
New Boron Containing Acridines: Synthesis and Preliminary Biological Study

 , , , ,
, , , ,  ,
,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
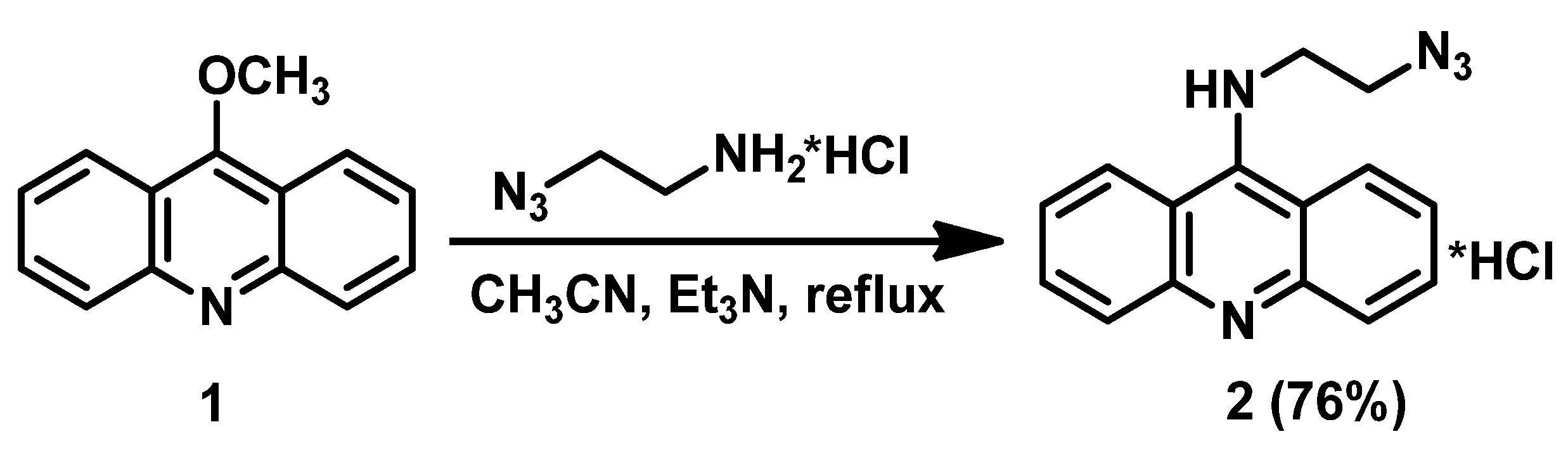
2.1. Synthesis of N9-Azidooacridine 2
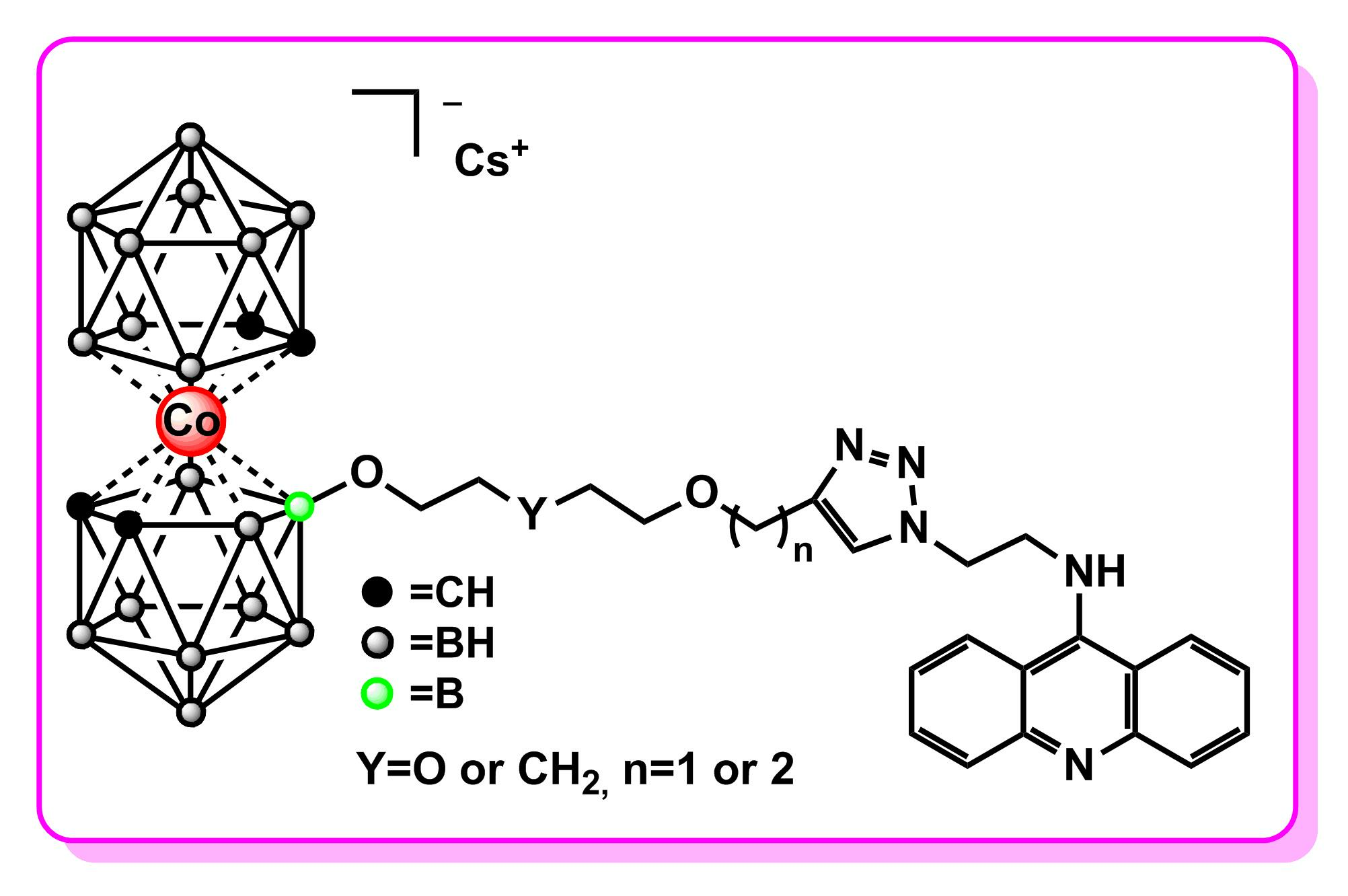
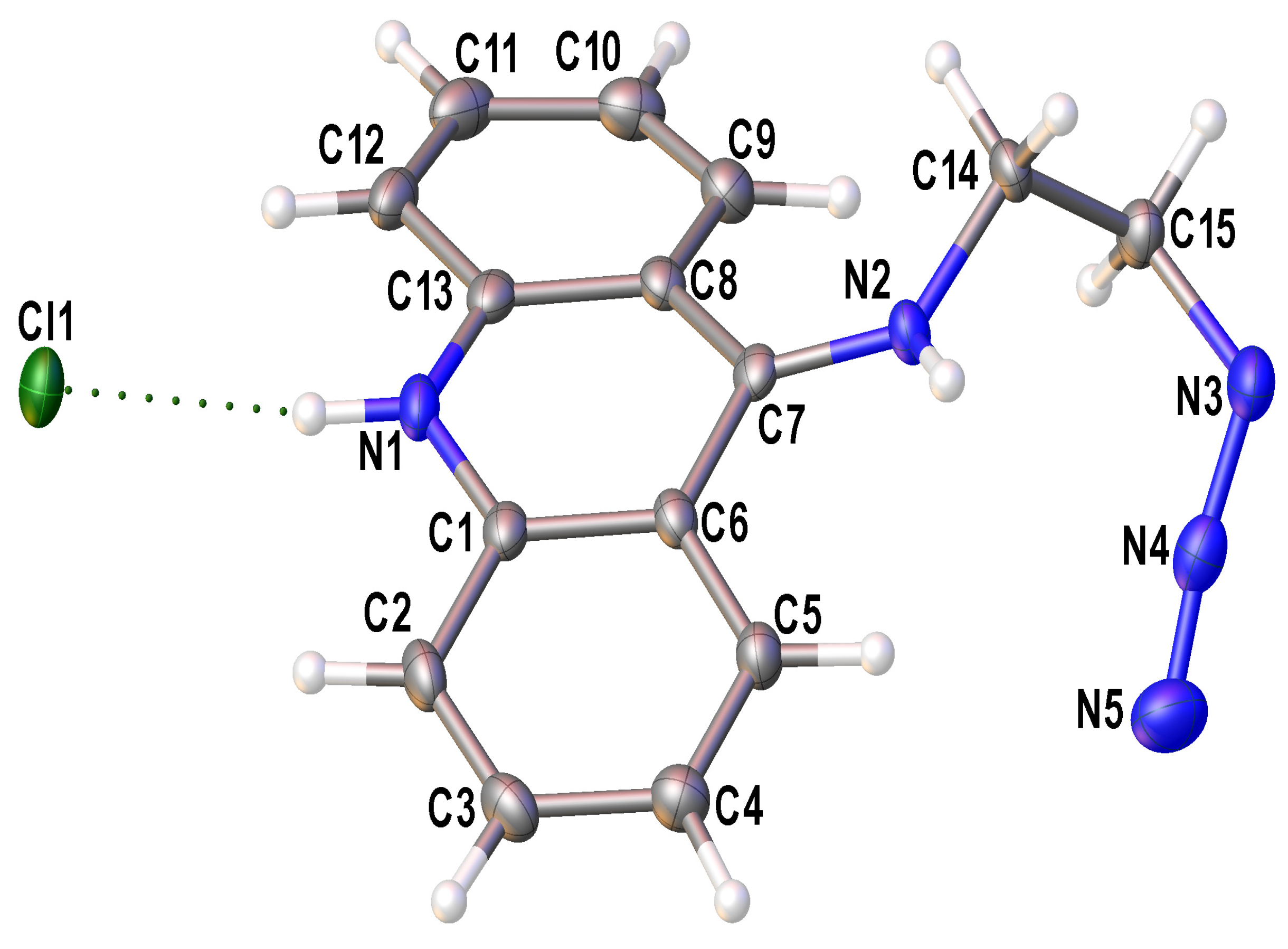
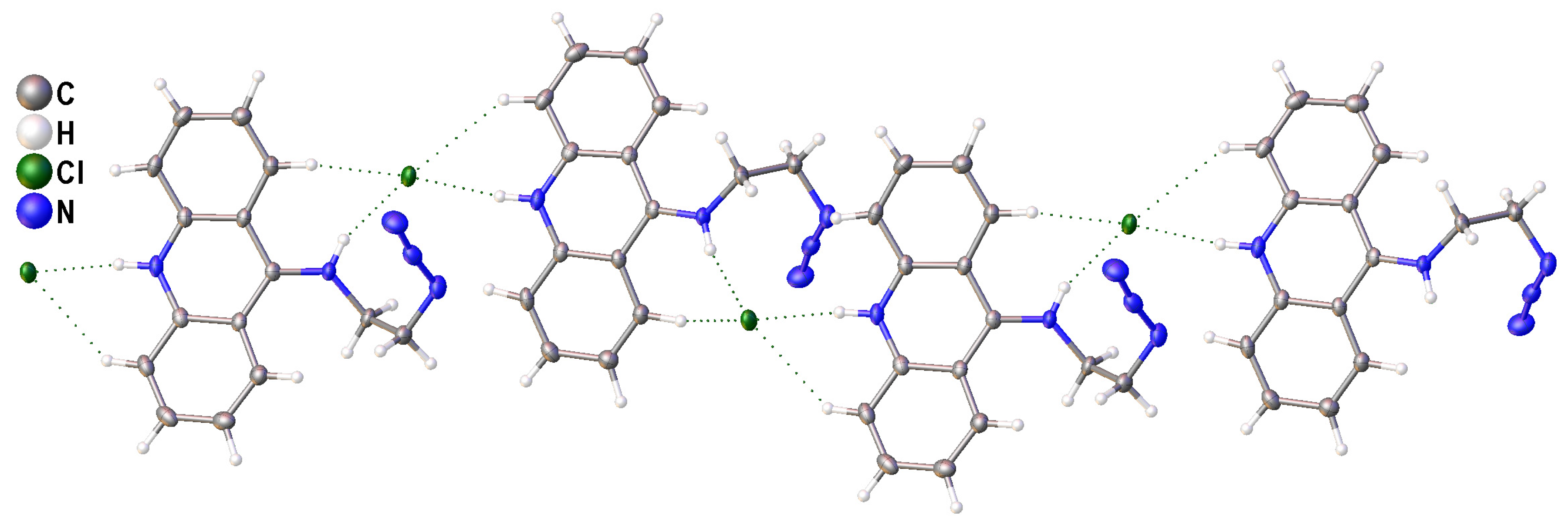
2.2. Single-Crystal X-ray Diffraction Studies of N9-Azidoacridine 2
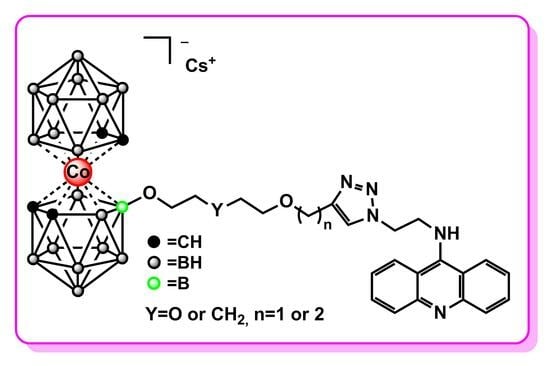
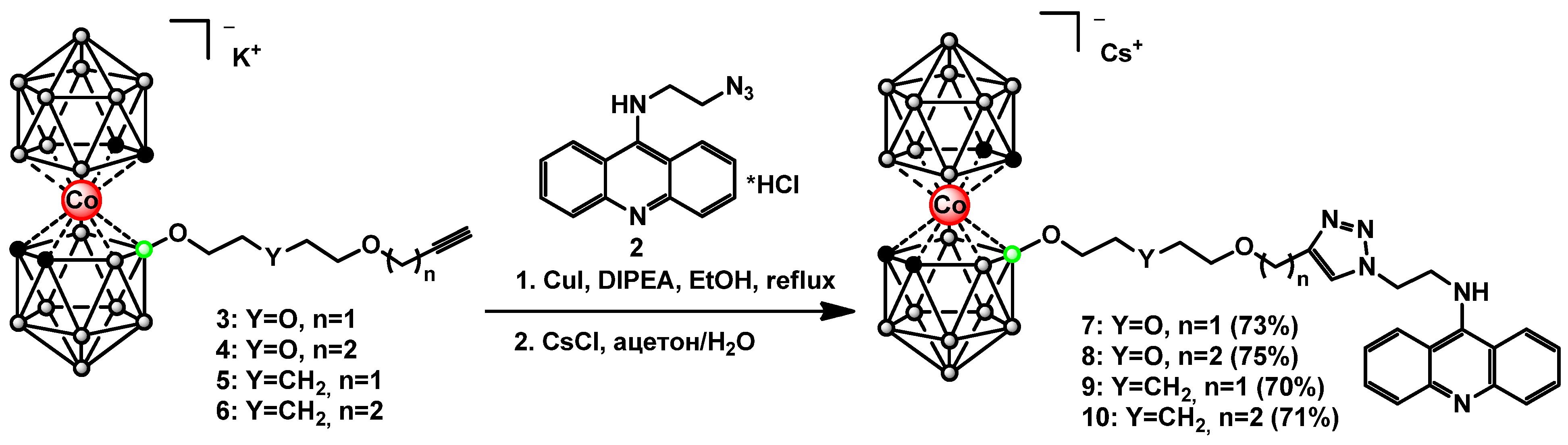
2.3. Synthesis of Cobalt Bis(dicarbollide)-Acridine Derivatives with 1,2,3-Triazoles 7–10
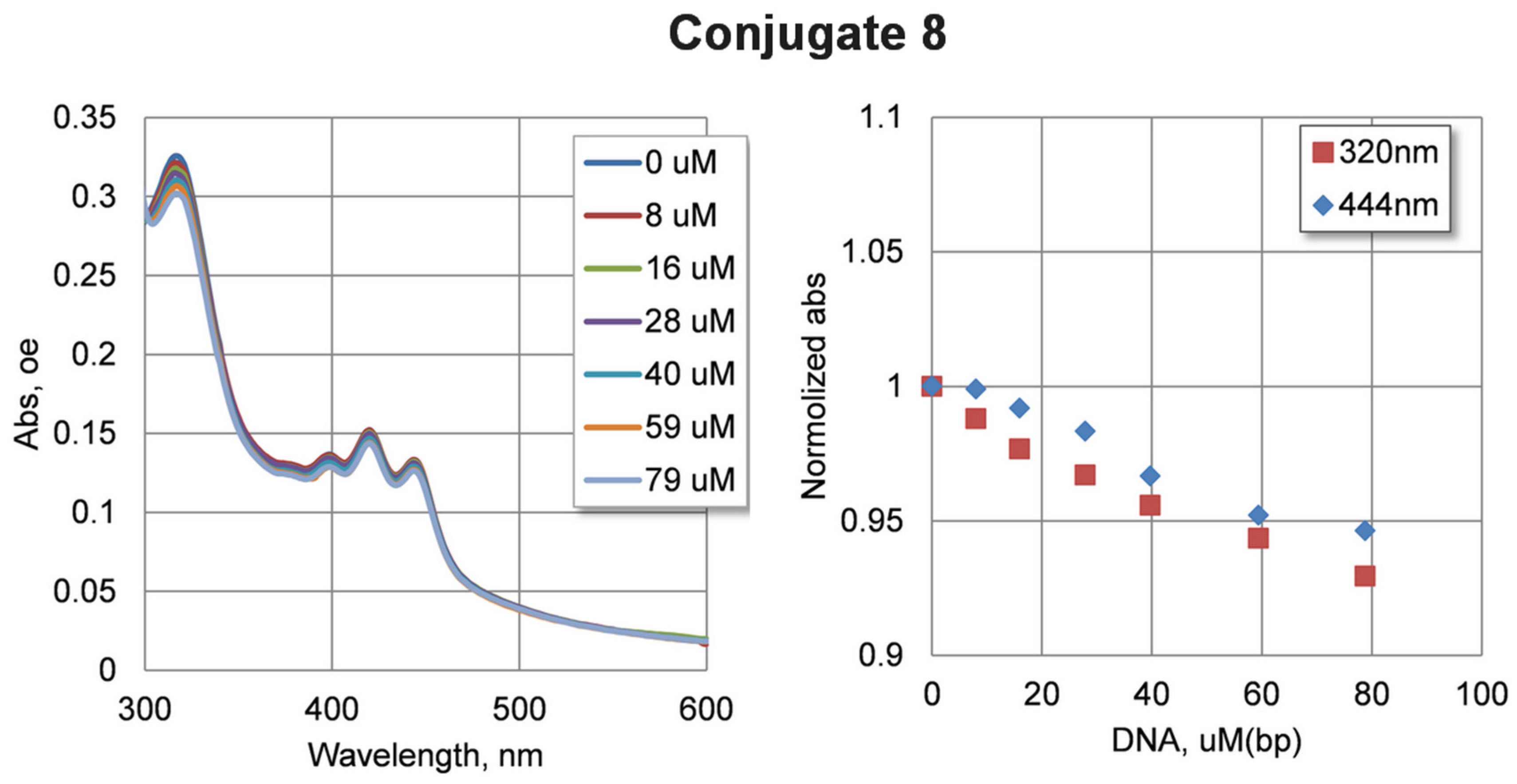
2.4. DNA Interaction Study
2.5. Antiproliferative Activity of Boronated Acridines 7–10
3. Materials and Methods
3.1. General Methods
3.1.1. Synthesis of N9-Azidoacridine 2
General Procedure for the Synthesis of the Conjugates of Cobalt Bis(Dicarbollide) with Acridine 7–10
3.1.2. Synthesis of Conjugate 7
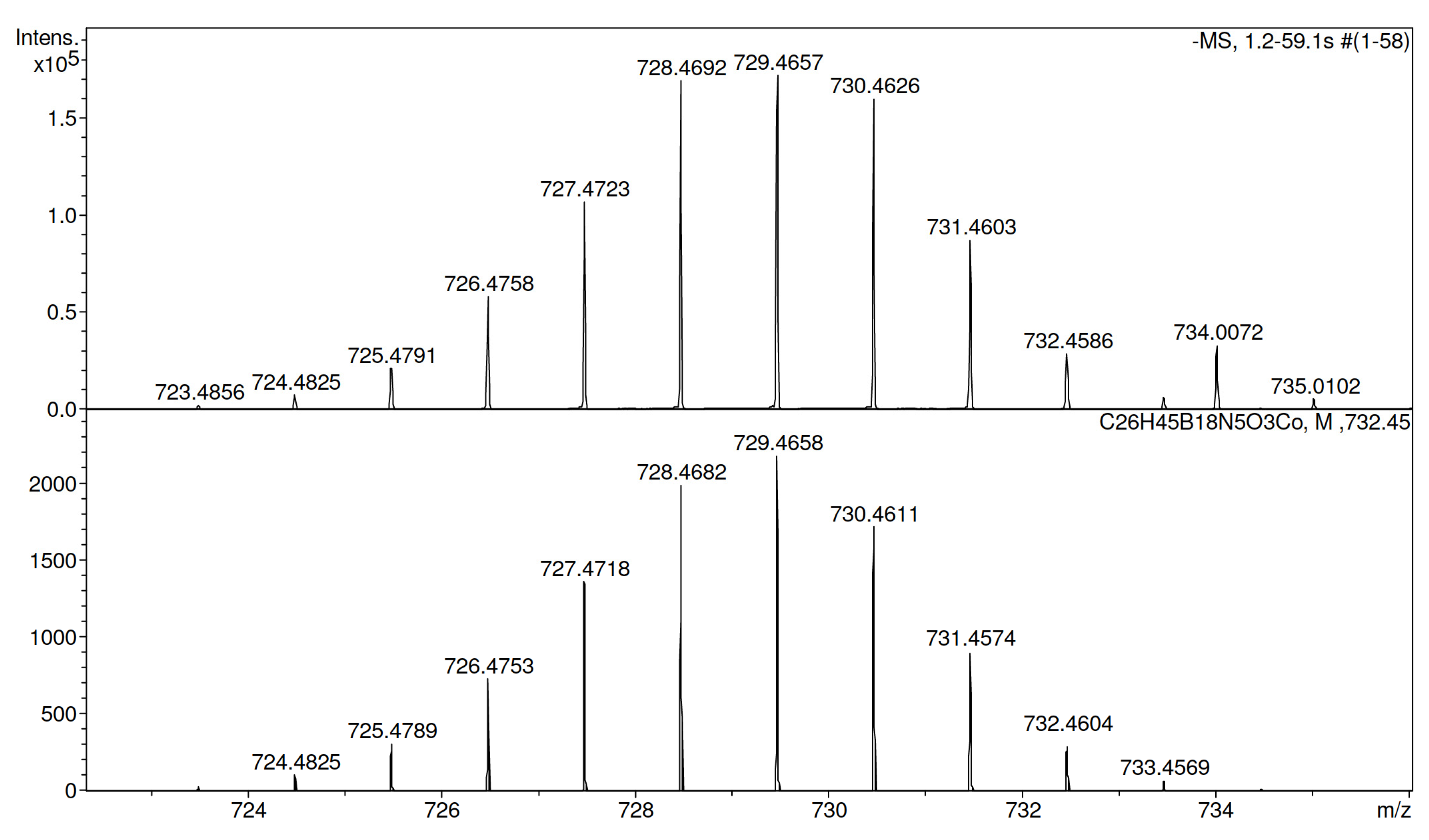
3.1.3. Synthesis of Conjugate 8
3.1.4. Synthesis of Conjugate 9
3.1.5. Synthesis of Conjugate 10
3.2. Absorbance Spectroscopy
3.3. Cells and MTT Assay
3.4. Crystallographic Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Miyatake, S.I.; Kawabata, S.; Goto, H.; Narita, Y.; Suzuki, M.; Hirose, K.; Takai, Y.; Ono, K.; Ohnishi, T.; Tanaka, H.; et al. Accelerator-based BNCT in rescue treatment of patients with recurrent GBM: A multicenter phase II study. J. Clin. Oncol. 2020, 38, 2536. [Google Scholar] [CrossRef]
- Kato, T.; Hirose, K.; Tanaka, H.; Mitsumoto, T.; Motoyanagi, T.; Arai, K.; Harada, T.; Takeuchi, A.; Kato, R.; Yajima, S.; et al. Design and construction of an accelerator-based boron neutron capture therapy (AB-BNCT) facility with multiple treatment rooms at the Southern Tohoku BNCT Research Center. Appl. Radiat. Isot. 2020, 156, 108961–108969. [Google Scholar] [CrossRef] [PubMed]
- Barth, R.F.; Coderre, J.A.; Vicente, M.G.; Blue, T.E. Boron neutron capture therapy of cancer: Current status and future prospects. Clin. Cancer Res. 2005, 11, 3987–4002. [Google Scholar] [CrossRef] [PubMed]
- Bregadze, V.I.; Sivaev, I.B. Boron Science: New Technologies and Applications; Hosmane, N.S., Ed.; CRC Press: Boca Raton, FL, USA, 2012; pp. 181–207. ISBN 9781439826621. [Google Scholar]
- Barth, R.F.; Zhang, Z.; Liu, T. A realistic appraisal of boron neutron capture therapy as a cancer treatment modality. Cancer Commun. 2018, 38, 36–43. [Google Scholar] [CrossRef]
- Barth, R.F.; Mi, P.; Yang, W. Boron delivery agents for neutron capture therapy of cancer. Cancer Commun. 2018, 38, 35–46. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Bregadze, V.I. Chemistry of cobalt bis(dicarbollides). A review. Collect. Czech. Chem. Commun. 1999, 64, 783–805. [Google Scholar] [CrossRef]
- Dash, B.P.; Satapathy, R.; Swain, B.R.; Mahanta, C.S.; Jena, B.B.; Hosmane, N.S. Cobalt bis(dicarbollide) anion and its derivatives. J. Organomet. Chem. 2017, 170, 849–850. [Google Scholar] [CrossRef]
- Fuentes, I.; Teixidor, F.; Viñas, C.; García-Mendiola, T.; Lorenzo, E.; Sato, S.; Nakamura, H.; Pita, M.; Marques, F. Metallacarboranes on the road to anticancer therapies: Cellular uptake, DNA interaction, and biological evaluation of cobaltabisdicarbollide [COSAN]−. Chem. Eur. J. 2018, 24, 17239–17254. [Google Scholar] [CrossRef]
- Tarrés, M.; Canetta, E.; Paul, E.; Forbes, J.; Azzouni, K.; Viñas, C.; Teixidor, F.; Harwood, A.J. Biological interaction of living cells with COSAN-based synthetic vesicles. Sci. Rep. 2015, 5, 7804–7811. [Google Scholar] [CrossRef]
- Spryshkova, R.A.; Karaseva, L.I.; Brattsev, V.A.; Serebriakov, N.G. Toxicity of functional derivatives of polyhedral carboranes. Med. Radiol. 1981, 26, 62–64. [Google Scholar]
- Rokitskaya, T.I.; Kosenko, I.D.; Sivaev, I.B.; Antonenko, Y.N.; Bregadze, V.I. Fast flip-flop of halogenated cobalt bis(dicarbollide) anion in a lipid bilayer membrane. Phys. Chem. Chem. Phys. 2017, 19, 25112–25128. [Google Scholar] [CrossRef] [PubMed]
- Verdiá-Báguena, C.; Alcaraz, A.; Aguilella, V.M.; Cioran, A.M.; Tachikawa, S.; Nakamura, H.; Teixidor, F.; Viñas, C. Amphiphilic COSAN and I2-COSAN crossing synthetic lipid membranes: Planar bilayers and liposomes. Chem. Commun. 2014, 50, 6700–6703. [Google Scholar] [CrossRef] [PubMed]
- Assaf, K.I.; Begaj, B.; Frank, A.; Nilam, M.; Mougharbel, A.S.; Kortz, U.; Nekvinda, J.; Grüner, B.; Gabel, D.; Nau, W.M. High-affinity binding of metallacarborane cobalt bis(dicarbollide) anions to cyclodextrins and application to membrane translocation. J. Org. Chem. 2019, 84, 11790–11798. [Google Scholar] [CrossRef] [PubMed]
- Hartman, T.; Carlsson, J. Radiation dose heterogeneity in receptor and antigen mediated boron neutron capture therapy. Radiother. Oncol. 1994, 31, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Gabel, D.; Foster, S.; Fairchild, R.G. The Monte Carlo simulation of the biological effect of the 10B(n,α)7Li reaction in cells and tissue and its implication for Boron Neutron Capture Therapy. Radiat. Res. 1987, 111, 14–25. [Google Scholar] [CrossRef]
- Crossley, E.L.; Ziolkowski, E.J.; Coderre, J.A.; Rendina, L.M. Boronated DNA-binding compounds as potential agents for Boron Neutron Capture Therapy. Mini-Rev. Med. Chem. 2007, 7, 303–313. [Google Scholar] [CrossRef]
- Tjarks, W.; Ghaneolhosseini, H.; Henssen, C.L.; Malmquist, J.; Sjöberg, S. Synthesis of para- and nido-carboranyl phenanthridinium compounds for neutron capture therapy. Tetrahedron Lett. 1996, 37, 6905–6908. [Google Scholar] [CrossRef]
- Ghaneolhosseini, H.; Tjarks, W.; Sjöberg, S. Synthesis of boronated phenanthridinium derivatives for potential use in boron neutron capture therapy (BNCT). Tetrahedron 1997, 53, 17519–17526. [Google Scholar] [CrossRef]
- Tjarks, W.; Malmquist, J.; Gedda, L.; Sjöberg, S.; Carlsson, J. Synthesis and initial biological evaluation of carborane-containing phenanthridinium derivatives. In Cancer Neutron Capture Therapy; Mishima, Y., Ed.; Springer: New York, NY, USA, 1996; pp. 121–126. [Google Scholar]
- Ghaneolhosseini, H.; Sjöberg, S. Synthesis of a boronated naphthalimide for potential use in boron neutron capture therapy (BNCT). Acta Chem. Scand. 1999, 53, 298–300. [Google Scholar] [CrossRef]
- Nekvinda, J.; Różycka, D.; Rykowski, S.; Wyszko, E.; Fedoruk-Wyszomirska, A.; Gurda, D.; Orlicka-Płocka, M.; Giel-Pietraszuk, M.; Kiliszek, A.; Rypniewski, W.; et al. Synthesis of naphthalimide-carborane and metallacarborane conjugates: Anticancer activity, DNA binding ability. Bioorg. Chem. 2020, 94, 103432. [Google Scholar] [CrossRef]
- Rykowski, S.; Gurda-Woźna, D.; Orlicka-Płocka, M.; Fedoruk-Wyszomirska, A.; Giel-Pietraszuk, M.; Wyszko, E.; Kowalczyk, A.; Stączek, P.; Bak, A.; Kiliszek, A.; et al. Design, synthesis, and evaluation of novel 3-carboranyl-1,8-naphthalimide derivatives as potential anticancer agents. Int. J. Mol. Sci. 2021, 22, 2772. [Google Scholar] [CrossRef] [PubMed]
- Laskova, J.; Kosenko, I.; Serdyukov, A.; Sivaev, I.; Bregadze, V.I. Synthesis of naphthalimide derivatives of closo-dodecaborate and nido-carborane. J. Organomet. Chem. 2021, 959, 122186. [Google Scholar] [CrossRef]
- Bogucka-Kocka, A.; Kołodziej, P.; Makuch-Kocka, A.; Różycka, D.; Rykowski, S.; Nekvinda, J.; Grüner, B.; Olejniczak, A.B. Nematicidal activity of naphthalimide–boron cluster conjugates. Chem. Commun. 2022, 58, 2528–2531. [Google Scholar] [CrossRef] [PubMed]
- Rykowski, S.; Gurda-Woźna, D.; Orlicka-Płocka, M.; Fedoruk-Wyszomirska, A.; Giel-Pietraszuk, M.; Wyszko, E.; Kowalczyk, A.; Stączek, P.; Biniek-Antosiak, K.; Rypniewski, W.; et al. Design of DNA intercalators based on 4-carboranyl-1,8-naphthalimides: Investigation of their DNA-binding ability and anticancer activity. Int. J. Mol. Sci. 2022, 23, 4598. [Google Scholar] [CrossRef]
- Rykowski, S.; Gurda-Wozna, D.; Fedoruk-Wyszomirska, A.; Orlicka-Płocka, M.; Kowalczyk, A.; Staczek, P.; Denel-Bobrowska, M.; Biniek-Antosiak, K.; Rypniewski, W.; Wyszko, E.; et al. Carboranyl-1,8-naphthalimide intercalators induce lysosomal membrane permeabilization and ferroptosis in cancer cell lines. J. Enzyme Inhib. Med. Chem. 2023, 38, 2171028. [Google Scholar] [CrossRef]
- Soloway, A.H. Boron compounds in cancer therapy. In Progress in Boron Chemistry; McCloskey, A.L., Steinberg, H., Eds.; Pergamon Press: New York, NY, USA, 1964; Volume 1, pp. 203–234. [Google Scholar]
- Davis, M.A.; Soloway, A.H. Carboranes. III. Boron-containing acridines. J. Med. Chem. 1967, 10, 730–732. [Google Scholar] [CrossRef]
- Ghaneolhosseini, H.; Tjarks, W.; Sjoberg, S. Synthesis of novel boronated acridines- and spermidines as possible agents for BNCT. Tetrahedron 1998, 54, 3877–3884. [Google Scholar] [CrossRef]
- Różycka, D.; Kowalczyk, A.; Bobrowska, M.D.; Kuźmycz, O.; Gapińska, M.; Stączek, P.; Olejniczak, A.B. Acridine/acridone–carborane conjugates as strong DNA-binding agents with anticancer potential. ChemMedChem 2023, 18, e202200666. [Google Scholar] [CrossRef]
- Cebula, J.; Fink, K.; Goldeman, W.; Szermer-Olearnik, B.; Nasulewicz-Goldeman, A.; Psurski, M.; Cuprych, M.; Kędziora, A.; Dudek, B.; Bugla-Płoskońska, G.; et al. Structural patterns enhancing the antibacterial activity of metallacarborane-based antibiotics. ChemRxiv 2023. [Google Scholar] [CrossRef]
- Howell, L.A.; Gulam, R.; Mueller, A.; O’Connell, M.A.; Searcey, M. Design and synthesis of threading intercalators to target DNA. Bioorg. Med. Chem. Lett. 2010, 20, 6956–6959. [Google Scholar] [CrossRef]
- Howell, L.A.; Bowater, R.A.; O’Connell, M.A.; Reszka, A.P.; Neidle, S.; Searcey, M. Synthesis of small molecules targeting multiple DNA structures using click chemistry. ChemMedChem 2012, 7, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Denny, W.A.; Cain, B.F.; Atwell, G.J.; Hansch, C.; Panthananickal, A.; Leo, A.J. Potential antitumor agents. Quantitative relationships between experimental antitumor activity, toxicity, and structure for the general class of 9-anilinoacridine antitumor agents. J. Med. Chem. 1982, 25, 276–315. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Vettoretti, G.; Tasso, B.; Novelli, F.; Boido, V.; Sparatore, F.; Busonera, B.; Ouhtit, A.; Farci, P.; Blois, S.; et al. Acridine derivatives as anti-BVDV agents. Antiv. Res. 2011, 91, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective ligation of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Rani, A.; Singh, G.; Singh, A.; Maqbool, U.; Kaur, G.; Singh, J. CuAAC-ensembled 1,2,3-triazole-linked isosteres as pharmacophores in drug discovery: Review. RSC Adv. 2020, 10, 5610–5635. [Google Scholar] [CrossRef] [PubMed]
- Smyshliaeva, L.A.; Varaksin, M.V.; Fomina, E.I.; Joy, M.N.; Bakulev, V.A.; Charushin, V.N.; Chupakhin, O.N. Cu(I)-catalyzed cycloaddition of vinylacetylene ortho-carborane and arylazides in the design of 1,2,3-triazolyl-modified vinylcarborane fluorophores. Organometalics 2020, 39, 3679–3688. [Google Scholar] [CrossRef]
- Bregadze, V.I.; Semioshkin, A.A.; Las´kova, J.N.; Berzina, M.Y.; Lobanova, I.A.; Sivaev, I.B.; Grin, M.A.; Titeev, R.A.; Brittal, D.I.; Ulybina, O.V.; et al. Novel types of boronated chlorine e6 conjugates via «click chemistry». Appl. Organometal. Chem. 2009, 23, 370–374. [Google Scholar] [CrossRef]
- Wojtczak, B.A.; Andrysiak, A.; Grüner, B.; Leśnikowski, Z.J. “Chemical ligation”: A versatile method for nucleoside modification with boron clusters. Chem. Eur. J. 2008, 14, 10675–10682. [Google Scholar] [CrossRef]
- Druzina, A.A.; Shmalko, A.V.; Sivaev, I.B.; Bregadze, V.I. Cyclic oxonium derivatives of cobalt and iron bis (dicarbollide)s and their use in organic synthesis. Russ. Chem. Rev. 2021, 90, 785–830. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Aghabozorg, H.; Ahmadvand, S.; Mirzaei, M.; Khavasi, H.R. Bis(9-aminoacridinium) bis(pyridine-2,6-dicarboxylato) cuprate(II) trihydrate. Acta Cryst. E 2010, 66, m1318–m1319. [Google Scholar] [CrossRef] [PubMed]
- Eshtiagh-Hosseini, H.; Mirzaei, M.; Eydizadeh, E.; Yousefi, Z.; Molčanov, K. Bis(9-aminoacridinium) bis(pyridine-2,6-dicarboxylato-κ3O2,N,O6)manganate(II) trihydrate. Acta Cryst. E 2011, 67, m1411–m1412. [Google Scholar] [CrossRef] [PubMed]
- Dhanabalan, N.; Thanigaimani, K.; Arshad, S.; Razak, I.A.; Santhanaraj, K.J. 9-Aminoacridin-10-ium 4-aminobenzoate dihydrate. Acta Cryst. E 2014, 70, o657–o658. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, M.; Eshtiagh-Hosseini, H.; Eydizadeh, E.; Yousefi, Z.; Molčanov, K. 9-Aminoacridinium bis(pyridine-2,6-dicarboxylato-κ3O2,N,O6)ferrate(III) tetrahydrate. Acta Cryst. E 2012, 68, m761–m762. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, M.; Eshtiagh-Hosseini, H.; Eydizadeh, E.; Yousefi, Z.; Molčanov, K. Bis(9-aminoacridinium) bis(pyridine-2,6-dicarboxylato)zincate(II) trihydrate. Acta Cryst. E 2012, 68, m355–m356. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Pendas, A.M.; Francisco, E.; Blanco, M.A.; Gatti, C. Bond paths as privileged exchange channels. Chem. Eur. J. 2007, 13, 9362–9371. [Google Scholar] [CrossRef]
- Anisimov, A.A.; Ananyev, I.V. Interatomic exchange-correlation interaction energy from a measure of quantum theory of atoms in molecules topological bonding: A diatomic case. J. Comput. Chem. 2020, 41, 2213–2222. [Google Scholar] [CrossRef]
- Romanova, A.; Lyssenko, K.; Ananyev, I. Estimations of energy of noncovalent bonding from integrals over interatomic zero-flux surfaces: Correlation trends and beyond. J. Comput. Chem. 2018, 39, 1607–1616. [Google Scholar] [CrossRef]
- Mishima, Y.; Ichihashi, M.; Hatta, S.; Honda, C.; Yamamura, K.; Nakagawa, T. New thermal neutron capture therapy for malignant melanoma: Melanogenesis-seeking 10B molecule-melanoma cell interaction from in vitro to first clinical trial. Pigm. Cell Res. 1989, 2, 226–234. [Google Scholar] [CrossRef]
- Kosenko, I.D.; Lobanova, I.A.; Chekulaeva, L.A.; Godovikov, I.A.; Bregadze, V.I. Synthesis of 1,4-disubstituted 1,2,3-triazoles based on cobalt bis (1,2-dicarbollide). Russ. Chem. Bull. 2013, 62, 497–503. [Google Scholar] [CrossRef]
- Laskova, J.; Serdyukov, A.; Kosenko, I.; Ananyev, I.; Titova, E.; Druzina, A.; Sivaev, I.; Antonets, A.A.; Nazarov, A.A.; Bregadze, V.I. New azido coumarins as potential agents for fluorescent labeling and their “click” chemistry reactions for the conjugation with closo-dodecaborate anion. Molecules 2022, 27, 8575–8589. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, X.; Zhang, Y.; Wang, Z.; Lasanajak, Y.; Song, X. Anthranilic acid as a versatile fluorescent tag and linker for functional glycomics. Bioconjugate Chem. 2018, 29, 3847–3855. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta. Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; PuschmannJ, H. OLEX2: A complete structure solution, refinement and analysis program. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. (Eds.) Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Adamo, C.; Vincenzo, B. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154124. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. Effect of the damping function in dispersion corrected density functional theory. J. Chem. Phys. 2011, 132, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Keith, T.A. (Ed.) AIMAll (Version 19.10.12); TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50, µM | ||||
|---|---|---|---|---|---|
| 7 | 8 | 9 | 10 | Cisplatin [55] | |
| WI38 | 21 ± 4 | 21 ± 5 | 33 ± 3 | 34 ± 8 | 8 ± 3 |
| A549 | >200 | >200 | >200 | >200 | 13 ± 3 |
| HCT116 | 49 ± 20 | 61 ± 25 | 43 ± 18 | 41 ± 1 | 13 ± 4 |
| MCF7 | 15 ± 3 | 25 ± 6 | 28 ± 3 | 13 ± 3 | 30 ± 9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Druzina, A.A.; Dudarova, N.V.; Ananyev, I.V.; Antonets, A.A.; Kaluzhny, D.N.; Nazarov, A.A.; Sivaev, I.B.; Bregadze, V.I. New Boron Containing Acridines: Synthesis and Preliminary Biological Study. Molecules 2023, 28, 6636. https://doi.org/10.3390/molecules28186636
Druzina AA, Dudarova NV, Ananyev IV, Antonets AA, Kaluzhny DN, Nazarov AA, Sivaev IB, Bregadze VI. New Boron Containing Acridines: Synthesis and Preliminary Biological Study. Molecules. 2023; 28(18):6636. https://doi.org/10.3390/molecules28186636
Chicago/Turabian StyleDruzina, Anna A., Nadezhda V. Dudarova, Ivan V. Ananyev, Anastasia A. Antonets, Dmitry N. Kaluzhny, Alexey A. Nazarov, Igor B. Sivaev, and Vladimir I. Bregadze. 2023. "New Boron Containing Acridines: Synthesis and Preliminary Biological Study" Molecules 28, no. 18: 6636. https://doi.org/10.3390/molecules28186636