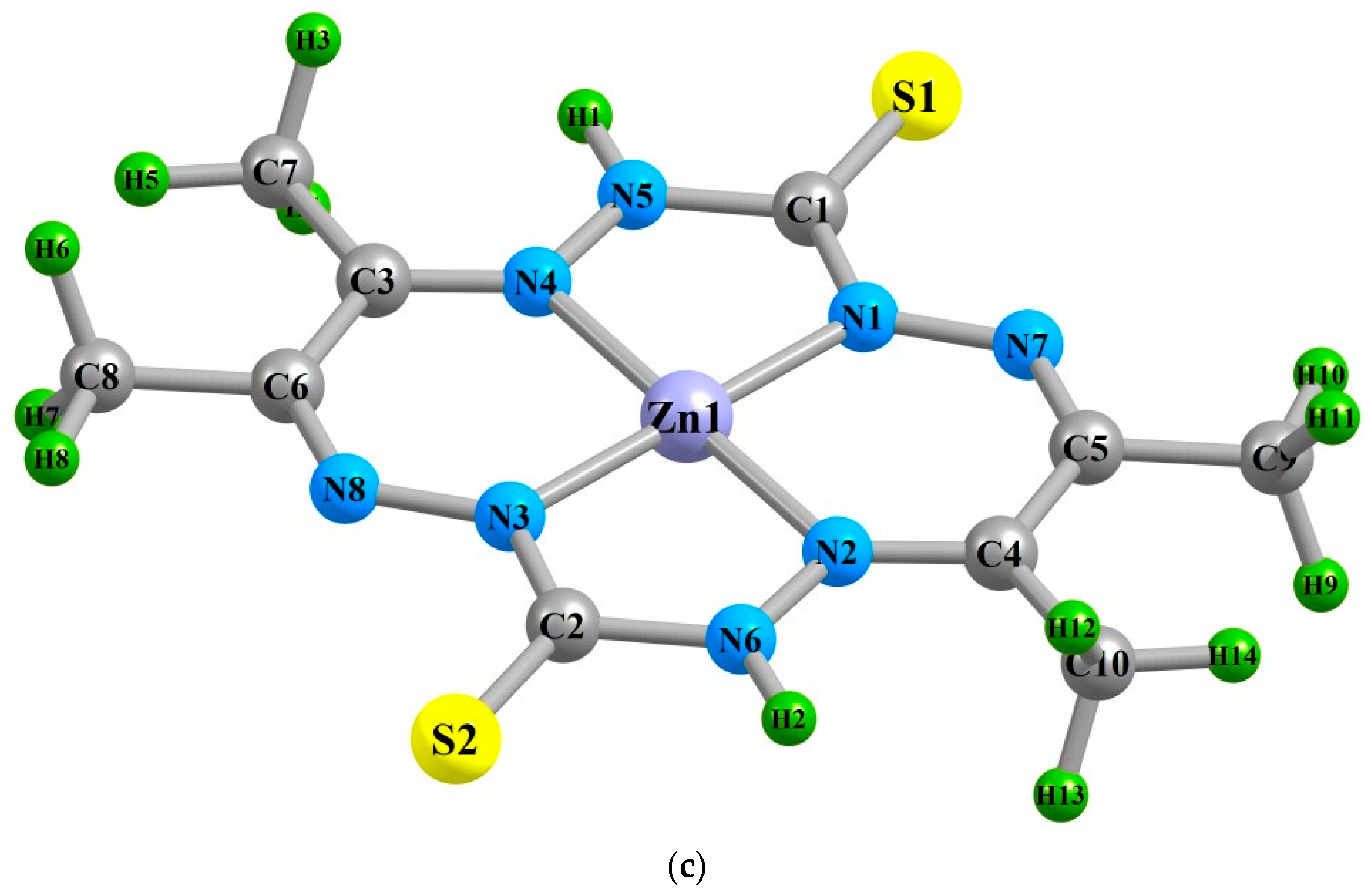
2. Results
3. Calculation Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Mikhailov, O.V.; Kazymova, M.A.; Shumilova, T.A.; Solovieva, S.E. Template synthesis in the M(II)–thiocarbohydrazide–diacetyl triple system (M = Ni, Cu) in a metal(II)hexacyanoferrate(II) gelatin-immobilized matrix. Transit. Metal Chem. 2004, 29, 732–736. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Self-assembly of supramolecular Ni(II) and Cu(II) metalmacrocyclic compounds with tetraazamacrocyclic ligand into a gelatin-immobilized matrix. J. Coord. Chem. 2010, 63, 4309–4318. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. Density Functional Theory Calculation of Molecular Structures of (5656) Macrotetracyclic 3d Metal Complexes with 4,12-Dithiooxo-1,8-dioxa-3,6,10,13-tetraazacyclotetradecanedione-5,11. Russ. J. Inorg. Chem. 2012, 57, 981–986. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. Structure of (5656) Macrotetracyclic Chelates in the Ternary Systems M(II)–Ethanedithioamide–Acetone (M = Mn, Fe, Co, Ni, Cu, Zn) According to DFT Calculations. Russ. J. Inorg. Chem. 2013, 58, 1073–1078. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. Quantum-Chemical Calculation of Molecular Structures of (5656) Macrotetracyclic 3d-Metal Complexes “Self-Assembled” in Quaternary Systems M(II) Ion– Ethanedithioamide–Formaldehyde– Ammonia by the Density Functional Theory Method. Russ. J. Inorg. Chem. 2014, 59, 218–223. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Macrotetracyclic Chelates of Doubly Charged 3d-Element Ions with 1,4,8,11-Tetraazacyclotetradecane-2,3,9,10-tetrathione and Their Molecular Structures according to Density Functional Theory Data. Russ. J. Inorg. Chem. 2014, 59, 1276–1282. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Molecular Structures of (5656) Macrotetracyclic Chelates Formed in the M(II) Ion– Ethanedithioamide– 2-Thiapropanediol-1,3 Systems according to Density Functional Theory Calculations. Russ. J. Inorg. Chem. 2015, 60, 1354–1359. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. DFT Analysis of Molecular Structure of 14-Membered Tetraaza-, Dioxotetraaza- and Hexaazamacroheterocyclic Ligands and Their Metal ComplexesRuss. J. Gen. Chem. 2016, 86, 1102–1107. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. DFT OPBE/TZVP Calculation of M olecular Structures of (5656) Macroheterocyclic Chelates of Double Charged 3d-Element Ions with 1,5,8,11-Tetraazacyclotetradecanetetrathione-2,3,9,10 and Its Dioxa- and Dithia Analogs. Macroheterocycles 2016, 9, 268–276. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Molecular Structures of (5656) Macrotetracyclic Chelates in M(II) Ion–Ethanedithioamide– Methanimine– Hydrogen Cyanide Quaternary Systems by DFT Calculations. Russ. J. Inorg. Chem. 2016, 61, 616–622. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Revs. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Revs. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, M.G.; Bushmarinov, I.S.; Sun, J.; Perdew, J.P.; Lyssenko, K.A. Density functional theory is straying from the path toward the exact functional. Science 2017, 355, 49–52. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Molecular Structure of M(N13) Compounds with 12-Membered Nitrogen-Containing Cycle and Axial Nitrogen Atom (M = Mn, Fe): Quantum-Chemical Design by DFT Method. Quantum Rep. 2023, 5, 282–293. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. Heteroligand Iron(V) Complexes Containing Porphyrazine, trans-Di[benzo]porphyrazine or Tetra[benzo]porphyrazine, Oxo and Fluoro Ligands: DFT Quantum-Chemical Study. Int. J. Mol. Sci. 2023, 24, 6442. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. DFT Method Used for Prediction of Molecular and Electronic Structures of Mn(VI) Macrocyclic Complexes with Porhyrazine/Phthalocyanine and Two Oxo Ligands. Materials 2023, 16, 2394. [Google Scholar] [CrossRef]
- Paulsen, H.; Duelund, L.; Winkler, H.; Toftlund, H.; Trautwein, A.X. Free Energy of Spin-Crossover Complexes Calculated with Density Functional Methods. Inorg. Chem. 2001, 40, 2201–2203. [Google Scholar] [CrossRef]
- Swart, M.; Groenhof, A.R.; Ehlers, A.W.; Lammertsma, K. Validation of Exchange−Correlation Functionals for Spin States of Iron Complexes. J. Phys. Chem. A 2004, 108, 5479–5483. [Google Scholar] [CrossRef]
- Swart, M.; Ehlers, A.W.; Lammertsma, K. Performance of the OPBE exchange-correlation functional. Mol. Phys. 2004, 102, 2467–2474. [Google Scholar] [CrossRef]
- Swart, M. Metal–ligand bonding in metallocenes: Differentiation between spin state, electrostatic and covalent bonding. Inorg. Chim. Acta 2007, 360, 179–189. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Weinhold, F.; Landis, C.R.; Glendening, E.D. What is NBO analysis and how is it useful? Int. Revs. Phys. Chem. 2016, 35, 399–440. [Google Scholar] [CrossRef]
- Ochterski, J.W. Thermochemistry in Gaussian; Gaussian, Inc.: Wallingford, CT, USA, 2000. [Google Scholar]
- Zhabanov, Y.A.; Tverdova, N.V.; Giricheva, N.I.; Girichev, G.V.; Stuzhin, P.A. DFT Study of molecular and electronic structure of magnesium (II) tetra(1,2,5-chalcogenadiazolo) porphyrazines, [TXDPzMg] (X = O, S, Se, Te). J. Porphyr. Phthalocyanines 2017, 21, 439–452. [Google Scholar] [CrossRef]
- Novakova, V.; Donzello, M.P.; Ercolani, C.; Zimcik, P.; Stuzhin, P.A. Tetrapyrazinoporphyrazines and their metal derivatives. Part II: Electronic structure, electrochemical, spectral, photophysical and other application related properties. Coord. Chem. Rev. 2018, 361, 1–73. [Google Scholar] [CrossRef]
- Sorokin, A.B. Recent progress on exploring µ-oxo bridged binuclear porphyrinoid complexes in catalysis and material science. Coord. Chem. Rev. 2019, 389, 141–160. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Onami, Y.; Koya, R.; Kawasaki, T.; Aizawa, H.; Nakagame, R.; Miyagawa, Y.; Haraguchi, T.; Akitsu, T.; Tsukiyama, K.; Palafox, M.A. Investigation by DFT Methods of the Damage of Human Serum Albumin including Amino Acid Derivative Schiff Base Zn(II) Complexes by IR-FEL Irradiation. Int. J. Mol. Sci. 2019, 20, 2846. [Google Scholar] [CrossRef]
- Otlyotov, A.A.; Ryzhov, I.V.; Kuzmin, I.A.; Zhabanov, Y.A.; Mikhailov, M.S.; Stuzhin, P.A. DFT Study of Molecular and Electronic Structure of Ca(II) and Zn(II) Complexes with Porphyrazine and tetrakis(1,2,5-thiadiazole)porphyrazine. Int. J. Mol. Sci. 2020, 21, 2923. [Google Scholar] [CrossRef]
- Martins, L.S.; Lameira, J.; Kruger, H.G.; Alves, C.N.; Silva, J.R.A. Evaluating the Performance of a Non-Bonded Cu2+ Model Including Jahn-Teller Effect into the Binding of Tyrosinase Inhibitors. Int. J. Mol. Sci. 2020, 21, 4783. [Google Scholar] [CrossRef] [PubMed]
- Latypov, S.K.; Kondrashova, S.A.; Polyancev, F.M.; Sinyashin, O.G. Quantum Chemical Calculations of 31P NMR Chemical Shifts in Nickel Complexes: Scope and Limitations. Organometallics 2020, 39, 1413–1422. [Google Scholar] [CrossRef]
- Zhou, Q.; Liu, Z.; Marks, T.J.; Darancet, P. Electronic Structure of Metallophthalocyanines, MPc (M = Fe, Co, Ni, Cu, Zn, Mg) and Fluorinated MPc. J. Phys. Chem. A 2021, 125, 4055–4061. [Google Scholar] [CrossRef] [PubMed]
- Zhabanov, Y.A.; Ryzhov, I.V.; Kuzmin, I.A.; Eroshin, A.V.; Stuzhin, P.A. DFT Study of Molecular and Electronic Structure of Y, La and Lu Complexes with Porphyrazine and Tetrakis(1,2,5-thiadiazole)porphyrazine. Molecules 2021, 26, 113. [Google Scholar] [CrossRef]
- Zhabanov, Y.A.; Eroshin, A.V.; Ryzhov, I.V.; Kuzmin, I.A.; Finogenov, D.N.; Stuzhin, P.A. Molecular Structure, Thermodynamic and Spectral Characteristics of Metal-Free and Nickel Complex of Tetrakis(1,2,5-thiadiazolo) porphyrazine. Molecules 2021, 26, 2945. [Google Scholar] [CrossRef]
- Eroshin, A.V.; Otlyotov, A.A.; Kuzmin, I.A.; Stuzhin, P.A.; Zhabanov, Y.A. DFT Study of the Molecular and Electronic Structure of Metal-Free Tetrabenzoporphyrin and Its Metal Complexes with Zn, Cd, Al, Ga, In. Int. J. Mol. Sci. 2022, 23, 939. [Google Scholar] [CrossRef]
- Ryzhov, I.V.; Eroshin, A.V.; Zhabanov, Y.A.; Finogenov, D.N.; Stuzhin, P.A. DFT Study of Molecular Structure, Electronic and Vibrational Spectra of Tetrapyrazinoporphyrazine, Its Perchlorinated Derivative and Their Al, Ga and In Complexes. Int. J. Mol. Sci. 2022, 23, 5379. [Google Scholar] [CrossRef]
- Eroshin, A.V.; Koptyaev, A.I.; Otlyotov, A.A.; Minenkov, Y.; Zhabanov, Y.A. Iron(II) Complexes with Porphyrin and Tetrabenzoporphyrin: CASSCF/MCQDPT2 Study of the Electronic Structures and UV–Vis Spectra by sTD-DFT. Int. J. Mol. Sci. 2023, 24, 7070. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
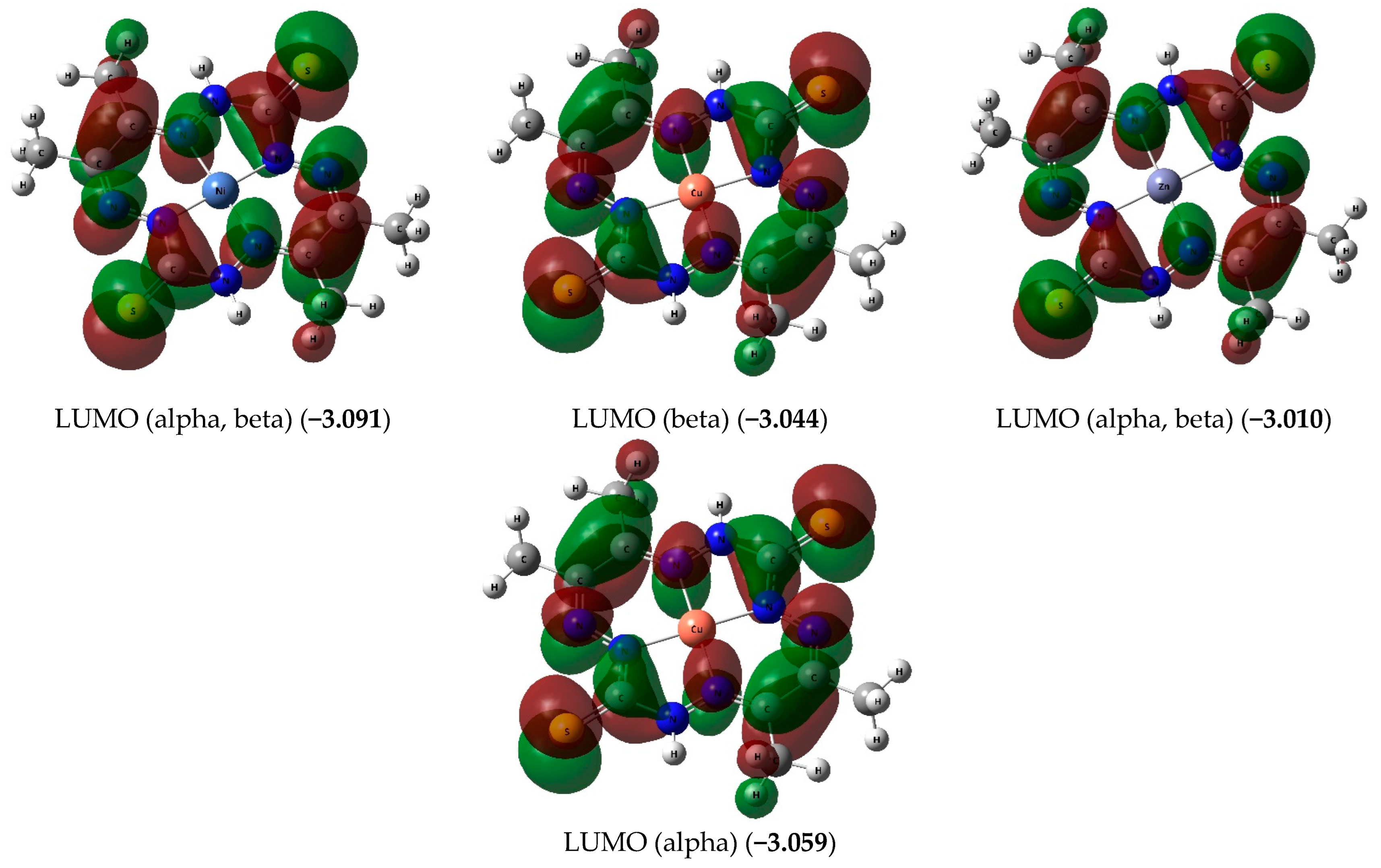{kind=link}
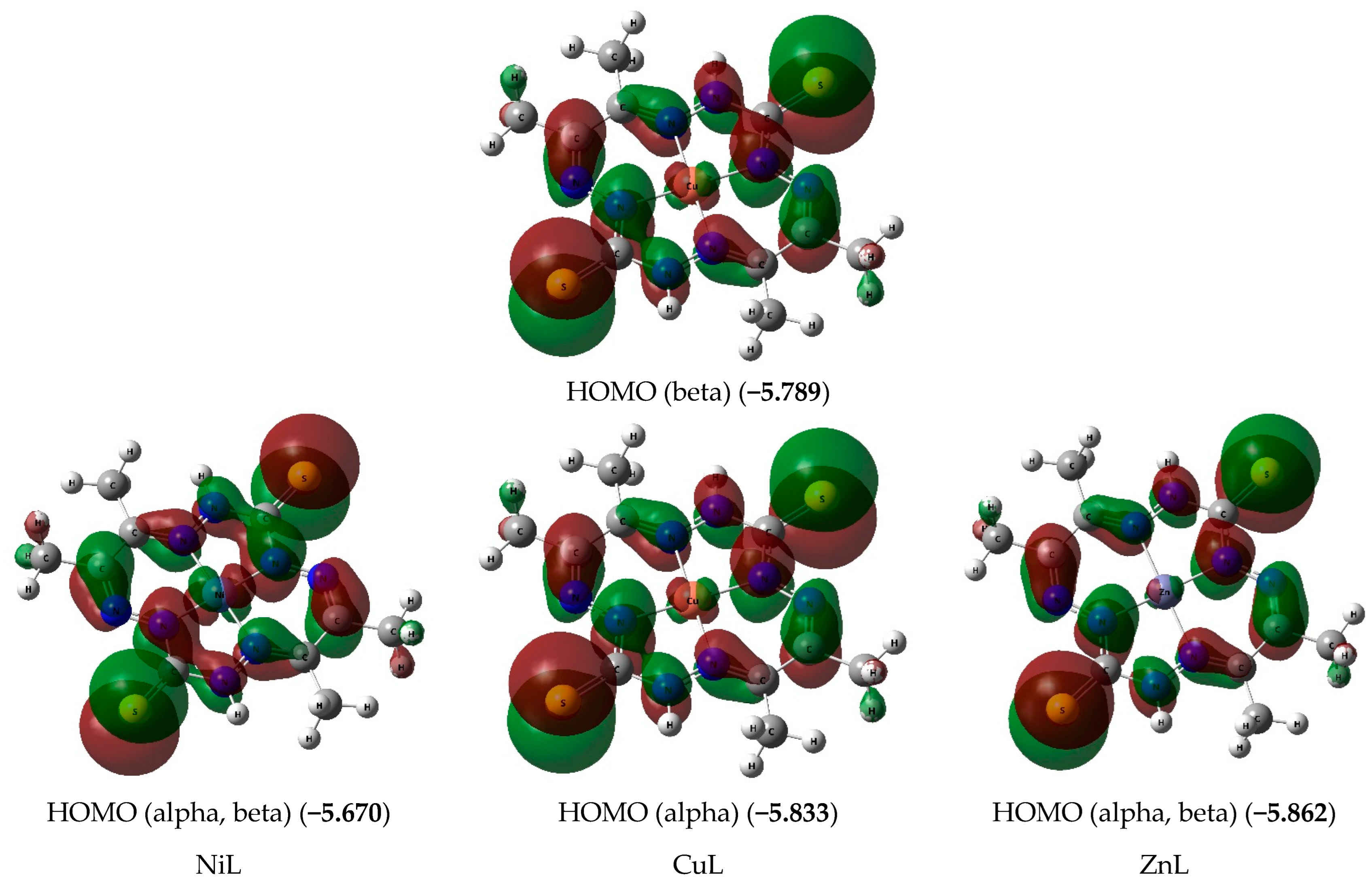{kind=link}
| Complex | NiL | CuL | ZnL | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Chemistry Model | Chemistry Model | Chemistry Model | |||||||
| Structural Parameter | B3PW91/TZVP | M06/TZVP | OPBE/TZVP | B3PW91/TZVP | M06/TZVP | OPBE/TZVP | B3PW91/TZVP | M06/TZVP | OPBE/TZVP |
| Bond lengths in the MN4 chelate node, pm | |||||||||
| (M1N1) | 183.9 | 184.2 | 183.4 | 188.9 | 188.8 | 189.4 | 191.0 | 190.5 | 191.4 |
| (M1N2) | 183.5 | 184.2 | 182.6 | 190.9 | 191.4 | 190.9 | 197.5 | 198.2 | 197.7 |
| (M1N3) | 183.9 | 184.2 | 183.4 | 188.9 | 188.8 | 189.4 | 191.0 | 190.5 | 191.3 |
| (M1N4) | 183.5 | 184.2 | 182.6 | 190.9 | 191.4 | 190.9 | 197.5 | 198.2 | 197.6 |
| Separate bond lengths outside the MN4 chelate node, pm | |||||||||
| (C1S1), (C2S2) | 165.9 | 165.7 | 165.9 | 166.0 | 165.7 | 166.1 | 166.0 | 165.7 | 166.0 |
| (C1N5), (C2N6) | 137.8 | 138.0 | 137.7 | 139.8 | 140.0 | 140.0 | 141.6 | 141.7 | 141.8 |
| (N2N6), (N4N5) | 135.6 | 135.7 | 135.1 | 134.9 | 134.9 | 134.2 | 134.3 | 134.3 | 133.8 |
| (C4C5), (C3C6) | 145.7 | 146.0 | 145.1 | 147.5 | 147.7 | 146.9 | 148.8 | 148.8 | 148.2 |
| (C5N7), (C6N8) | 130.1 | 129.5 | 131.2 | 130.4 | 129.8 | 131.6 | 130.7 | 130.0 | 132.0 |
| (C6C8), (C5C9) | 150.6 | 150.1 | 150.6 | 150.8 | 150.3 | 150.9 | 151.0 | 150.4 | 151.1 |
| (C4C10), (C7C8) | 149.9 | 149.4 | 149.9 | 150.0 | 149.5 | 150.1 | 150.1 | 150.0 | 150.2 |
| Bond angles in the MN4 chelate node, deg | |||||||||
| (N1M1N2) | 94.1 | 94.1 | 94.1 | 94.7 | 94.7 | 94.7 | 95.1 | 95.2 | 95.0 |
| (N2M1N3) | 85.9 | 85.9 | 85.9 | 85.3 | 85.3 | 85.3 | 84.9 | 84.8 | 85.0 |
| (N3M1N4) | 94.1 | 94.1 | 94.1 | 94.7 | 94.7 | 94.7 | 95.1 | 95.2 | 95.0 |
| (N4M1N1) | 85.9 | 85.9 | 85.9 | 85.3 | 85.3 | 85.3 | 84.9 | 84.8 | 85.0 |
| Bond angles sum (BAS) | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 |
| Non-bond angles in the N4 grouping, deg | |||||||||
| (N1N2N3) | 90.1 | 90.0 | 90.2 | 89.4 | 89.2 | 89.5 | 88.1 | 87.7 | 88.2 |
| (N2N3N4) | 89.9 | 90.0 | 89.8 | 90.6 | 90.8 | 90.5 | 91.9 | 92.3 | 91.8 |
| (N3N4N1) | 90.1 | 90.0 | 90.2 | 89.4 | 89.2 | 89.5 | 88.1 | 87.7 | 88.2 |
| (N4N1N2) | 89.9 | 90.0 | 89.8 | 90.6 | 90.8 | 90.5 | 91.9 | 92.3 | 91.8 |
| Non-bond angles sum (NBAS) | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 |
| Bond angles in the 5-numbered (M1N4N5C1N1) chelate ring, deg | |||||||||
| (M1N4N5) | 110.5 | 110.3 | 110.9 | 109.0 | 108.9 | 109.1 | 107.9 | 107.6 | 107.8 |
| (N4N5C1) | 119.1 | 119.2 | 119.3 | 120.5 | 120.5 | 120.9 | 121.1 | 121.0 | 121.6 |
| (N5C1N1) | 109.7 | 109.8 | 109.1 | 111.1 | 111.1 | 110.7 | 111.9 | 112.0 | 111.6 |
| (C1N1M1) | 114.8 | 114.8 | 114.8 | 114.1 | 114.2 | 114.0 | 114.2 | 114.5 | 114.0 |
| (N1M1N4) | 85.9 | 85.9 | 85.9 | 85.3 | 85.3 | 85.3 | 84.9 | 84.8 | 85.0 |
| Bond angles sum (VAS51) | 540.0 | 540.0 | 540.0 | 540.0 | 540.0 | 540.0 | 540.0 | 540.0 | 540.0 |
| Bond angles in the 5-numbered (M1N2N6C2N3) chelate ring, deg | |||||||||
| (M1N2N6) | 110.5 | 110.3 | 110.9 | 109.0 | 108.9 | 109.1 | 107.9 | 107.6 | 107.8 |
| (N2N6C2) | 119.1 | 119.2 | 119.3 | 120.5 | 120.5 | 120.9 | 121.1 | 121.0 | 121.6 |
| (N6C2N3) | 109.7 | 109.8 | 109.1 | 111.1 | 111.1 | 110.7 | 111.9 | 112.0 | 111.6 |
| (C2N3M1) | 114.8 | 114.8 | 114.8 | 114.1 | 114.2 | 114.0 | 114.2 | 114.5 | 114.0 |
| (N3M1N2) | 85.9 | 85.9 | 85.9 | 85.3 | 85.3 | 85.3 | 84.9 | 84.8 | 85.0 |
| Bond angles sum (VAS52) | 540.0 | 540.0 | 540.0 | 540.0 | 540.0 | 540.0 | 540.0 | 540.0 | 540.0 |
| Bond angles in the 6-numbered (M1N1N7C5C4N2) chelate ring, deg | |||||||||
| (M1N1N7) | 128.5 | 128.3 | 128.8 | 127.0 | 126.8 | 127.1 | 126.1 | 125.9 | 126.6 |
| (N1N7C5) | 122.2 | 122.4 | 122.3 | 122.3 | 122.5 | 122.4 | 122.2 | 122.3 | 121.7 |
| (N7C5C4) | 127.0 | 127.2 | 126.8 | 129.5 | 129.5 | 129.6 | 131.6 | 131.6 | 132.1 |
| (C5C4N2) | 120.3 | 120.4 | 119.9 | 120.4 | 120.5 | 120.0 | 120.0 | 120.1 | 119.4 |
| (C4N2M1) | 127.9 | 127.6 | 128.1 | 126.1 | 126.0 | 126.2 | 125.0 | 124.4 | 125.2 |
| (N2M1N1) | 94.1 | 94.1 | 94.1 | 94.7 | 94.7 | 94.7 | 95.1 | 95.2 | 95.0 |
| Bond angles sum (VAS61) | 720.0 | 720.0 | 720.0 | 720.0 | 720.0 | 720.0 | 720.0 | 719.5 | 720.0 |
| Bond angles in the 6-numbered (M1N3N8C6C3N4) chelate ring, deg | |||||||||
| (M1N3N8) | 128.5 | 128.3 | 128.8 | 127.0 | 126.8 | 127.1 | 126.1 | 125.9 | 126.6 |
| (N3N8C6) | 122.2 | 122.4 | 122.3 | 122.3 | 122.5 | 122.4 | 122.2 | 122.3 | 121.7 |
| (N8C6C3) | 127.0 | 127.2 | 126.8 | 129.5 | 129.5 | 129.6 | 131.6 | 131.6 | 132.1 |
| (C6C3N4) | 120.3 | 120.4 | 119.9 | 120.4 | 120.5 | 120.0 | 120.0 | 120.1 | 119.4 |
| (C3N4M1) | 127.9 | 127.6 | 128.1 | 126.1 | 126.0 | 126.2 | 125.0 | 124.4 | 125.2 |
| (N4M1N3) | 94.1 | 94.1 | 94.1 | 94.7 | 94.7 | 94.7 | 95.1 | 95.2 | 95.0 |
| Bond angles sum (VAS62) | 720.0 | 720.0 | 720.0 | 720.0 | 720.0 | 720.0 | 720.0 | 719.5 | 720.0 |
| Bond angles outside chelate rings, deg | |||||||||
| (N1C1S1), (N3C2S2) | 130.6 | 130.6 | 130.9 | 130.7 | 130.6 | 131.1 | 130.8 | 130.7 | 131.4 |
| (N5N4C3), (N6N2C4) | 121.6 | 122.1 | 121.0 | 124.8 | 125.2 | 124.6 | 127.2 | 127.6 | 127.1 |
| (N4C3C7), (N2C4C10) | 118.8 | 118.7 | 119.1 | 118.6 | 118.5 | 118.9 | 118.8 | 119.3 | 119.0 |
| (C3C6C8), (C4C5C9) | 118.8 | 118.4 | 119.6 | 117.3 | 117.0 | 117.9 | 116.1 | 115.6 | 116.6 |
| (C8C6N8), (C9C5N7) | 114.1 | 114.4 | 113.6 | 113.2 | 113.5 | 112.4 | 112.2 | 112.8 | 111.3 |
| (C6C3C7), (C5C4C10) | 120.9 | 120.8 | 121.0 | 121.0 | 121.0 | 121.1 | 121.3 | 120.6 | 121.5 |
| Complex | Chemistry Model | The Charges on the Atoms, in Electron Charge Units (ē) | <S**2> | ||||
|---|---|---|---|---|---|---|---|
| M1 | N1 | N2 | N3 | N4 | |||
| NiL | B3PW91/TZVP | +0.379 | −0.316 | −0.187 | −0.316 | −0.187 | 0.0000 |
| M06/TZVP | +0.382 | −0.335 | −0.198 | −0.335 | −0.198 | 0.0000 | |
| OPBE/TZVP | +0.314 | −0.265 | −0.173 | −0.265 | −0.173 | 0.0000 | |
| CuL | B3PW91/TZVP | +0.729 | −0.411 | −0.257 | −0.411 | −0.257 | 0.7500 |
| M06/TZVP | +0.711 | −0.425 | −0.262 | −0.425 | −0.262 | 0.7500 | |
| OPBE/TZVP | +0.672 | −0.364 | −0.245 | −0.364 | −0.245 | 0.7500 | |
| ZnL | B3PW91/TZVP | +1.088 | −0.508 | −0.321 | −0.509 | −0.321 | 0.0000 |
| M06/TZVP | +1.071 | −0.524 | −0.326 | −0.524 | −0.326 | 0.0000 | |
| OPBE/TZVP | +1.085 | −0.475 | −0.318 | −0.475 | −0.318 | 0.0000 | |
| Complex | Chemistry Model | ΔfH0298, kJ/mol | Sf0298, J/mol K | ΔfG0298, kJ/mol |
|---|---|---|---|---|
| NiL | B3PW91/TZVP | 769.1 | 745.5 | 966.6 |
| M06/TZVP | 891.0 | 742.7 | 1089.4 | |
| OPBE/TZVP | 550.1 | 753.4 | 745.3 | |
| CuL | B3PW91/TZVP | 916.4 | 748.5 | 1114.1 |
| M06/TZVP | 1052.2 | 753.7 | 1248.3 | |
| OPBE/TZVP | 753.3 | 760.5 | 947.4 | |
| ZnL | B3PW91/TZVP | 834.8 | 772.5 | 1027.9 |
| M06/TZVP | 983.9 | 766.1 | 1178.9 | |
| OPBE/TZVP | 669.8 | 771.5 | 863.2 |
| Complex | Chemistry Model | ΔrH0 298, kJ | ΔrSr0298, J/K | ΔrG0 298, kJ |
|---|---|---|---|---|
| NiL | B3PW91/TZVP | −411.6 | 46.6 | −425.5 |
| M06/TZVP | −393.1 | 65.6 | −412.7 | |
| CuL | B3PW91/TZVP | −204.7 | 64.1 | −223.8 |
| M06/TZVP | −191.2 | 91.2 | −218.4 | |
| ZnL | B3PW91/TZVP | −77.0 | 80.5 | −101.0 |
| M06/TZVP | −66.7 | 96.0 | −95.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikhailov, O.V.; Chachkov, D.V. Molecular and Electronic Structures of Macrocyclic Compounds Formed at Template Synthesis in the M(II)—Thiocarbohydrazide—Diacetyl Triple Systems: A Quantum-Chemical Analysis by DFT Methods. Molecules 2023, 28, 4383. https://doi.org/10.3390/molecules28114383
Mikhailov OV, Chachkov DV. Molecular and Electronic Structures of Macrocyclic Compounds Formed at Template Synthesis in the M(II)—Thiocarbohydrazide—Diacetyl Triple Systems: A Quantum-Chemical Analysis by DFT Methods. Molecules. 2023; 28(11):4383. https://doi.org/10.3390/molecules28114383
Chicago/Turabian StyleMikhailov, Oleg V., and Denis V. Chachkov. 2023. "Molecular and Electronic Structures of Macrocyclic Compounds Formed at Template Synthesis in the M(II)—Thiocarbohydrazide—Diacetyl Triple Systems: A Quantum-Chemical Analysis by DFT Methods" Molecules 28, no. 11: 4383. https://doi.org/10.3390/molecules28114383