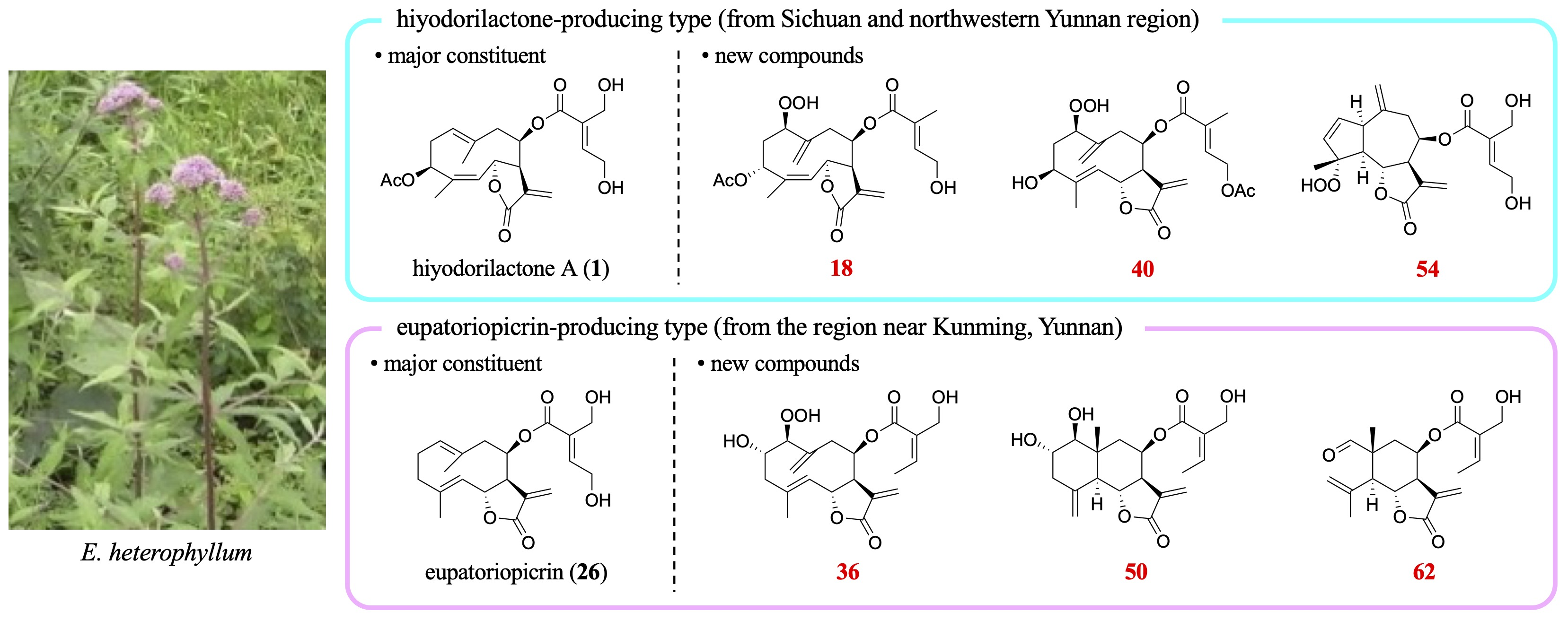
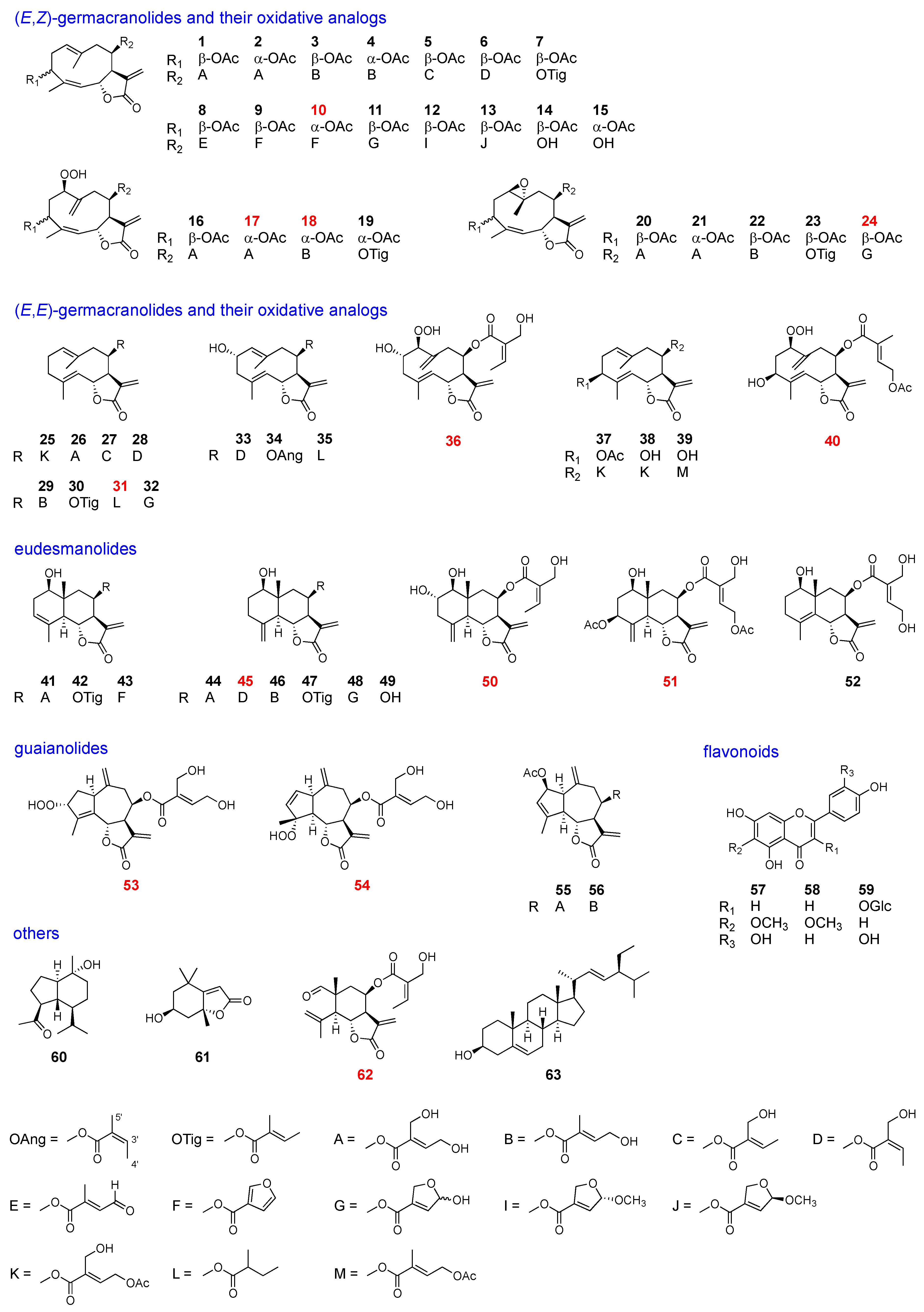
2.2. Isolation and Structural Elucidation of Leaf Chemicals
Dried leaves of each sample were extracted with MeOH, and the compounds were separated using silica-gel column chromatography and normal phase HPLC to yield 63 compounds, 13 of which were previously unreported. The isolated compounds were categolized into six types: (
E,
Z)-germacranolides and their oxidative analogs, (
E,
E)-germacranolides and their oxidative analogs, eudesmanolides, guaianolides, flavonoids, and others, as listed in
Figure 3 and
Table 1.
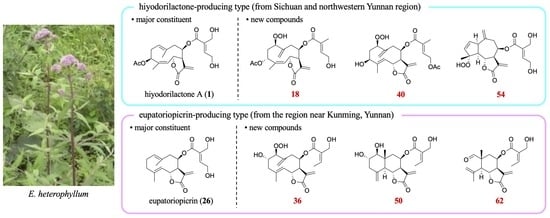
Among the isolated compounds, the following were known: hiyodorilactone A (
1) [
8], eupaformosanin (
2) [
9], hiyodorilactone B (
3) [
8], 20-desoxyeupaformasanin (
4) [
10], eupasimplicins B (
5) [
16,
17], eupachinsin B (
6) [
18], 3β-acetoxy-8β-tigloyloxyheliangolide (
7) [
19], 4′-dehydrochromolaenide (
8) [
20], santhemoidin A (
9) [
21], 20-dehydroeucannabinolide-semi acetal (
11) [
10], santhemoidin B (
12) [
21], 4′-epi-santhemoidin B (
13) [
19], hiyodorilactone C (
14) [
8], eupaformonin (
15) [
22], hydroperoxyheterophyllin A (
16) [
6], hydroperoxyheterophyllin H (
19) [
6], epoxyeucannabinolid (
20) [
23], 1β,10α-epoxyeupaformosanin (
21) [
9], eupalinin B (
22) [
24], heliangin-3-
O-acetate (
23) [
25], 8β-(4′-acetoxy-5′-hydroxytigloyloxy)-costunolide (
25) [
12], eupatoriopicrin (
26) [
11], 8β-(5′-hydroxytigloyloxy)-costunolide (
27) [
14], eupaglehnin C (
28) [
13], 20-desoxyeupatoriopicrin (
29) [
10], 8β-tigloyloxycostunolide (
30) [
15], 20-dehydroeupatoriopicrin-semi acetal (
32) [
10], deacetyleupaserrin (
33) [
26], 2α-hydroxyeupatolide-8-
O-angelate (
34) [
27], 2α-hydroxy-8β-(2-methylbutyryloxy)-germacra-1(10)
E,4
E,11(13)-trien-12,6α-olide (
35) [
28], 8β-(4′-acetoxy-5′-hydroxytigloyloxy)-novanin (
37) [
19], hiyodorilactone D (
38) [
7], 4
E-deacetyl chromolaenide-4′-
O-acetate (
39) [
29], 1-hydroxy-8-(4′,5′-dihydroxytigloyloxy)-3,11(13)-eudesmadien-6,12-olide (
41) [
12], 1β-hydroxy-8β-tiglinoyloxyarbusculin B (
42) [
25], 1-hydroxy-8-furoyloxy-eudesma-3,11(13)-dien-6,12-olide (
43) [
30], 1-hydroxy-8-(4,5-dihydroxytiglyloxy)-eudesma-4(15),11(13)-dien-6,12-olide (
44) [
30], l-hydroxy-8-sarracenyloxyeudesma-4(15),11(13)-dien-6,12-olide (
46) [
31], 8β-tiglinoyloxyreynosin (
47) [
25], 1-hydroxy-8-(3-[2,5-dihydro-5-hydroxy]-furoyloxy)-eudesma-4(15),11(13)-dien-6,12-olide (
48) [
30], 8β-hydroxyreynosin (
49) [
32], 1-hydroxy-8-(4′,5′-dihydroxytigloyloxy)-4,11(13)-eudesmadien-6,12-olide (
52) [
12], eupahakonesin (
55) [
33], eupachifolin C (
56) [
34], eupafolin (
57) [
35], hispidulin (
58) [
36], quercetin-3-glucoside (
59) [
37], oplopanone (
60) [
38], loliolide (
61) [
39], and stigmasterol (
63) [
40]. The structures of the new compounds (
10,
17,
18,
24,
31,
36,
40,
45,
50,
51,
53,
54, and
62) were elucidated as follows.
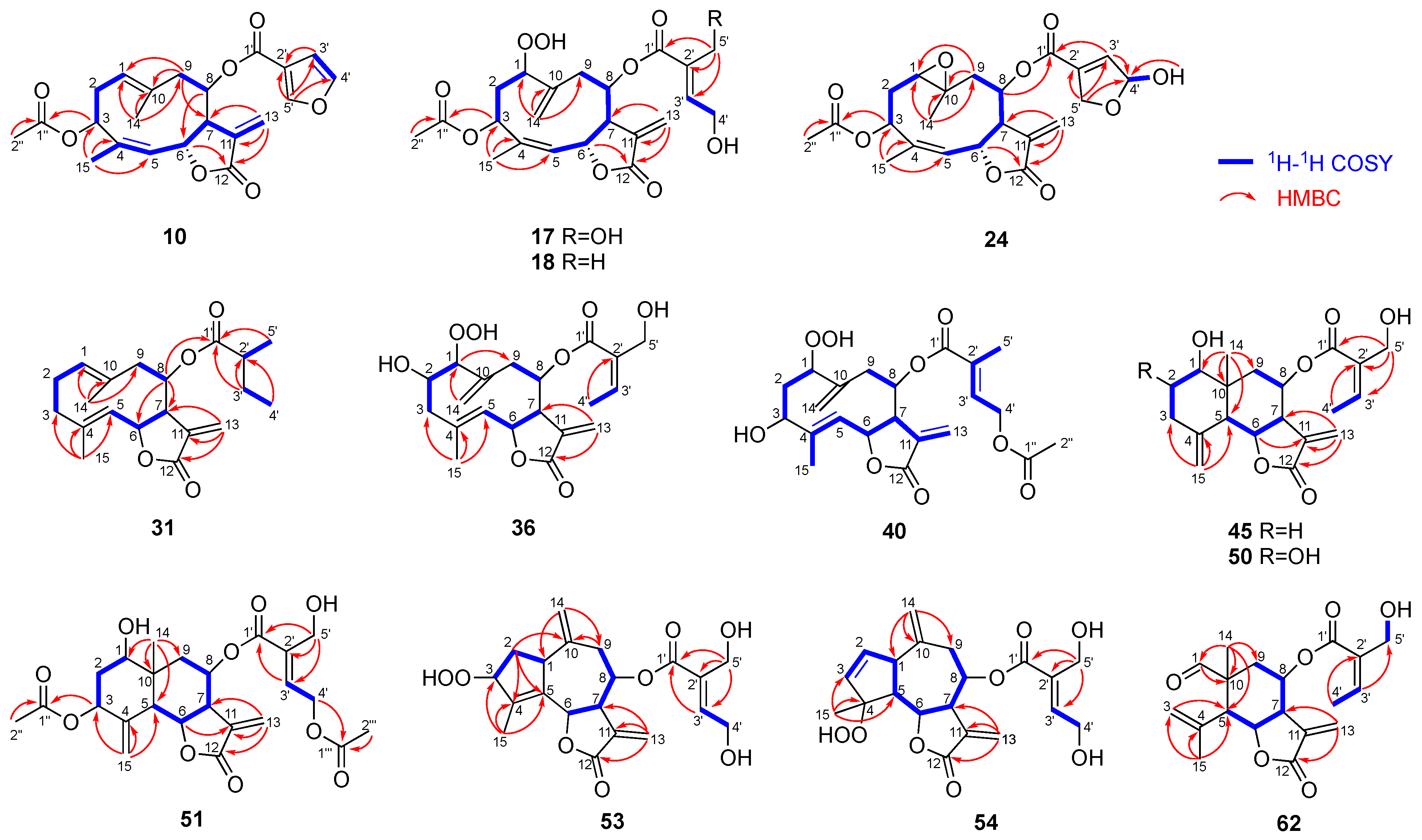
Compound
10 was obtained as a colorless oil. Its HREIMS spectrum showed the molecular ion peak at
m/
z 400.1518 to establish the molecular formula of C
22H
24O
7 with 11 degrees of unsaturation. The IR spectrum of
10 exhibited absorptions at 1765 and 1743 cm
−1, suggesting the presence of a γ-lactone and an ester group. The
1H and
13C NMR spectra of
10 (
Table 2 and
Table 3) were similar to those of santhemoidin A (
9) [
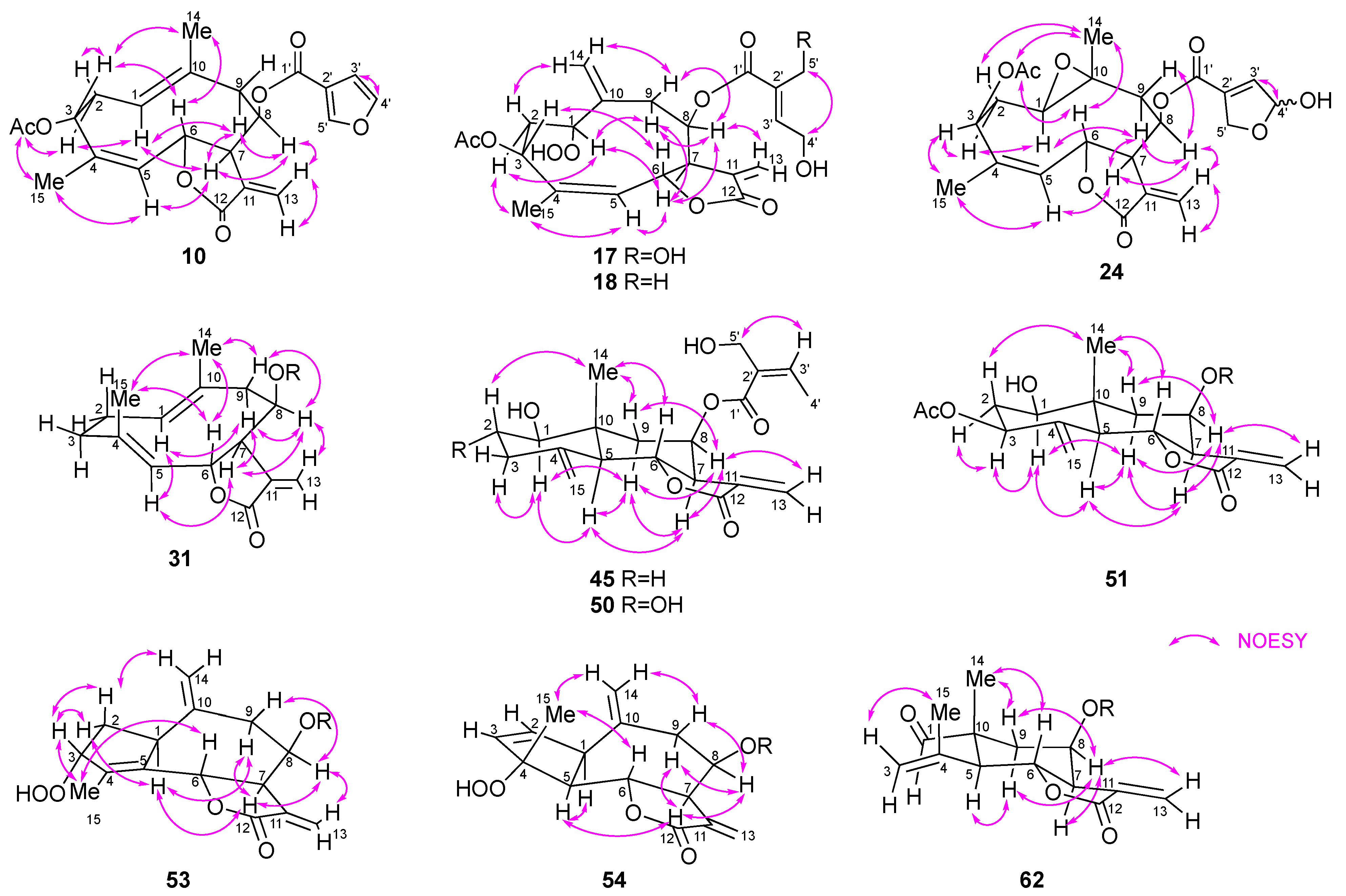
21]. In addition, HMBC correlations from H-3 to C-1″ and from H-6 to C-12 and a NOESY correlation between H-8 and H-13b (δ
H 5.80) (
Figure 4 and
Figure 5) indicated that
10 was a (4
Z)-germacranolide with an acetoxy group at C-3, a 3-furoyloxy group at C-8, and a γ-lactone between C-12 and C-6, respectively. Thus,
10 has the same planar structure as
9. However, the signals attributable to H-3 and H-6 were observed at δ
H 5.63 and 5.31 in the
1H NMR spectrum of
10, whereas the corresponding signals of
9 were at δ
H 5.25 and 5.92, respectively. This characteristic is found between the C-3 epimers of (4
Z)-germacranolide, hiyodorilactone A (
1) [
8], and eupaformosanin (
2) [
9], which suggests that
10 is the C-3 epimer of
9. This was confirmed by the NOESY correlations among H-3, H-6, and H
3-14 (
Figure 5). Thus, the structure of
10 was identified as (4
Z)-3α-acetoxy-8β-(3-furoyloxy)germacra-1(10),4,11(13)-trien-(12,6α)-olide. The absolute configuration was determined to be (3
R,6
R,7
R,8
R)-
10 because the experimental ECD spectrum of
10 was in good agreement with the theoretical ECD spectrum (
Figure S79).
Compound
17 showed a quasimolecular ion [M + H]
+ at
m/
z 453.1764 in its HRFABMS, which suggests a molecular formula of C
22H
28O
10. The
1H and
13C NMR spectra of
17 (
Table 2 and
Table 3) resembled those of hydroperoxyheterophyllin A (
16) [
6], suggesting that
17 was a 1β-hydroperoxyheliangolide related to
16. The major differences between their
1H NMR spectra were observed in the chemical shifts of H-3 (
17: δ
H 5.75;
16: δ
H 5.41) and H-6 (
17: δ
H 5.69;
16: δ
H 6.15). These observations were similar to the above-mentioned case of
9 and
10, indicating an α-orientation of the acetoxy group at C-3 in
17. This conclusion was supported by the NOE between H-3 and H-6 (
Figure 5). Thus,
17 was identified as (4
Z)-3α-acetoxy-8β-(4′,5′-dihydroxytigloyloxy)-1β-hydroperoxygermacra-4,10(14),11(13)-trien-(12,6α)-olide. In a similar manner,
18 was determined to be a 5′-deoxy derivative of
17. Its molecular formula C
22H
28O
9 with one less oxygen atom than that of
17, and the HMBC correlations from H
3-5′ (δ
H 1.82) to C-1′/C-2′/C-3′ support this inference (
Figure 4).
The molecular formula of
24 was determined to be C
22H
26O
9 via HRFABMS. Its
1H NMR spectrum is similar to that of 1β,10α-epoxyeucannabinolide (
20) [
23] (
Table 2), differing only in the signals attributable to the ester group at C-8. The signals δ
H 6.66 (m, H-3′), 6.18 (m, H-4′), 4.89 (m, H-5′a), and 4.69 (m, H-5′b) suggested the presence of a 4′,5′-epoxy-4′-hydroxytigloyl group in
24 [
10,
30]. Moreover, a pair of 4′-OH signals at δ
H 3.03/2.94 (each 0.5H, d,
J = 8.4 Hz) indicated that
24 was a mixture of hemiacetal isomers (
ca. 1:1). The NOESY correlations shown in
Figure 5 suggest that the stereochemistry of the heliangolide core is the same as that of
20. Thus,
24 was characterized as a C-4′ epimer of (4
Z)-3β-acetoxy-1β,10α-epoxy-8β-(4′,5′-epoxy-4′-hydroxytigloyloxy)germacra-4,11(13)-dien-(12,6α)-olide.
Compound
31 had the molecular formula of C
20H
28O
4 as indicated by the quasimolecular ion peak at
m/
z 333.2059 [M + H]
+ in its HRCIMS. The
1H and
13C NMR spectra showed signals corresponding to a 2-methylbutanoyloxy group (
Table 2 and
Table 3). The remaining signals of
31 were nearly identical to those of the terpene scaffold of 8β-tigloyloxycostunolide (
30) [
15]. The NOESY correlations of H-1/H-5, H-5/H-7, H-7/H-8, H-6/H
3-14, and H-6/H
3-15 established the relative configuration of the germacranolide moiety as illustrated in
Figure 5. Based on these observations,
31 was identified as 8β-(2′-methylbutanoyloxy)germacra-1(10),4,11(13)-trien-(12,6α)-olide.
Compound
36 showed a [M + K]
+ peak at
m/
z 433.1241 in HRFABMS, confirming its molecular formula as C
20H
26O
8. The IR absorptions at 3380, 1745, and 1715 cm
−1 suggested the presence of a hydroxy group, γ-lactone, and ester group, respectively. The 1D and 2D NMR spectra of
36 were recorded at 233 K because its
1H NMR spectrum exhibited broad signals at room temperature, suggesting conformational flexibility. The
1H and
13C NMR spectra of
36 (
Table 4) were similar to those of deacetyleupaserrin (
33) [
26] except that the signals corresponding to an olefinic methine and a methyl group in
33 were replaced with those of an oxygen-bearing methine [δ
H 4.13 (H-1); δ
C 97.9 (C-1)] and an exomethylene group [δ
H 5.44 and 5.12 (H
2-14); δ
C 120.0 (C-14)], respectively, implying that
36 was a C-1 hydroperoxy analog of deacetyleupaserrin (
33). This was confirmed by the COSY correlations of H-1/H-2/H
2-3, H-5/H-6/H-7, H-8/H
2-9, and H-3′/H-4′, along with the HMBC correlations from H
3-15 to C-3, C-4, and C-5; from H
2-14 to C-1; from H-1 to C-9; from H
2-13 to C-7 and C-12; and from H
3-4′ to C-2′ (
Figure 4). Therefore, the planar structure of
36 was determined as shown in
Figure 4. Unfortunately, the NOE correlations required for determining the relative configurations of
36 were not observed; nevertheless, considering the stereochemistry of
33 and other 1-hydroperoxy germacranolides found in this plant,
36 was concluded as 1β-hydroperoxy-2α-hydroxy-8β-(5′-hydroxyangeloyloxy)germacra-4,10(14),11(13)-trien-(12,6α)-olide.
The HRESIMS spectrum of
40 showed a [M + Na]
+ peak at
m/
z 459.1632 to establish a molecular formula of C
22H
28O
9 with nine degrees of unsaturation. The IR absorptions at 3400 cm
−1 corresponded to a hydroxy group and those at 1743, 1735, and 1715 cm
−1 are attributed to carbonyl groups. Similar to the case of
36, the
1H NMR spectrum of
40 also afforded broad signals at room temperature. Even at 233 K, the quality of the
13C NMR spectrum remained insufficient owing to the small amount of
40 obtained; however, the
1H NMR spectrum clearly showed pairs of signals (in a ratio of 2:3 based on the integration), suggesting the coexistence of two conformers. A careful analysis of the
1H NMR (
Table 4) and the
1H-
1H COSY spectra (
Figure 4) of both conformers suggested a structural similarity of
40 with 4
E-deacetyl chromolaenide-4′-
O-acetate (
39) [
29] as well as the presence of a hydroperoxy group [δ
H 8.56 (major) and 8.43 (minor)] and an additional exomethylene [δ
H 5.44/5.13 (major) and 5.35/5.02 (minor)]. The differences in the
1H NMR spectrum of
40 with that of
39 were attributable to a 1β-hydroperoxy-10(14)-ene structure, as is the case with
36 and
33. Therefore,
40 was identified as 8β-(4′-acetoxytigloyloxy)-1β-hydroperoxy-3β-hydroxygermacra-4,10(14),11(13)-trien-(12,6α)-olide.
Compound
45 showed a quasimolecular ion [M + H]
+ at
m/
z 363.1808 in its HRFABMS, which suggests a molecular formula of C
20H
26O
6. The
1H NMR data suggested a structural similarity of
45 to that of the known eudesmanolide
44 [
30] (
Table 5); however, the COSY correlation between H-3′ [δ
H 6.40 (q,
J = 7.3 Hz)] and H
3-4′ [δ
H 2.04 (d,
J = 7.3 Hz)] and the NOESY correlation between H-3′ and H
2-5′ indicated that the 4′,5′-dihydroxytigloyl group in
44 was replaced with a 5′-hydroxyangeloyl group in
45 (
Figure 4 and
Figure 5). Therefore,
45 was identified as 1β-hydroxy-8β-(5′-hydroxyangeloyloxy)eudesma-4(15),11(13)-dien-(12,6α)-olide.
The
1H and
13C NMR spectra of
50 revealed that it is also an eudesmanolide similar to
45. Its molecular formula was determined to be C
20H
26O
7, one more oxygen atom than
45, using HRFABMS. In addition, the COSY correlations between H-1 (δ
H 3.19)/H-2 (δ
H 3.62)/H
2-3 (δ
H 2.66 and 2.11) and the NOE correlation between H-2β and H
3-14 suggested the presence of another hydroxy group at C-2α in
50 compared to that in
45 (
Figure 4 and
Figure 5). Thus,
50 was identified as 1β,2α-dihydroxy-8β-(5′-hydroxyangeloyloxy)eudesma-4(15),11(13)-dien-(12,6α)-olide.
The molecular formula of
51 was determined to be C
24H
30O
10 using HRFABMS. Its
1H and
13C NMR data (
Table 5) were closely related to those of eupakirunsin H [
41], suggesting that
51 was also a eudesmanolide. The downfield shift of H-3 (δ
H 5.20) and H-8 (δ
H 5.83) in the
1H NMR spectrum as well as the COSY and HMBC correlations shown in
Figure 4 indicated that the hydroxy group at C-3 and tigloyloxy group at C-8 in eupakirunsin H were replaced by acetoxy and 4′-acetoxy-5′-hydroxytigloyl groups, respectively, in
51.
The HRESIMS spectrum of compound
53 showed a [M + Na]
+ peak at
m/
z 415.1368, which suggests the molecular formula C
20H
24O
8 with nine degrees of unsaturation. The
1H and
13C NMR spectra revealed the presence of one methyl, two oxymethylenes, three oxymethines, two exocyclic double bonds, one trisubstituted double bond, one tetrasubstituted double bond, and two carbonyls (
Table 6). The above spectroscopic data accounted for six degrees of unsaturation, and therefore,
53 should be tricyclic. Compound
53 was deduced to be a guaianolide with oxygen-functionalities at C-3, C-6, and C-8, one of which is a 4′,5′-dihydroxytigloyloxy group, as evidenced by the COSY and HMBC correlations shown in
Figure 4. A significant downfield shift of H-8 (δ
H 5.72) as well as the NOESY correlation between H-13b and H-8 suggested the presence of a 4′,5′-dihydroxytigloyloxy moiety at C-8 and a γ-lactone between C-12 and C-6. Moreover, the molecular formula of
53 and the chemical shift of C-3 (δ 94.2) suggested the presence of a hydroperoxy group at this position. The elucidated planar structure of
53 is shown in
Figure 4. The NOESY spectrum showed a cross-peak between H-7 and H-1, H-8, and H-9α, indicating that these hydrogens were in the same orientation (
Figure 5). H-3 showed NOE correlations with H-2a and H-2b, but not with H-1, indicating the α-orientation of hydroperoxy group. Finally, H-6 was assigned a β-orientation owing to its coupling constant (
J6,7 = 10.5 Hz). Therefore,
53 was identified as 8β-(4′,5′-dihydroxytigloyloxy)-3α-hydroperoxyguaia-4,10(14),11(13)-trien-(12,6α)-olide.
HRFABMS and 1D/2D NMR spectra of
54 revealed that it is also a guaianolide with the same molecular formula as that of
53 (
Table 6 and
Figure 4 and
Figure 5). A COSY correlation between two olefinic protons at C-2 (δ
C 133.0; δ
H 5.77) and C-3 (δ
C 137.2; δ
H 6.01) indicated a disubstituted double bond. The chemical shift of C-4 observed at δ
C 95.2 in the
13C NMR spectrum and NOE correlation between H
3-15 and H-6 indicated the presence of a hydroperoxy group at C-4α. Thus,
54 was concluded to be a 4α-hydroperoxy-2-ene isomer of
53.
Compound
62 was isolated as a colorless oil. A [M + Na]
+ peak was observed at
m/
z 371.1472 in its HRESIMS, corresponding to the molecular formula of C
19H
24O
6 with eight degrees of unsaturation. The IR spectrum suggested the presence of hydroxy (3501 cm
−1), γ-lactone (1769 cm
−1), aldehyde (1726 cm
−1), and α,β-unsaturated carbonyl (1715 cm
−1) groups. The
1H and
13C NMR spectra showed the characteristic signals for a 5′-hydroxyangeloyl moiety (
Table 6), which implied that
62 was a norsesquiterpenoid. The
1H-
1H COSY spectrum exhibited a spin system from H-5 to H
2-9 (
Figure 4). Furthermore, in the HMBC spectrum, H
3-14 was correlated with C-1/C-5/C-9/C-10 and H
3-15 with C-3/C-4/C-5, indicating that
62 was a 2-norelemanolide. A downfield shift of H-8 (δ
H 5.87) as well as the NOE between H-8 and H-13b (δ
H 5.57) as shown in
Figure 5 confirmed the position of an α-methylene-γ-lactone and 5′-hydroxyangeloyl group. The relative stereochemistry of
62 is similar to that of
45 based on NOE correlations and coupling constants. Thus,
62 was identified as 8β-(5′-hydroxyangeloyloxy)-1-oxo-2-norelema-3,11(13)-dien-(12,6α)-olide.
The experimental ECD spectra of
17,
18,
24,
45,
50,
51,
54, and
62 showed a similar trend to that of
10, especially the negative Cotton effect around 210 nm mainly owing to the α-methylene-γ-lactone moiety. This indicated that the absolute configurations at C-6 and C-7 of these compounds are the same as those of
10 while the other chromophore might have a weaker contribution to their experimental ECD spectra [
18]. In addition, considering the biosynthesis of sesquiterpenoids in higher plants, the other new compounds,
31,
36,
40, and
53, would have the same stereochemistry.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}