

The Synthesis, Characterization, and Fluxional Behavior of a Hydridorhodatetraborane

Abstract
:1. Introduction
2. Results and Discussion
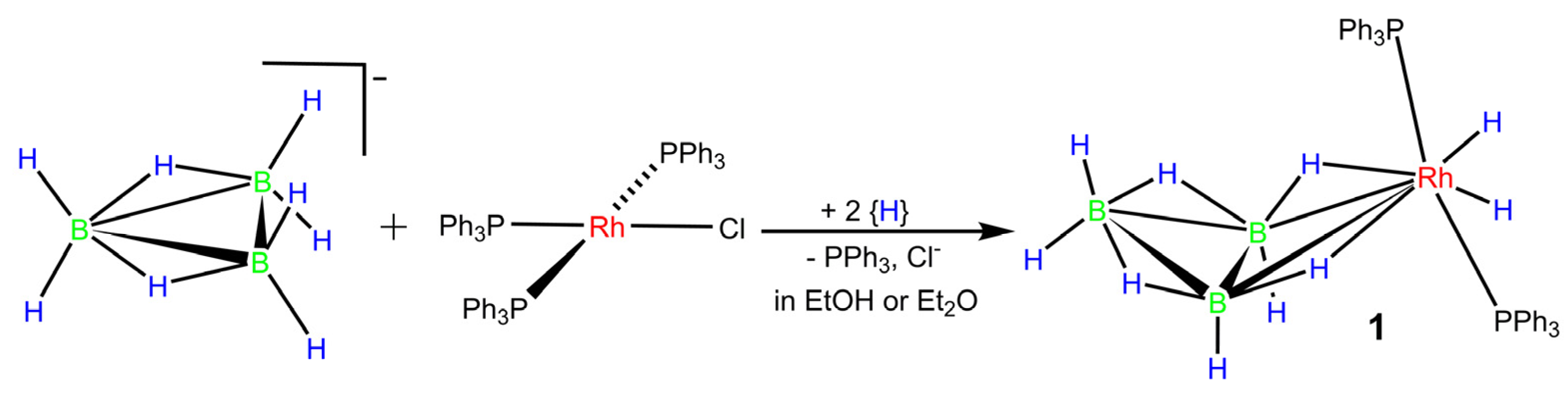
2.1. Synthesis of [Rh(η2-B3H8)(H)2(PPh3)2] (1)
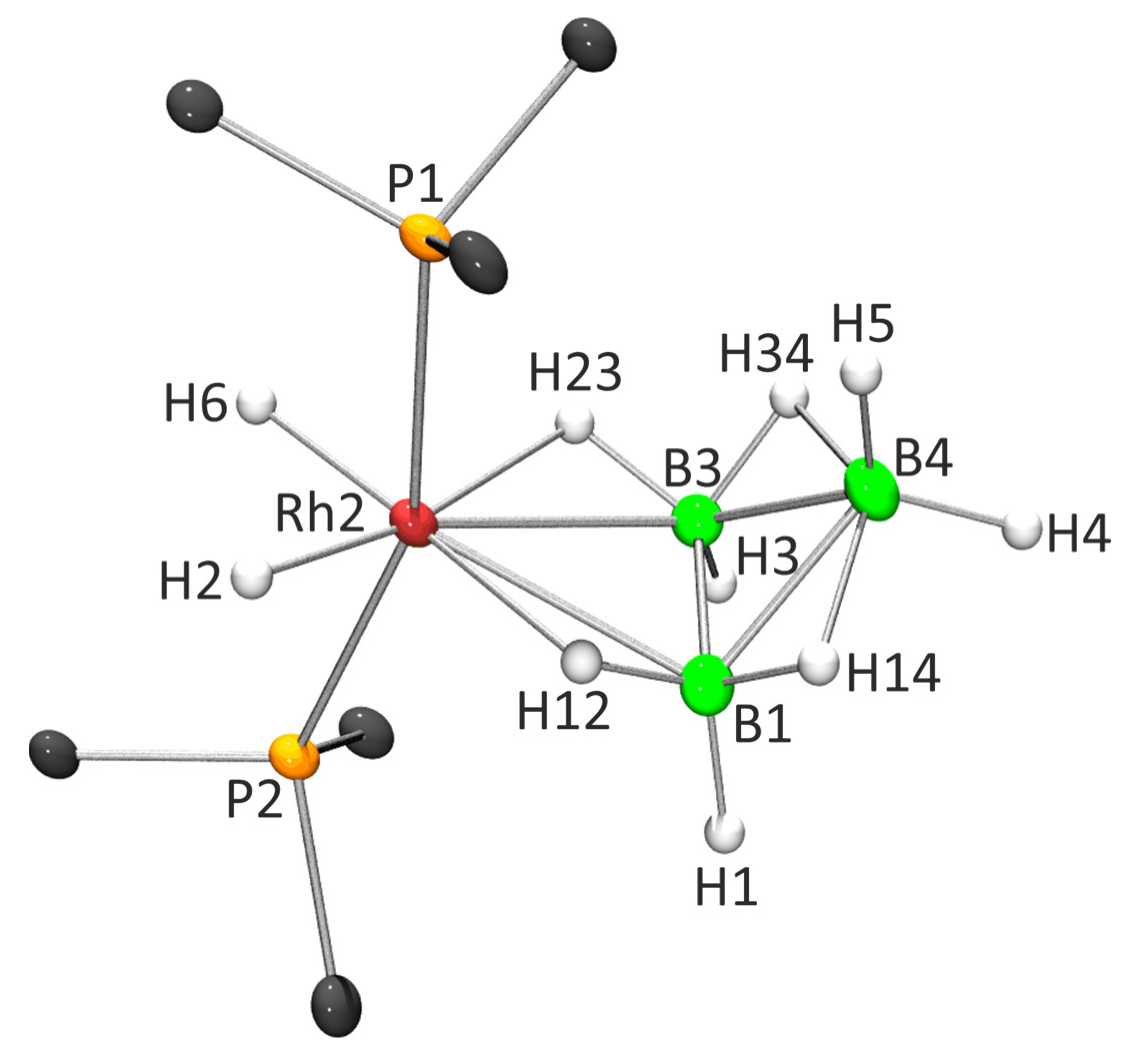
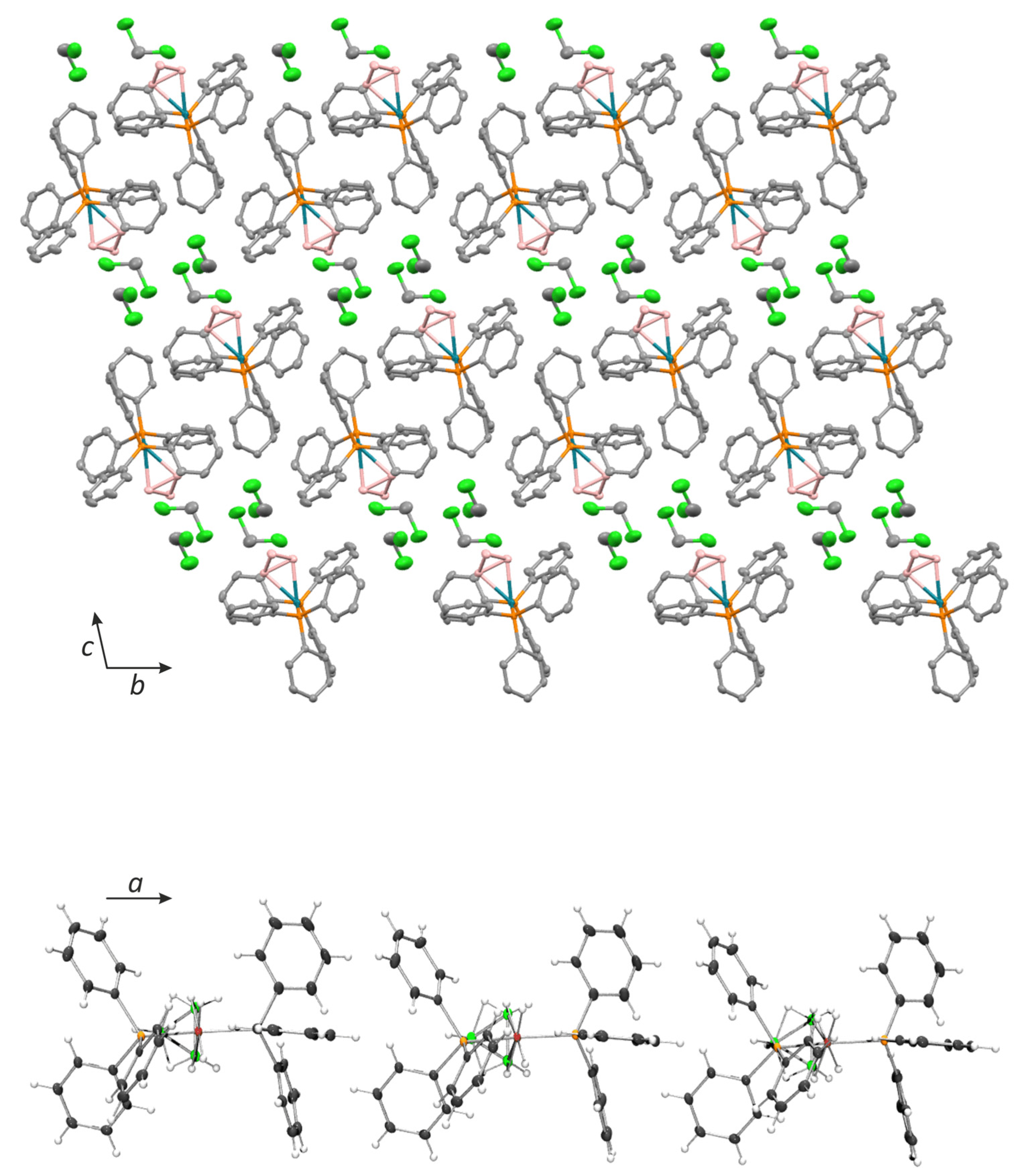
2.2. X-ray Diffraction Analysis
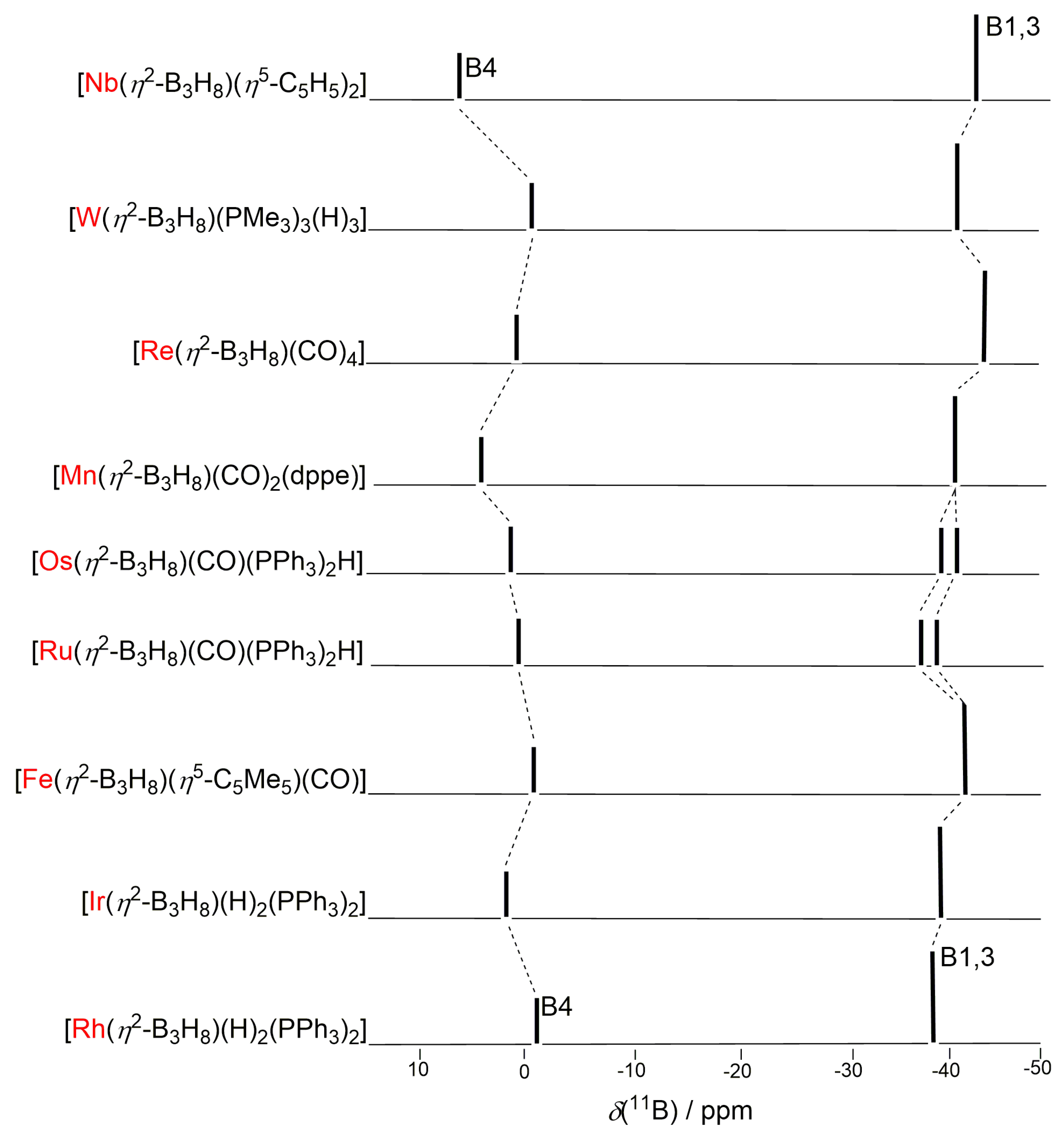
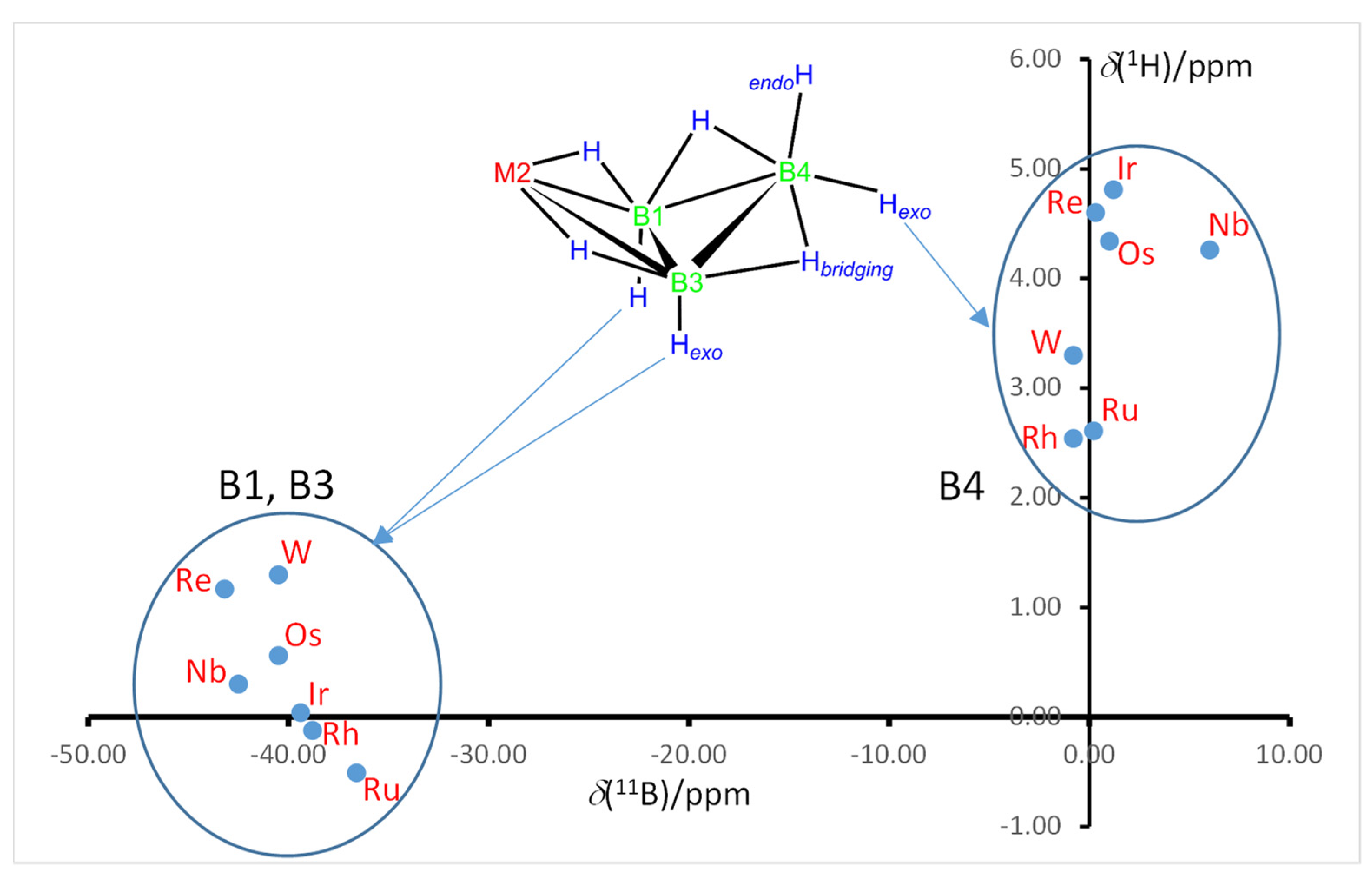
2.3. NMR Characterization and Comparison
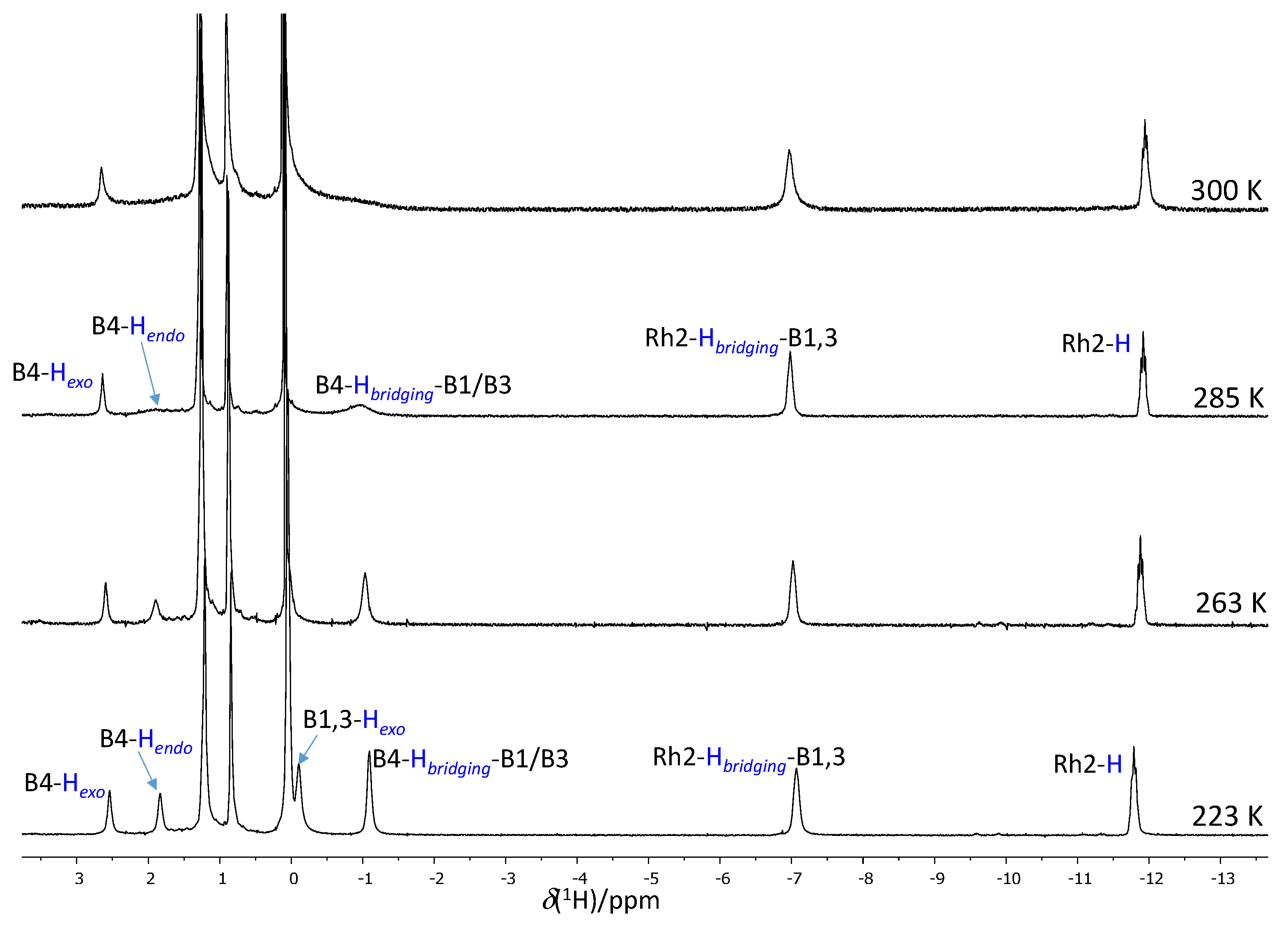
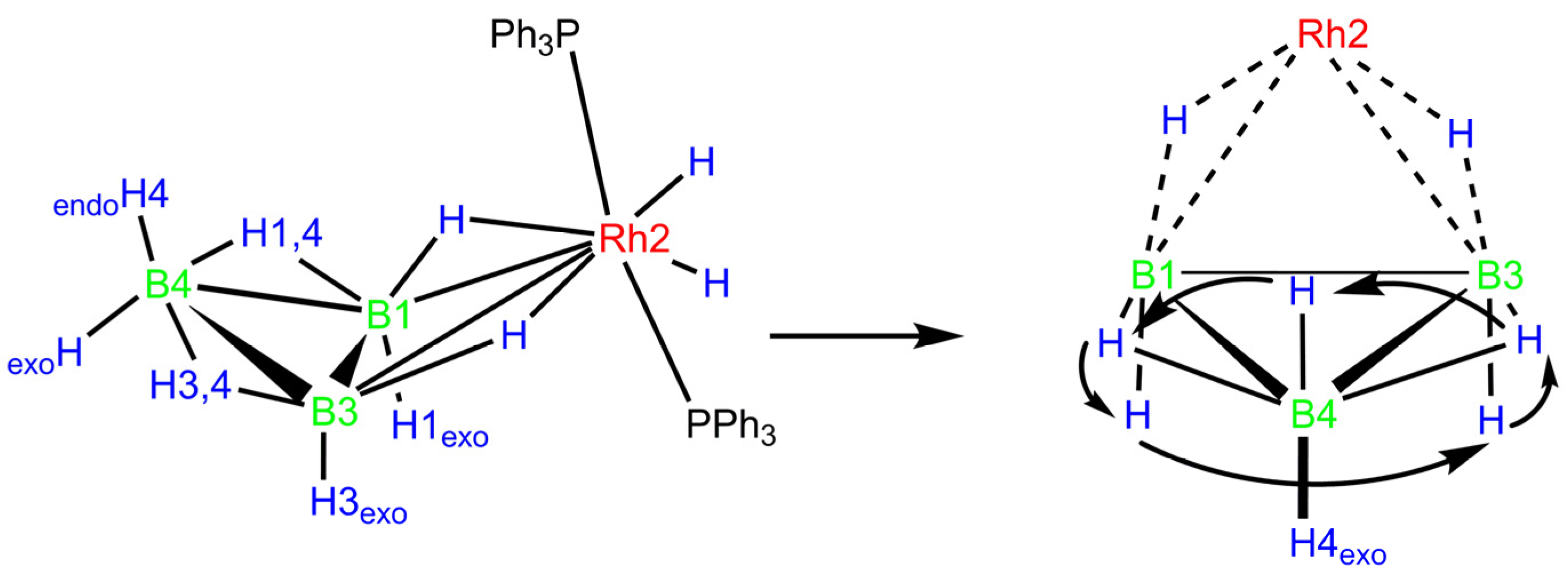
2.4. Fluxional Behavior
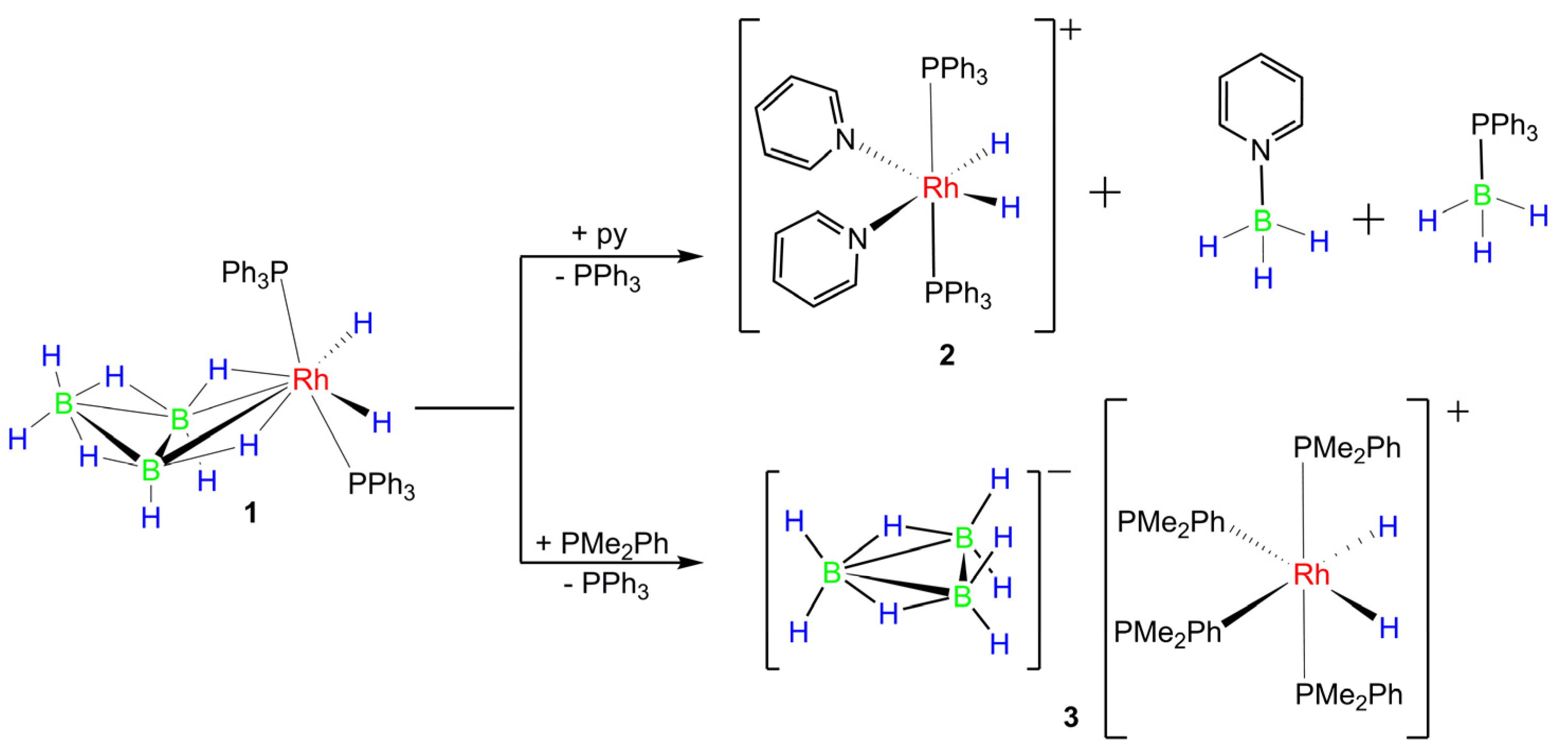
2.5. Reactions of 1 with Lewis Bases
3. Conclusions
4. Materials and Methods
4.1. General
4.2. Crystal Structure Determination
4.3. Mass Spectrometry
4.4. Computational Details
4.5. Preparation of [Rh(η2-B3H8)(H)2(PPh3)2] (1)
4.6. Reactions of [Rh(η2-B3H8)(H)2(PPh3)2] (1) with Lewis Bases
- Reaction of [Rh(η2-B3H8)(H)2(PPh3) 2] (1) with py. 10.2 mg of 1 (1.50 × 10−2 mmol) treated with 1.20 μg of pyridine (1.50 × 10−2 mmol), in a Schlenk tube immersed in a bath of isopropanol at −30 °C. The resulting yellow solution was stirred, under an atmosphere of argon for 5 h; during this time, the temperature was raised to +10 °C. The reaction was stirred for another 20 min at room temperature. The solvent was evaporated under vacuum to give an orange solid, which was dissolved in deuterated dichloromethane and studied using NMR spectroscopy. 31P-{1H} (162 MHz, 233 K): δ +47.2 ppm [1J(31P-103Rh) = 118 Hz], together with the signal of O=PPh3. 1H-{31P} (400 MHz, 233 K): δ +8.59 (d, J = 5.0 Hz, ortho-NC5H5, 2H), +8.59 (d, J = 5.0 Hz, o-NC5H5, 2H), +8.34 (br. s, o-NC5H5, 2H), +7.95 (t., p-NC5H5, 2H), +7.95 (t., p-NC5H5, 2H), +6.54 (br. s, m-NC5H5, 2H), between +7.73 and +6.80 (m, C6H5-rings and NC5H5), −16.89 (t, 1J(103Rh-1H2,6) + 2J(1H2-1H2,3) = 2J(1H6-1H1,2) = 12.9 Hz, 2H) and -17.96 (dd, 1J(103Rh-1H2,6) = 23.4, 2J(1H2-1H2,3) = 2J(1H6-1H1,2) = 10.9 Hz, 2H). 1H-{11B} (400 MHz, 233 K): δ −16.89 (app. quintet, 1J(103Rh-1H2,6) + 2J(31P-31P) + 2J(1H2-1H2,3) = 12.9 Hz, 2H) and −17.96 (td, J = 25.5, 13.1 Hz, Rh-H2). δ 11B (400 MHz, 298 K): δ +18.8 (br.), +1.47 (s), −1.5 (t), −9.31 (t, 95 Hz), −12.0 (q, 1J(11B-1H) = 98 Hz, py-BH3), −37.8 (dq, 62 Hz, Ph3PBH3), −27.4 (t, 98 Hz).
- In situ characterization of [Rh(H)2(PMe2Ph)4][B3H8] (3). 12.6 mg of 1 (1.88 × 10−2 mmol), dissolved in CD2Cl2, in a NMR tube, which was immersed in an isopropanol bath at −30 °C, and 2.6 mg (2.7 μL) of PMe2Ph (1.88 × 10−2 mmol) was added into the NMR tube, under a flow of argon. The reaction was studied using NMR spectroscopy, starting at 233 K and then heating the sample to room temperature. 31P-{1H} (162 MHz, 233 K): δ +26.7 ppm [s, O=PPh3], +19.7 (very br., PhMe2P-BH3), +0.3 [dt, 1J(103Rh–31P) = 97 Hz, 2J(31P1–31P2) = 24 Hz], −7.2 (s, PPh3), −10.6 p.pm [dt, 1J(103Rh–31P) = 86 Hz, 2J(31P1–31P2) = 24 Hz], together with the signals of 1 (Table 1). 1H-{31P} (400 MHz, 233 K): δ +7.84 to 6.94 (m, aromatics, C6H5), +1.56 (s, CH3), +1.46 (s, CH3), −10.25 (d, 1J(103Rh-1H) = 13.8 Hz. 1H-{11B} (400 MHz, 233 K): +1.2 (d, 2J(31P–1H), PhMe2-BH3), +0.23(s, B3H8−), −10.25 (d of pseudo-quintets, second order, 2J(11P-1Htrans) = 147.9 Hz, 1J(103Rh-1H) = 14.0 Hz, 2J(11P-1Hcis) = 17.2 Hz, 2H) and −17.96 (dd, 1J(103Rh-1H2,6) = 23.4, 2J(1H2-1H2,3) = 2J(1H6-1H1,2) = 10.9 Hz, 2H) ppm. 11B (400 MHz, 298 K): δ −16.4 (br. s), −30.5 (sept, B3H8−), −37.7 (quartet of d, 1J(11B-31P) = 59 Hz, 1J(11B-1H) = 102 Hz, PhMe2P–BH3), −45.2 (br. s).
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Barton, L.; Strivastava, D.K. Comprehensive Organometallic Chemistry II; Abel, E.W., Stone, F.G.A., Wilkinson, G., Eds.; Pergamon: New York, NY, USA, 1995; Volume 1, pp. 275–372. [Google Scholar]
- Cotton, F.A.; Wilkinson, G.; Murillo, C.A.; Bochmann, M. Advanced Inorganic Chemistry, 6th ed.; Wiley: New York, NY, USA, 1999. [Google Scholar]
- Kennedy, J.D. The Polyhedral Metalloboranes. 1. Metalloborane Clusters with 7 Vertices And Fewer. In Progress in Inorganic Chemistry; John Wiley & Sons: New York, NY, USA, 1984; Volume 32, pp. 519–679. [Google Scholar]
- Weller, A.S. d- and f-Block Metallaboranes. In Comprehensive Organometallic Chemistry III; Crabtree, R.H., Mingos, D.M.P., Eds.; Elsevier: Oxford, UK, 2007; Volume 3, pp. 133–174. [Google Scholar]
- Bould, J.; Greenwood, N.N.; Kennedy, J.D. The first osmaboranes and a new iridatetraborane. J. Organomet. Chem. 1983, 249, 11–21. [Google Scholar] [CrossRef]
- Grebenik, P.D.; Leach, J.B.; Green, M.L.H.; Walker, N.M. Transition metal mediated homologation of BH3·THF: Synthesis and crystal structure of [WH3(PMe3)3B3H8]. J. Organomet. Chem. 1988, 345, C31–C34. [Google Scholar] [CrossRef]
- Grebenik, P.D.; Leach, J.B.; Pounds, J.M.; Green, M.L.H.; Mountford, P. Niobium metallaboranes: A novel metallaborane analogue of pentaborane(11). J. Organomet. Chem. 1990, 382, C1–C5. [Google Scholar] [CrossRef]
- Ghosh, S.; Beatty, A.M.; Fehlner, T.P. The Reaction of Cp*ReH6, Cp* = C5Me5, with Monoborane to Yield a Novel Rhenaborane. Synthesis and Characterization of arachno-Cp*ReH3B3H8. Collect. Czech. Chem. Commun. 2002, 67, 808–812. [Google Scholar] [CrossRef]
- Fehlner, T.P. Systematic Metallaborane Chemistry. Organometallics 2000, 19, 2643–2651. [Google Scholar] [CrossRef]
- Geetharani, K.; Kumar Bose, S.; Pramanik, G.; Kumar Saha, T.; Ramkumar, V.; Ghosh, S. An Efficient Route to Group 6 and 8 Metallaborane Compounds: Synthesis of arachno-[Cp*Fe(CO)B3H8] and closo-[(Cp*M)2B5H9] (M = Mo, W). Eur. J. Inorg. Chem. 2009, 2009, 1483–1487. [Google Scholar] [CrossRef]
- Green, M.L.H.; Leach, J.B.; Kelland, M.A. Synthesis and Interconversion of Some Small Ruthenaboranes: Reaction of a Ruthenium Borohydride with Pentaborane(9) to Form Larger Ruthenaboranes. Organometallics 2007, 26, 4031–4037. [Google Scholar] [CrossRef]
- Guggenberger, L.J. Crystal structure of the tetramethylammonium salt of the octahydrotriborotetracarbonylchromium anion, (CO)4CrB3H8. Inorg. Chem. 1970, 9, 367–373. [Google Scholar] [CrossRef]
- Beckett, M.A.; Brassington, D.S.; Coles, S.J.; Gelbrich, T.; Hursthouse, M.B. Synthesis and characterisation of a series of Group 7 metal 2,2,2,2-dicarbonylbis(triorganophosphine)-arachno-2-metallatetraboranes, [M(CO)2L2(B3H8)] (M=Re, Mn); crystal and molecular structures of [Re(CO)2(dppf)(B3H8)] and [Mn(CO)2(dppe)(B3H8)]. Polyhedron 2003, 22, 1627–1632. [Google Scholar] [CrossRef]
- Burns, I.D.; Hill, A.F.; Williams, D.J. Ruthenatetraboranes: Molecular Structure of [Ru(B3H8)(PPh3){κ3-HB(pz)3}] (pz = Pyrazol-1-yl). Inorg. Chem. 1996, 35, 2685–2687. [Google Scholar] [CrossRef]
- Bown, M.; Ingham, S.L.; Norris, G.E.; Waters, J.M. exo-2-([eta]6-Hexamethylbenzene)-endo-2-chloro-2-ruthena-arachno-tetraborane(8). Acta Crystallogr. Sect. C 1995, 51, 1503–1505. [Google Scholar] [CrossRef]
- Bould, J.; Rath, N.P.; Barton, L. [(CO)H(PPh3)2-arachno-OsB3H8]. Acta Crystallogr. Sect. C 1996, 52, 1388–1390. [Google Scholar] [CrossRef]
- Lippard, S.J.; Melmed, K.M. Transition metal borohydride complexes. III. Structure of octahydrotriboratobis(triphenylphosphine)copper(I). Inorg. Chem. 1969, 8, 2755–2762. [Google Scholar] [CrossRef]
- Williams, R.E. Coordination number pattern recognition theory of carborane structures. Adv. Inorg. Chem. Radiochem. 1976, 18, 67–142. [Google Scholar]
- Olah, G.A.; Wade, K.; Williams, R.E. Electron Deficient Boron and Carbon Clusters; John Wiley & Sons: New York, NY, USA, 1991. [Google Scholar]
- Wade, K. Structural and bonding patterns in cluster chemistry. Adv. Inorg. Chem. Radiochem. 1976, 18, 1–66. [Google Scholar]
- Dance, I.; Scudder, M. The sextuple phenyl embrace, a ubiquitous concerted supramolecular motif. J. Chem. Soc. Chem. Commun. 1995, 10, 1039–1040. [Google Scholar] [CrossRef]
- Dance, I.; Scudder, M. Molecules embracing in crystals. CrystEngComm 2009, 11, 2233–2247. [Google Scholar] [CrossRef]
- Hore, P.J. Nuclear Magnetic Resonance; Oxford University Press: Oxford, UK, 2011. [Google Scholar]
- Dhubhghaill, O.N.; Spalding, T.R.; Ferguson, G.; Kaitner, B.; Fontaine, X.L.R.; Kennedy, J.D.; Reed, D. Metallaheteroborane Chemistry. 4. The Synthesis of closo-[2,2-(PPh3)2-2-H-1,2-XMB10H10] (X = Se Or Te, M = Rh Or Ir) Compounds, their Characterization by Nuclear Magnetic-Resonance Techniques, and the crystal and molecular structure of the X = Te, M = Rh complex. J. Chem. Soc. Dalton Trans. 1988, 11, 2739–2745. [Google Scholar]
- Ferguson, G.; Kennedy, J.D.; Fontaine, X.L.R.; Faridoon; Spalding,, T.R. Metallaheteroborane chemistry.3. Synthesis of [2,2-(PEt3)2-1,2-TePtB10H10], [2,2-(PBun3)2-1,2-TePTtB10H10], [2,2-(PMe2Ph)2-1,2-TePtB10H10], their characterization by nuclear magnetic-resonance spectroscopy, and the crystal and molecular-structure of [2,2-(PEt3)2-1,2-TePtB10H10]. J. Chem. Soc. Dalton Trans. 1988, 10, 2555–2564. [Google Scholar]
- O’Connell, D.; Patterson, J.C.; Spalding, T.R.; Ferguson, G.; Gallagher, J.F.; Li, Y.; Kennedy, J.D.; Macías, R.; Thornton-Pett, M.; Holub, J. Conformational polymorphism and fluxional behaviour of M(PR3)2 units in closo-twelve-atom metallaheteroboranes with MX2B9 (X = C or As) and MZB10 cages (Z = S, Se or Te). J. Chem. Soc. Dalton Trans. 1996, 15, 3323–3333. [Google Scholar] [CrossRef]
- McGrath, M.; Spalding, T.R.; Fontaine, X.L.R.; Kennedy, J.D.; Thornton-Pett, M.J. Metallaheteroborane chemistry. Part 9. Syntheses and spectroscopy of platinum and palladium phosphine complexes containing η5-{As2B9}-based cluster ligands. Crystal structures of [3,3-L2-closo-3,1,2-PtAs2B9H9] (L = PPh3 or PMe2Ph) and [3-Cl-3,8-(PPh3)2-closo-3,1,2-PdAs2B9H8]. J. Chem. Soc. Dalton Trans. 1991, 3223–3233. [Google Scholar]
- Fontaine, X.L.R.; Kennedy, J.D.; McGrath, M.; Spalding, T.R. Metallheteroborane Chemistry. 8. NMR study of some arsena- and stibaboranes and of the rhodadiarsenaboranes [3,3-(PPh3)2-3-(H)-closo-3,1,2-RhAs2B9H9] and [3-(η5-C5Me5)-closo-3,1,2-RhAs2B9H9]. Magn. Reson. Chem. 1991, 29, 711–720. [Google Scholar] [CrossRef]
- Fontaine, X.L.R.; Greenwood, N.N.; Kennedy, J.D.; Nestor, K.; Thorntonpett, M.; Hermanek, S.; Jelinek, T.; Stibr, B. Polyhedral Metallacarbaborane Chemistry—Preparation, Molecular-Structure, And Nuclear Magnetic-Resonance Investigation of [3-(η5-C5Me5)-closo-3,1,2-M2B9H11] (M = Rh Or Ir). J. Chem. Soc. Dalton Trans. 1990, 2, 681–689. [Google Scholar] [CrossRef]
- Ferguson, G.; Gallagher, J.F.; McGrath, M.; Sheehan, J.P.; Spalding, T.R.; Kennedy, J.D. Metallaheteroborane chemistry. Part 11. Selective syntheses of the palladium heteroborane complexes [2,2-(PR3)2-closo-2,1-PdEB10H10] (R3= Ph3, MePh2 or Me2Ph; E = Se or Te) and [2-X-2-(PPh3)-closo-2,1-PdTeB10H9(PPh3)] (X = Cl, Br, I, CN, SCN or O2CMe). J. Chem. Soc. Dalton Trans. 1993, 27–34. [Google Scholar] [CrossRef]
- Shanan-Atidi, H.; Bar-Eli, K. A convenient method for obtaining free energies of activation by the coalescence temperature of an unequal doublet. J. Phys. Chem. 1970, 74, 961–963. [Google Scholar] [CrossRef]
- Chen, M.W.; Calabrese, J.C.; Gaines, D.F.; Hillenbrand, D.F. Low-temperature crystal and molecular structure of tetracarbonyl[2-bromoheptahydrotriborato(1-)]manganese, (CO)4MnB3H7Br, and a proton NMR study of the kinetics of its intramolecular hydrogen exchange in solution. J. Am. Chem. Soc. 1980, 102, 4928–4933. [Google Scholar] [CrossRef]
- Gaines, D.F.; Walsh, J.L.; Morris, J.H.; Hillenbrand, D.F. The chemistry of beryllaboranes. Characterization and reactions of beryllium bis(tetrahydroborate), Be(BH4)2, and beryllium bis(octahydrotriborate), Be(B3H8)2. Inorg. Chem. 1978, 17, 1516–1522. [Google Scholar] [CrossRef]
- Bushweller, C.H.; Beall, H.; Dewkett, W.J. Stereodynamics of L2CuB3H8. Rate of triborohydride ion (B3H8−) rearrangement as a function of L. Inorg. Chem. 1976, 15, 1739–1740. [Google Scholar] [CrossRef]
- Beall, H.; Bushweller, C.H. Dynamical processes in boranes, borane complexes, carboranes, and related compounds. Chem. Rev. 1973, 73, 465–486. [Google Scholar] [CrossRef]
- Serrar, C.; Es-sofi, A.; Boutalib, A.; Ouassas, A.; Jarid, A.; Nebot-Gil, I.; Tomás, F. Theoretical Study of the Structural and Fluxional Behavior of Copper(I)-Octahydrotriborate Complex. J. Phys. Chem. A 2001, 105, 9776–9780. [Google Scholar] [CrossRef]
- Borlin, J.; Gaines, D.F. Internal exchange in new Group III metalloborane derivatives, dimethylaluminum ((CH3)2AlB3H8), and dimethylgallium triborane(8) (CH3)2GaB3H8). J. Am. Chem. Soc. 1972, 94, 1367–1369. [Google Scholar] [CrossRef]
- Bruker-AXS SAINT. Area-Detector Integration Software, version 6.01; Bruker-AXS SAINT: Madison, WI, USA, 2001. [Google Scholar]
- Bruker-AXS SADABS. Area Detector Absorption Program; Bruker-AXS SADABS: Madison, WI, USA, 1996. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Chen, X.-M.; Ma, N.; Liu, X.-R.; Wei, C.; Cui, C.-C.; Cao, B.-L.; Guo, Y.; Wang, L.-S.; Gu, Q.C. Facile Synthesis of Unsolvated Alkali Metal Octahydrotriborate Salts MB3H8 (M = K, Rb, and Cs), Mechanisms of Formation, and the Crystal Structure of KB3H8. Angew. Chem. 2019, 131, 2746–2750. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) Cluster Data: | |||
| Assignment 1 | δ (11B) 2 | Assignment 1 | δ (1H) 3 |
| B4 | −1.0 [−3.8] | exo-H4 | +2.54 [+2.74] |
| B1,3 | −38.9 [−41.0] | endo-H5 | +1.82 [+2.33] |
| exo-H1, exo-H3 | −0.11 [+1.02] | ||
| H1,4; H3,4 (B–H–B) | −1.11 [−0.65] | ||
| H1,2; H3,2 (Rh–H–B) | −7.07 [−5.10] | ||
| H2, H6 (Rh–H) | −11.79 4 [−6.37] | ||
| (b) Phosphorous-31 Data: | |||
| Assignment | δ (31P) 3 | 1J(103Rh–31P)/Hz | 2J(31P1–31P2)/Hz |
| P1 | 39.9 | 111 | 367 |
| P2 | 44.7 | 111 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diaw-Ndiaye, F.; Sanz Miguel, P.J.; Rodríguez, R.; Macías, R. The Synthesis, Characterization, and Fluxional Behavior of a Hydridorhodatetraborane. Molecules 2023, 28, 6462. https://doi.org/10.3390/molecules28186462
Diaw-Ndiaye F, Sanz Miguel PJ, Rodríguez R, Macías R. The Synthesis, Characterization, and Fluxional Behavior of a Hydridorhodatetraborane. Molecules. 2023; 28(18):6462. https://doi.org/10.3390/molecules28186462
Chicago/Turabian StyleDiaw-Ndiaye, Fatou, Pablo J. Sanz Miguel, Ricardo Rodríguez, and Ramón Macías. 2023. "The Synthesis, Characterization, and Fluxional Behavior of a Hydridorhodatetraborane" Molecules 28, no. 18: 6462. https://doi.org/10.3390/molecules28186462