
Novel Potential Janus Kinase Inhibitors with Therapeutic Prospects in Rheumatoid Arthritis Addressed by In Silico Studies
 ,
,  , ,
, ,
Abstract
:
1. Introduction
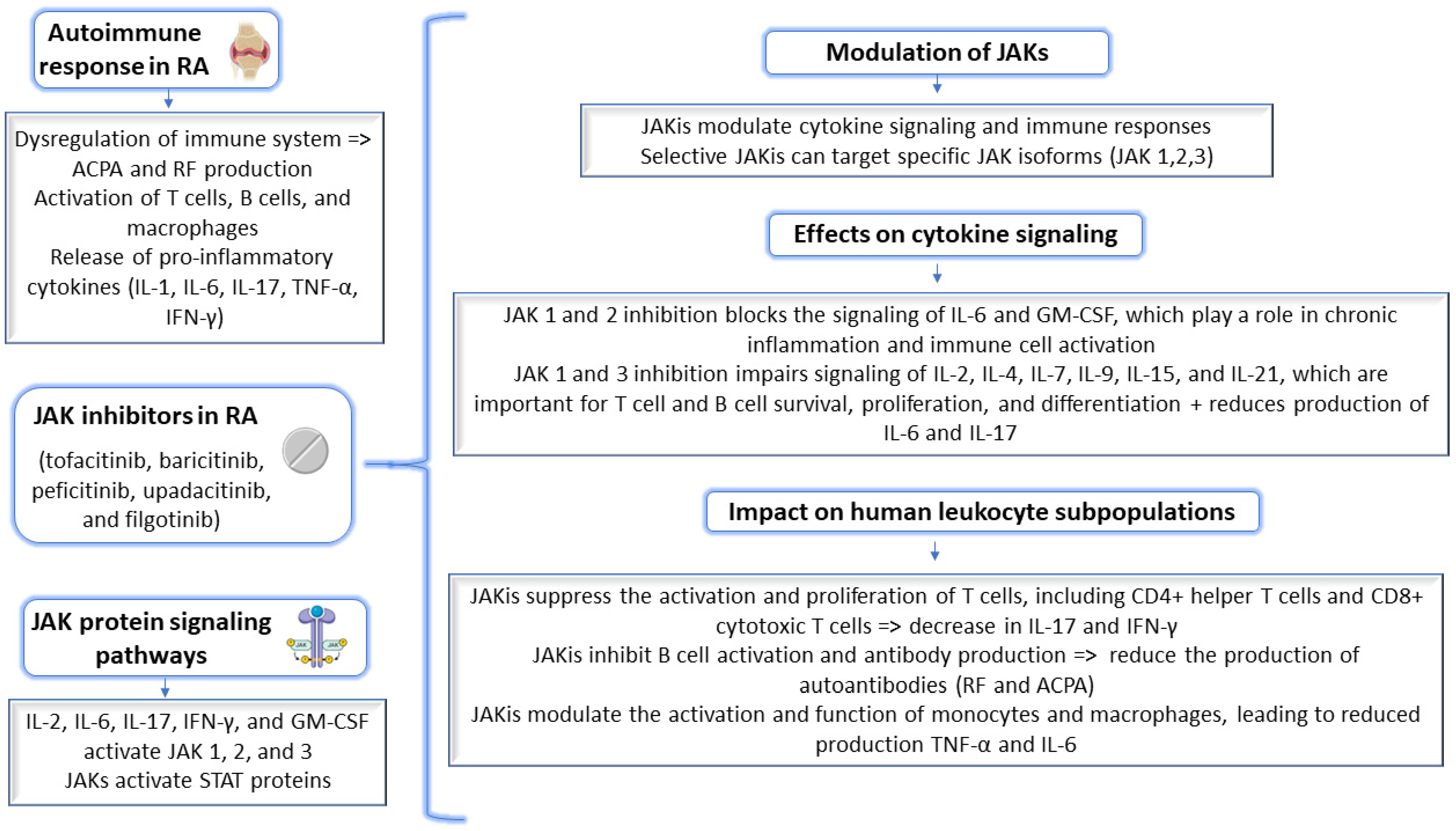
2. Results and Discussion
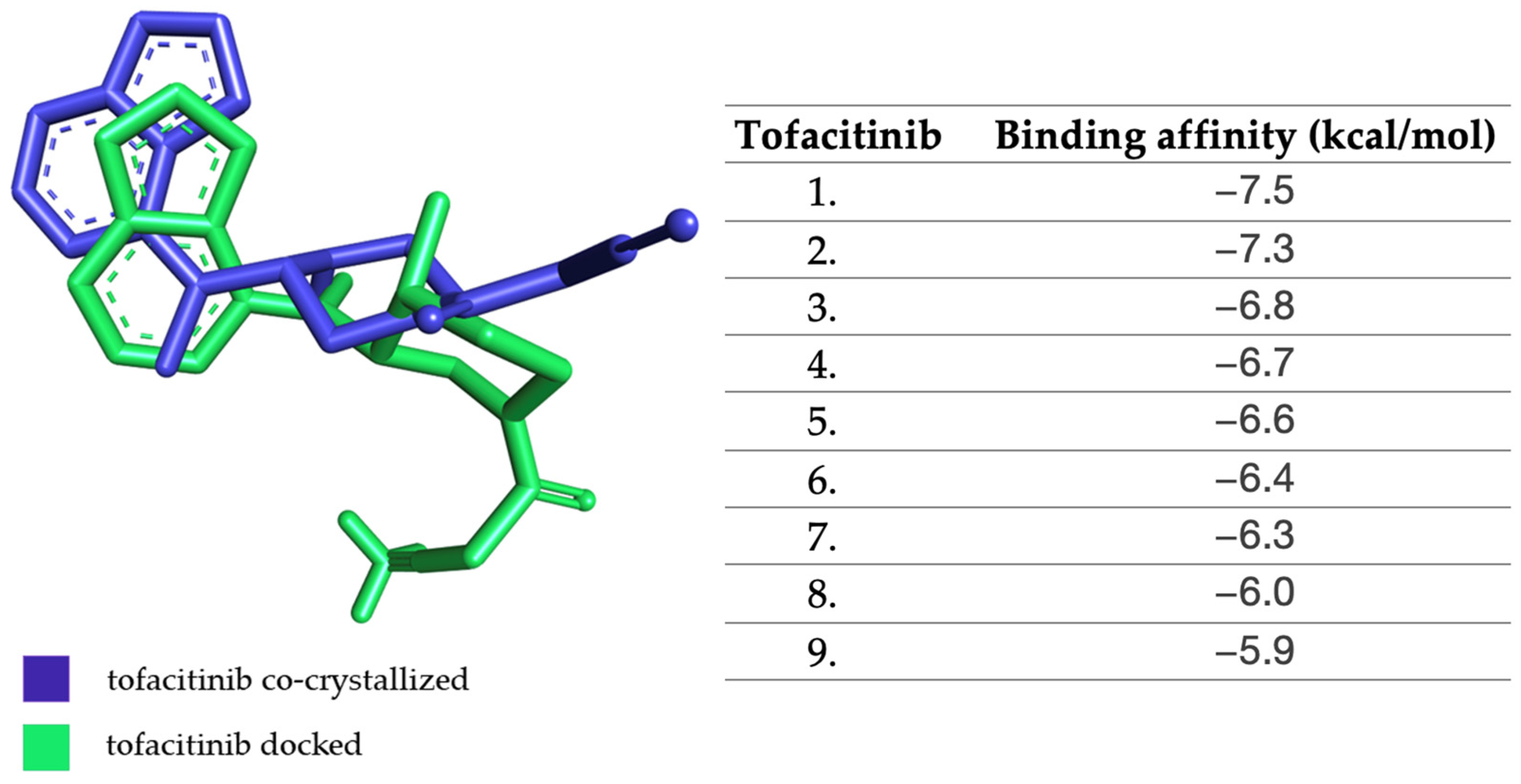
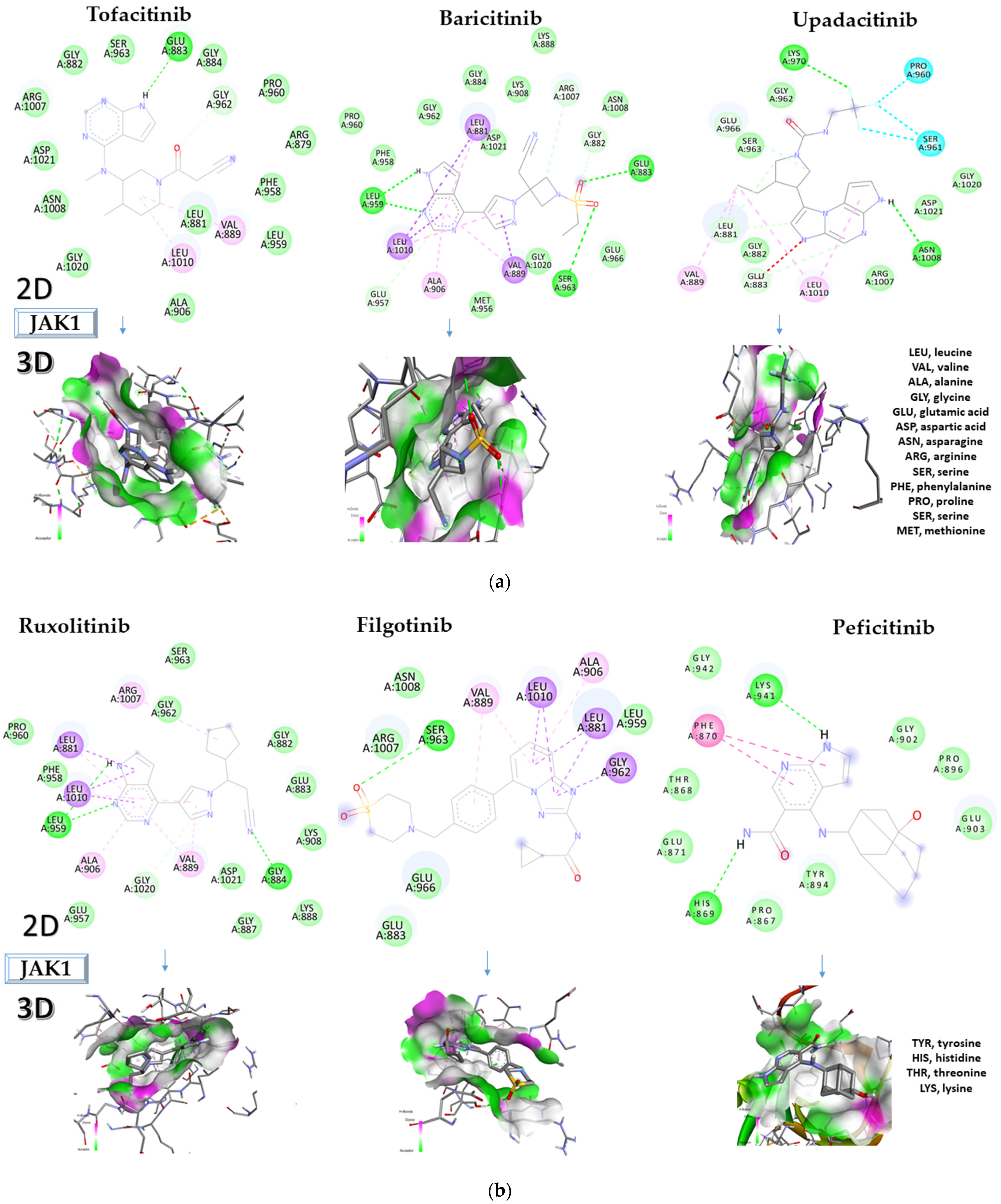
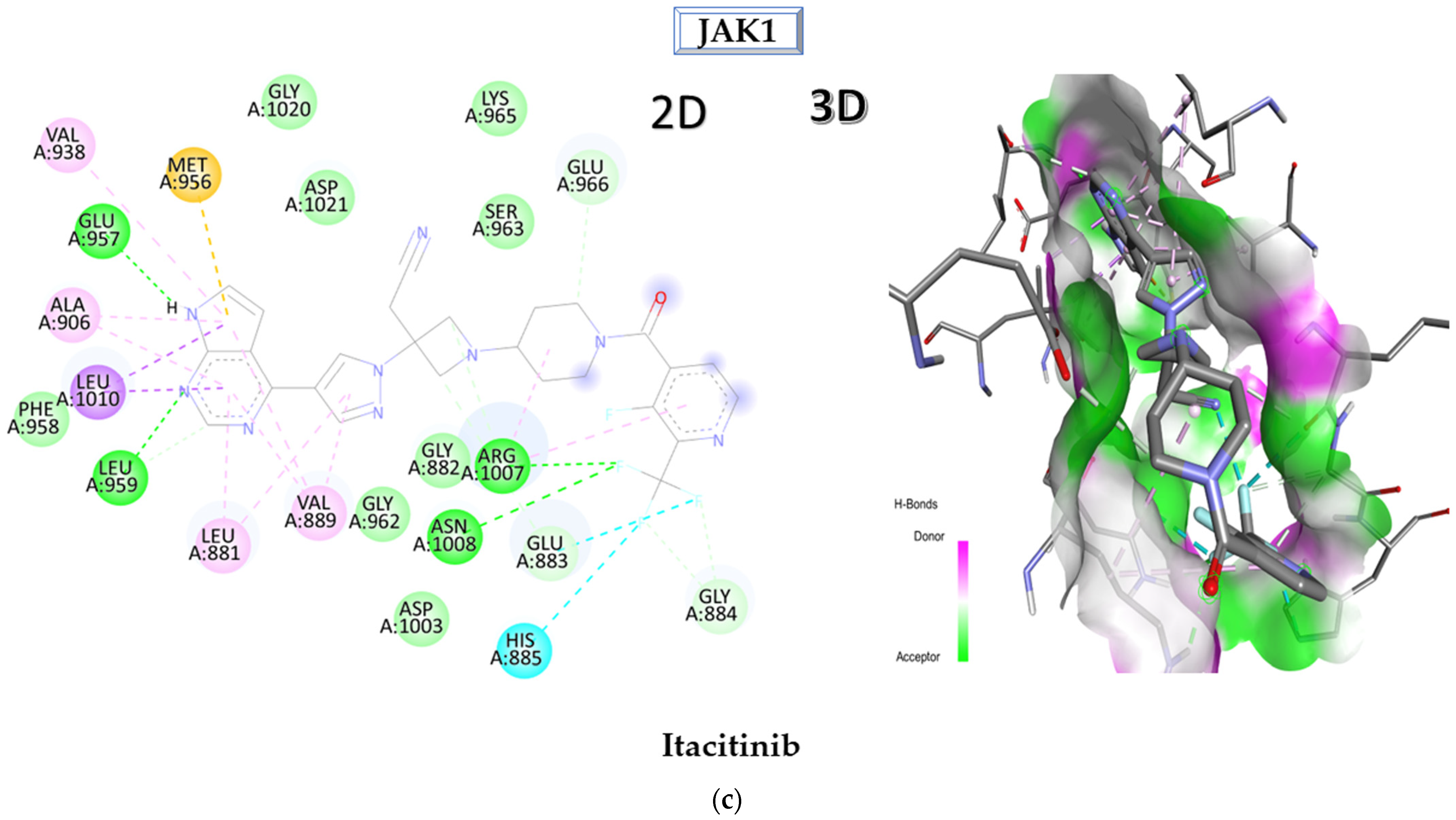
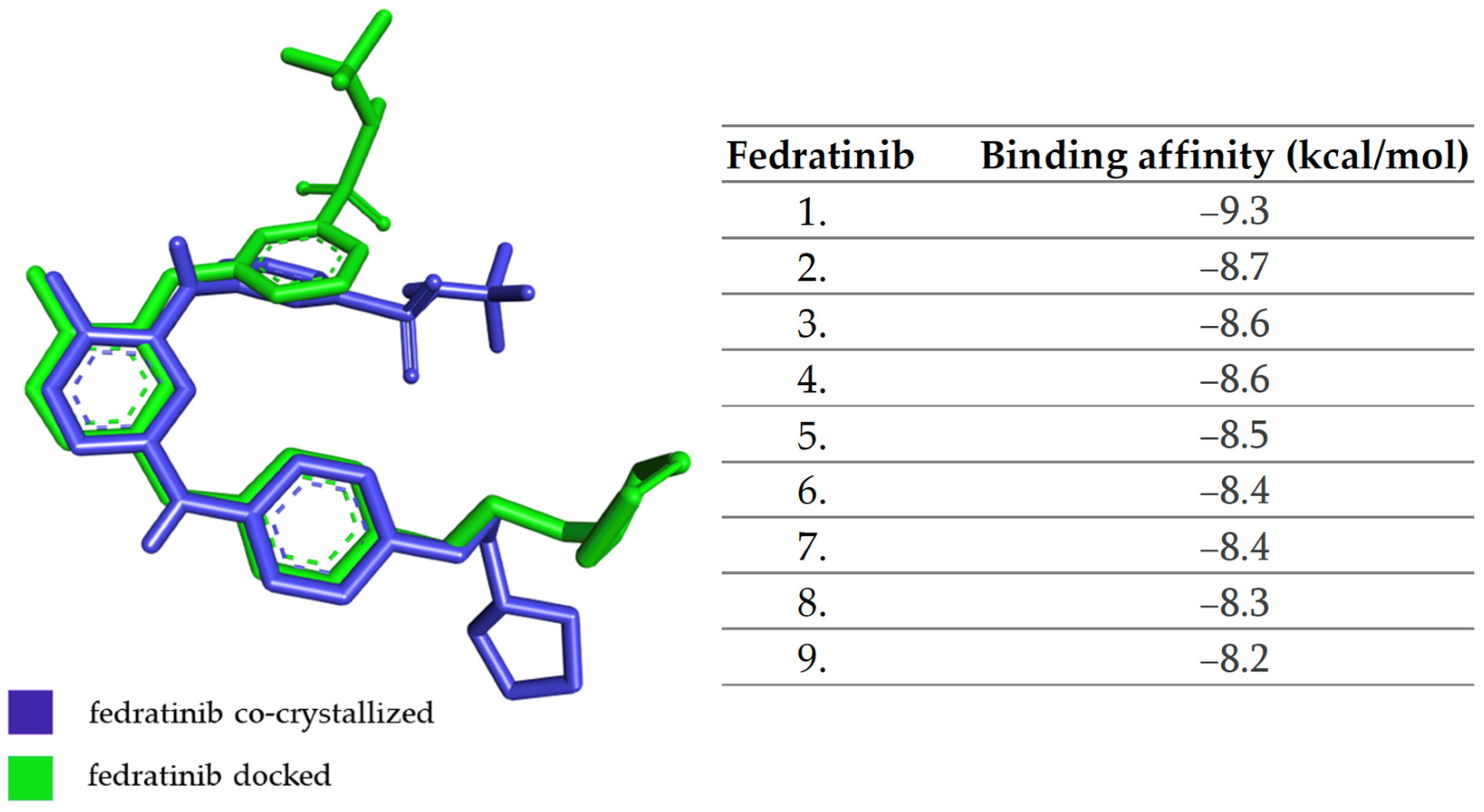
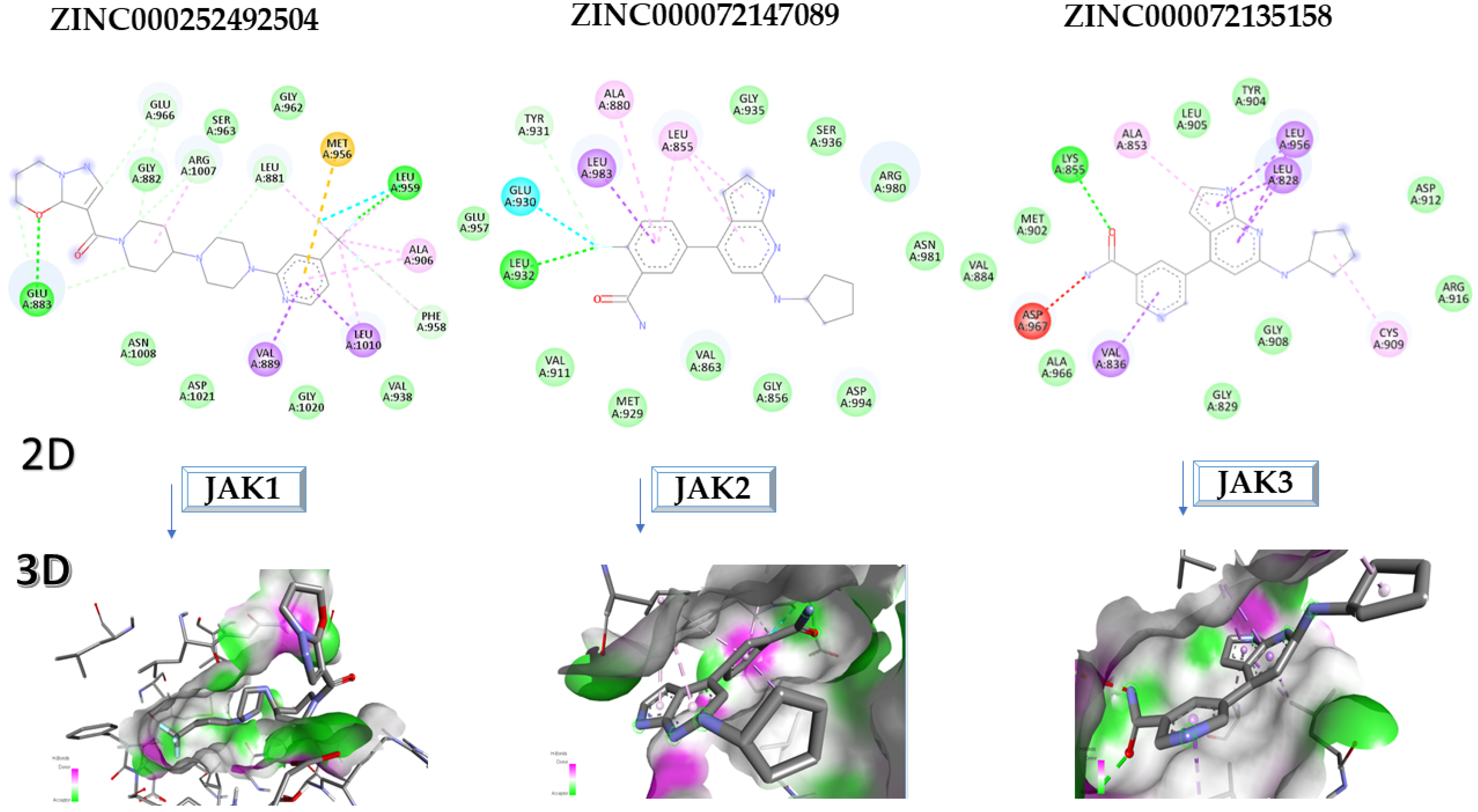
2.1. Molecular Docking Approach Targeting JAK1
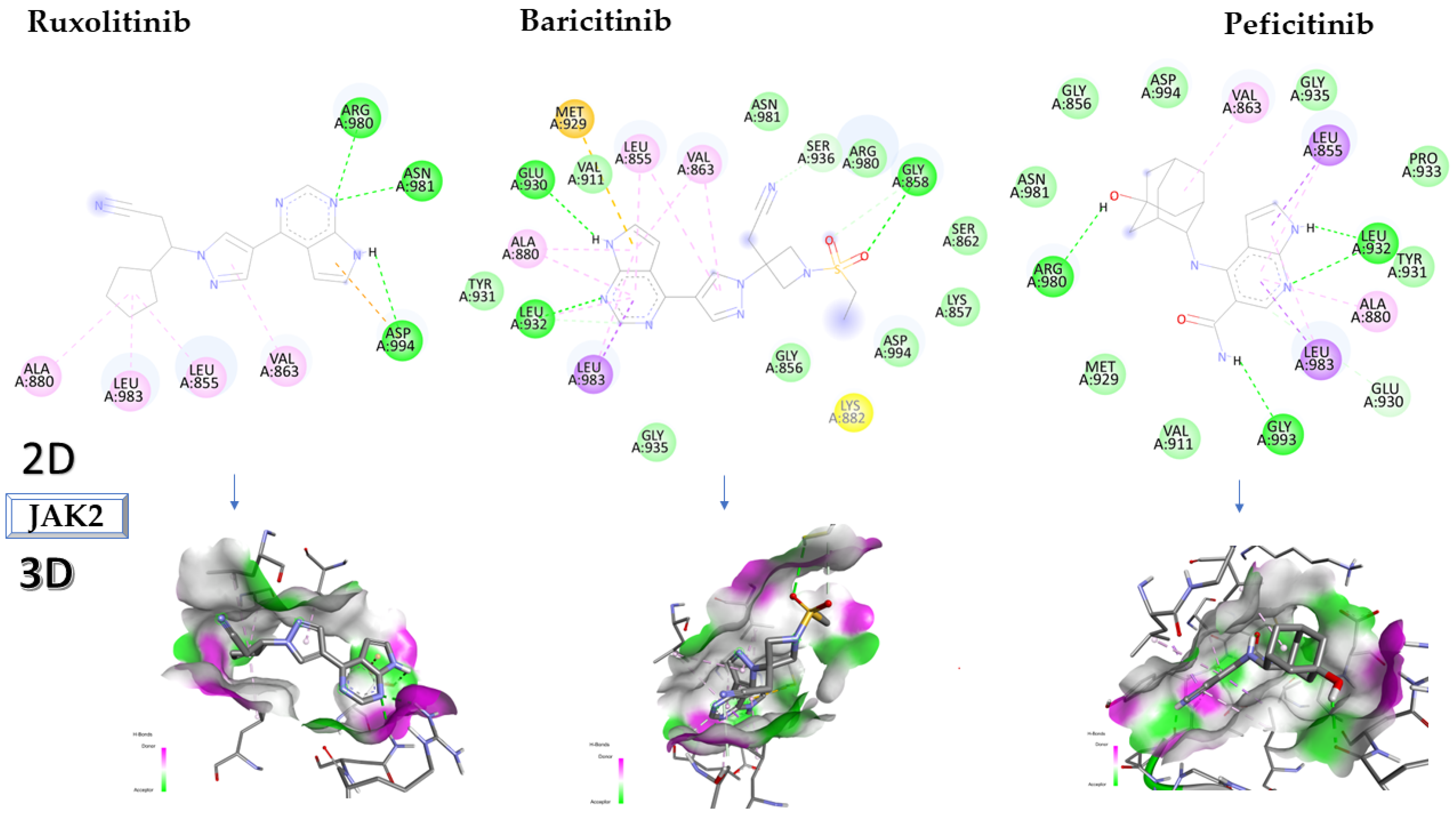
2.2. Molecular Docking Approach Targeting JAK2
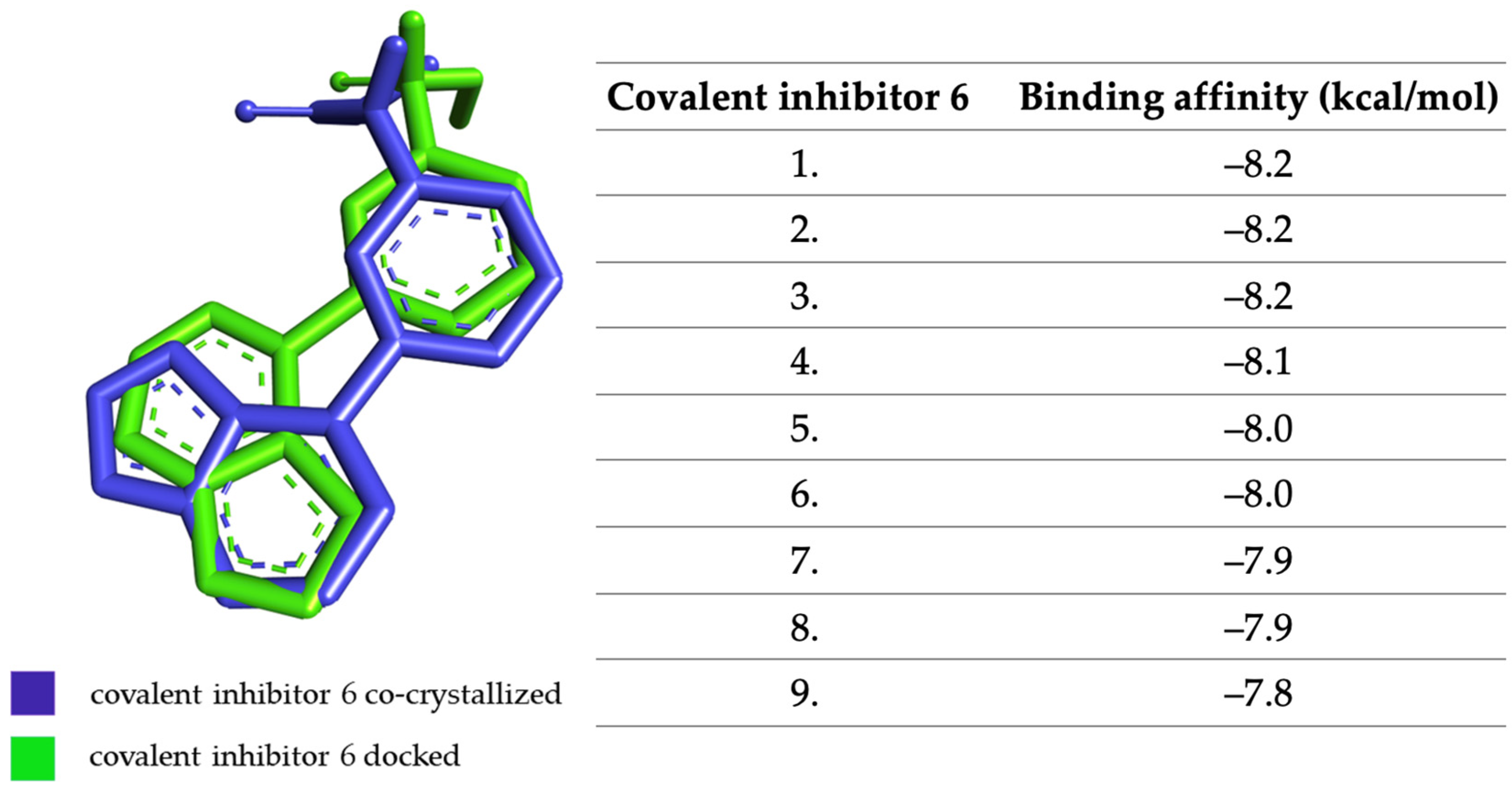
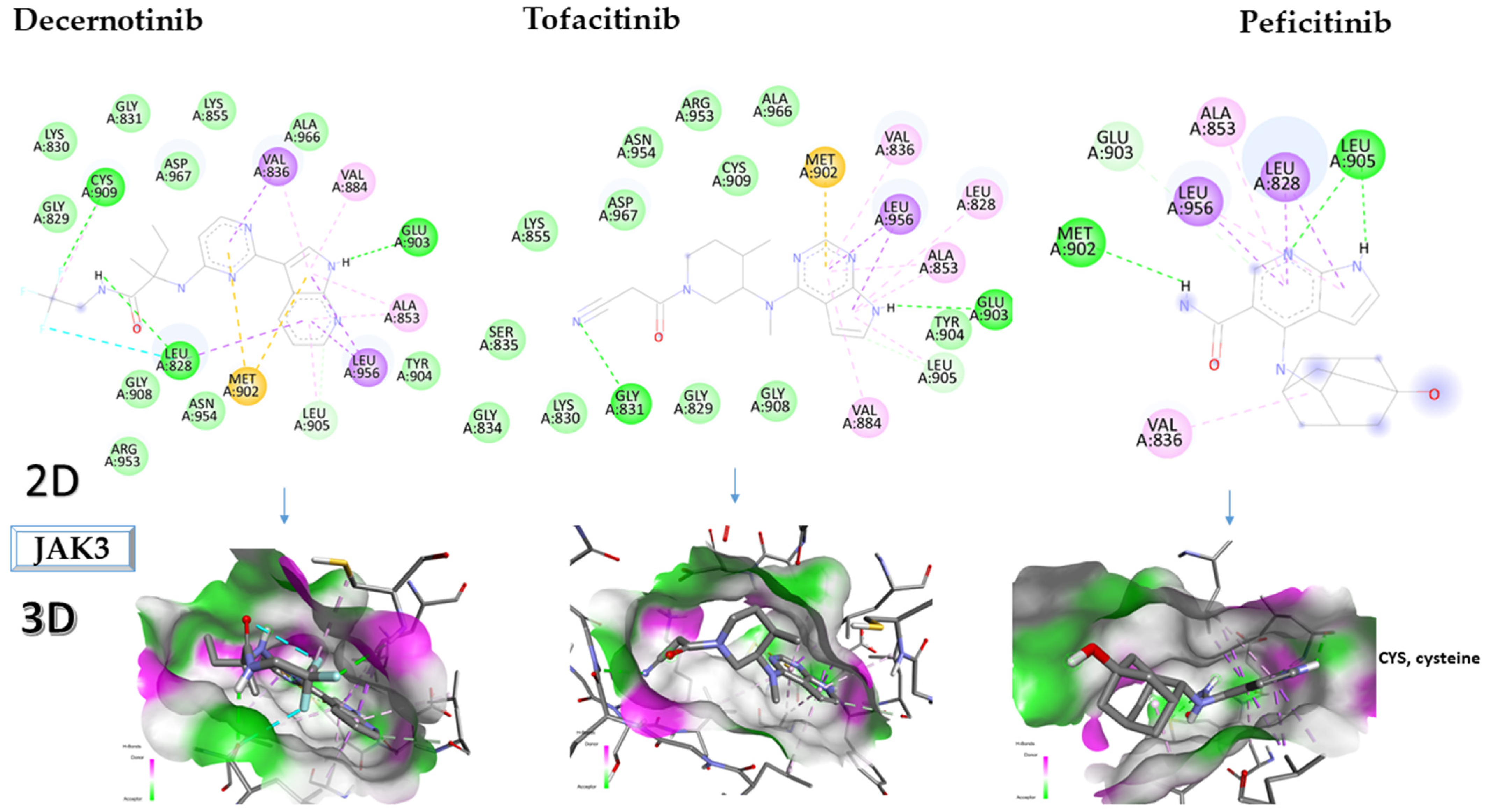
2.3. Molecular Docking Approach Targeting JAK3
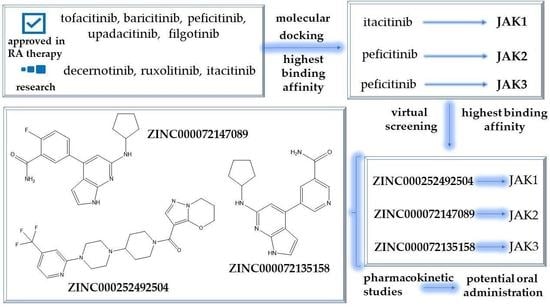
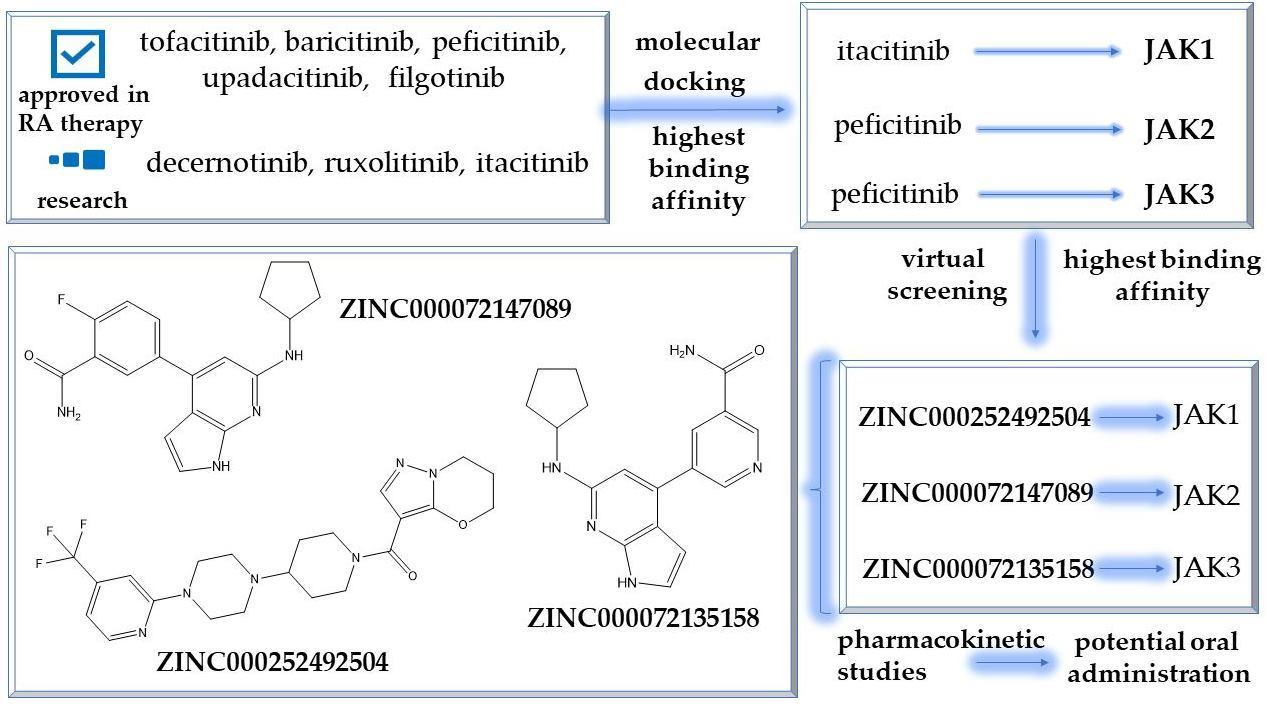
2.4. Virtual Screening of the Most Promising Compounds
2.5. In Silico Evaluation of the Pharmacokinetic Profile of Newly Identified Compounds
3. Materials and Methods
3.1. Identification and Selection of Target Proteins
3.2. Ligand Preparation
3.3. Protein Preparation
3.4. Molecular Docking Analysis
3.5. Virtual Screening
3.6. Pharmacokinetic Profile Predictions
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, J.; Guo, S.; Schrodi, S.J.; He, D. Molecular and Cellular Heterogeneity in Rheumatoid Arthritis: Mechanisms and Clinical Implications. Front. Immunol. 2021, 12, 790122. [Google Scholar] [CrossRef]
- Lin, Y.J.; Anzaghe, M.; Schülke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells 2020, 9, 880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInnes, I.B.; Schett, G. The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrovská, N.; Prajzlerová, K.; Vencovský, J.; Šenolt, L.; Filková, M. The pre-clinical phase of rheumatoid arthritis: From risk factors to prevention of arthritis. Autoimmun. Rev. 2021, 20, 102797. [Google Scholar] [CrossRef]
- van Drongelen, V.; Holoshitz, J. Human Leukocyte Antigen-Disease Associations in Rheumatoid Arthritis. Rheum. Dis. Clin. N. Am. 2017, 43, 363–376. [Google Scholar] [CrossRef]
- Singh, A.; Behl, T.; Sehgal, A.; Singh, S.; Sharma, N.; Naved, T.; Bhatia, S.; Al-Harrasi, A.; Chakrabarti, P.; Aleya, L.; et al. Mechanistic insights into the role of B cells in rheumatoid arthritis. Int. Immunopharmacol. 2021, 99, 108078. [Google Scholar] [CrossRef]
- Radu, A.-F.; Bungau, S.G. Nanomedical approaches in the realm of rheumatoid arthritis. Ageing Res. Rev. 2023, 87, 101927. [Google Scholar] [CrossRef]
- Tanaka, Y.; Luo, Y.; O’Shea, J.J.; Nakayamada, S. Janus kinase-targeting therapies in rheumatology: A mechanisms-based approach. Nat. Rev. Rheumatol. 2022, 18, 133–145. [Google Scholar] [CrossRef]
- Huang, J.; Fu, X.; Chen, X.; Li, Z.; Huang, Y.; Liang, C. Promising Therapeutic Targets for Treatment of Rheumatoid Arthritis. Front. Immunol. 2021, 12, 686155. [Google Scholar] [CrossRef]
- Gadina, M.; Chisolm, D.A.; Philips, R.L.; McInness, I.B.; Changelian, P.S.; O’Shea, J.J. Translating JAKs to Jakinibs. J. Immunol. 2020, 204, 2011–2020. [Google Scholar] [CrossRef] [PubMed]
- Bungau, S.G.; Behl, T.; Singh, A.; Sehgal, A.; Singh, S.; Chigurupati, S.; Vijayabalan, S.; Das, S.; Palanimuthu, V.R. Targeting Probiotics in Rheumatoid Arthritis. Nutrients 2021, 13, 3376. [Google Scholar] [CrossRef]
- Bullock, J.; Rizvi, S.A.A.; Saleh, A.M.; Ahmed, S.S.; Do, D.P.; Ansari, R.A.; Ahmed, J. Rheumatoid Arthritis: A Brief Overview of the Treatment. Med. Princ. Pract. 2019, 27, 501–507. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewé, R.; Bijlsma, J.; Burmester, G.; Chatzidionysiou, K.; Dougados, M.; Nam, J.; Ramiro, S.; Voshaar, M.; Van Vollenhoven, R.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann. Rheum. Dis. 2017, 76, 960–977. [Google Scholar] [CrossRef] [PubMed]
- Radu, A.F.; Bungau, S.G.; Negru, P.A.; Marcu, M.F.; Andronie-Cioara, F.L. In-depth bibliometric analysis and current scientific mapping research in the context of rheumatoid arthritis pharmacotherapy. Biomed. Pharmacother. 2022, 154, 113614. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewé, R.B.M.; Bergstra, S.A.; Kerschbaumer, A.; Sepriano, A.; Aletaha, D.; Caporali, R.; Edwards, C.J.; Hyrich, K.L.; Pope, J.E.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann. Rheum. Dis. 2023, 82, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.H. Clinical significance of Janus Kinase inhibitor selectivity. Rheumatology 2019, 58, 953–962. [Google Scholar] [CrossRef] [Green Version]
- Markham, A.; Keam, S.J. Peficitinib: First Global Approval. Drugs 2019, 79, 887–891. [Google Scholar] [CrossRef]
- McInnes, I.B.; Byers, N.L.; Higgs, R.E.; Lee, J.; Macias, W.L.; Na, S.; Ortmann, R.A.; Rocha, G.; Rooney, T.P.; Wehrman, T.; et al. Comparison of baricitinib, upadacitinib, and tofacitinib mediated regulation of cytokine signaling in human leukocyte subpopulations. Arthritis Res. Ther. 2019, 21, 183. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Traves, P.G.; Murray, B.; Campigotto, F.; Galien, R.; Meng, A.; Di Paolo, J.A. JAK selectivity and the implications for clinical inhibition of pharmacodynamic cytokine signalling by filgotinib, upadacitinib, tofacitinib and baricitinib. Ann. Rheum. Dis. 2021, 80, 865–875. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kavanaugh, A.; Wicklund, J.; McInnes, I.B. Filgotinib, a novel JAK1-preferential inhibitor for the treatment of rheumatoid arthritis: An overview from clinical trials. Mod. Rheumatol. 2022, 32, 1–11. [Google Scholar] [CrossRef]
- Parmentier, J.M.; Voss, J.; Graff, C.; Schwartz, A.; Argiriadi, M.; Friedman, M.; Camp, H.S.; Padley, R.J.; George, J.S.; Hyland, D.; et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol. 2018, 2, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowty, M.E.; Jesson, M.I.; Ghosh, S.; Lee, J.; Meyer, D.M.; Krishnaswami, S.; Kishore, N. Preclinical to clinical translation of tofacitinib, a Janus kinase inhibitor, in rheumatoid arthritis. J. Pharmacol. Exp. Ther. 2014, 348, 165–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmroth, M.; Kuuliala, K.; Peltomaa, R.; Virtanen, A.; Kuuliala, A.; Kurttila, A.; Kinnunen, A.; Leirisalo-Repo, M.; Silvennoinen, O.; Isomäki, P. Tofacitinib Suppresses Several JAK-STAT Pathways in Rheumatoid Arthritis In Vivo and Baseline Signaling Profile Associates With Treatment Response. Front. Immunol. 2021, 12, 3809. [Google Scholar] [CrossRef]
- Harrington, R.; Al Nokhatha, S.A.; Conway, R. JAK Inhibitors in Rheumatoid Arthritis: An Evidence-Based Review on the Emerging Clinical Data. J. Inflamm. Res. 2020, 13, 519–531. [Google Scholar] [CrossRef]
- Angelini, J.; Talotta, R.; Roncato, R.; Fornasier, G.; Barbiero, G.; Cin, L.D.; Brancati, S.; Scaglione, F. JAK-Inhibitors for the Treatment of Rheumatoid Arthritis: A Focus on the Present and an Outlook on the Future. Biomolecules 2020, 10, 1002. [Google Scholar] [CrossRef]
- Burmester, G.R.; Kremer, J.M.; Van den Bosch, F.; Kivitz, A.; Bessette, L.; Li, Y.; Zhou, Y.; Othman, A.A.; Pangan, A.L.; Camp, H.S. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease-modifying anti-rheumatic drugs (SELECT-NEXT): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2018, 391, 2503–2512. [Google Scholar] [CrossRef]
- Li, N.; Gou, Z.P.; Du, S.Q.; Zhu, X.H.; Lin, H.; Liang, X.F.; Wang, Y.S.; Feng, P. Effect of JAK inhibitors on high- and low-density lipoprotein in patients with rheumatoid arthritis: A systematic review and network meta-analysis. Clin. Rheumatol. 2022, 41, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.R.; Lee, E.B.; Kaplan, I.V.; Kwok, K.; Geier, J.; Benda, B.; Soma, K.; Wang, L.; Riese, R. Tofacitinib, an oral Janus kinase inhibitor: Analysis of malignancies across the rheumatoid arthritis clinical development programme. Ann. Rheum. Dis. 2016, 75, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.M.; Genovese, M.C.; Enejosa, J.V.; Mysler, E.; Bessette, L.; Peterfy, C.; Durez, P.; Ostor, A.; Li, Y.; Song, I.H. Safety and effectiveness of upadacitinib or adalimumab plus methotrexate in patients with rheumatoid arthritis over 48 weeks with switch to alternate therapy in patients with insufficient response. Ann. Rheum. Dis. 2019, 78, 1454–1462. [Google Scholar] [CrossRef] [Green Version]
- Jain, D.; Udhwani, T.; Sharma, S.; Gandhe, A.; Reddy, P.B.; Nayarisseri, A.; Singh, S.K. Design of novel JAK3 Inhibitors towards Rheumatoid Arthritis using molecular docking analysis. Bioinformation 2019, 15, 68–78. [Google Scholar] [CrossRef]
- Kaltsonoudis, E.; Pelechas, E.; Voulgari, P.V.; Drosos, A.A. Unmet needs in the treatment of rheumatoid arthritis. An observational study and a real-life experience from a single university center. Semin. Arthritis Rheum. 2019, 48, 597–602. [Google Scholar] [CrossRef]
- Genovese, M.C.; Van Vollenhoven, R.F.; Pacheco-Tena, C.; Zhang, Y.; Kinnman, N. VX-509 (Decernotinib), an Oral Selective JAK-3 Inhibitor, in Combination with Methotrexate in Patients with Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Bragina, M.E.; Daina, A.; Perez, M.A.S.; Michielin, O.; Zoete, V. The SwissSimilarity 2021 Web Tool: Novel Chemical Libraries and Additional Methods for an Enhanced Ligand-Based Virtual Screening Experience. Int. J. Mol. Sci. 2022, 23, 811. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Itteboina, R.; Ballu, S.; Sivan, S.K.; Manga, V. Molecular modeling-driven approach for identification of Janus kinase 1 inhibitors through 3D-QSAR, docking and molecular dynamics simulations. J. Recept. Signal Transduct. Res. 2017, 37, 453–469. [Google Scholar] [CrossRef]
- Sk, M.F.; Jonniya, N.A.; Roy, R.; Kar, P. Unraveling the Molecular Mechanism of Recognition of Selected Next-Generation Antirheumatoid Arthritis Inhibitors by Janus Kinase 1. ACS Omega 2022, 7, 6195–6209. [Google Scholar] [CrossRef]
- Galvez-Llompart, M.; Ocello, R.; Rullo, L.; Stamatakos, S.; Alessandrini, I.; Zanni, R.; Tuñón, I.; Cavalli, A.; Candeletti, S.; Masetti, M.; et al. Targeting the JAK/STAT Pathway: A Combined Ligand- and Target-Based Approach. J. Chem. Inf. Model. 2021, 61, 3091–3108. [Google Scholar] [CrossRef]
- Achutha, A.S.; Pushpa, V.L.; Manoj, K.B. Comparative molecular docking studies of phytochemicals as Jak2 inhibitors using Autodock and ArgusLab. Mater. Today Proc. 2021, 41, 711–716. [Google Scholar] [CrossRef]
- Balupuri, A.; Balasubramanian, P.K.; Cho, S.J. 3D-QSAR, docking, molecular dynamics simulation and free energy calculation studies of some pyrimidine derivatives as novel JAK3 inhibitors. Arab. J. Chem. 2020, 13, 1052–1078. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Cayot, A.; Laroche, D.; Disson-Dautriche, A.; Arbault, A.; Maillefert, J.F.; Ornetti, P. Cytochrome P450 interactions and clinical implication in rheumatology. Clin. Rheumatol. 2014, 33, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Maia, E.H.B.; Assis, L.C.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Reichel, A.; Lienau, P. Pharmacokinetics in Drug Discovery: An Exposure-Centred Approach to Optimising and Predicting Drug Efficacy and Safety. Handb. Exp. Pharmacol. 2016, 232, 235–260. [Google Scholar]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided. Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligands | Binding Affinity (kcal/mol) | Structure |
|---|---|---|
| Baricitinib | −9.0 |  |
| Upadacitinib | −6.4 |  |
| Ruxolitinib | −9.0 |  |
| Itacitinib | −9.7 | |
| Peficitinib | −6.5 |  |
| Filgotinib | −9.1 |  |
| Ligands | Binding Affinity (kcal/mol) |
|---|---|
| Baricitinib | −8.3 |
| Ruxolitinib | −8.5 |
| Peficitinib | −9.5 |
| Ligands | Binding Affinity (kcal/mol) |
|---|---|
| Decernotinib | −8.5 |
| Tofacitinibului | −8.4 |
| Peficitinib | −9.5 |
| Compound | Structure | Binding Affinity (kcal/mol) | Similarity Score |
|---|---|---|---|
| ZINC000252492504 |  | −9.0 | 0.303 |
| ZINC000272517982 |  | −8.8 | 0.301 |
| ZINC000584884024 |  | −7.8 | 0.442 |
| ZINC000530112879 |  | −7.7 | 0.304 |
| ZINC000013974879 |  | −7.4 | 0.337 |
| Compound | Structure | Binding Affinity (kcal/mol) | Similarity Score |
|---|---|---|---|
| ZINC000072147089 |  | −8.6 | 0.325 |
| ZINC000239174350 |  | −8.5 | 0.400 |
| ZINC000016384670 |  | −8.5 | 0.338 |
| ZINC000072135158 |  | −8.5 | 0.313 |
| ZINC000263818685 |  | −8.3 | 0.342 |
| Compound | Structure | Binding Affinity (kcal/mol) | Similarity Score |
|---|---|---|---|
| ZINC000072135158 |  | −8.6 | 0.313 |
| ZINC000072147089 |  | −8.5 | 0.325 |
| ZINC000072149208 |  | −8.5 | 0.319 |
| ZINC000239174350 |  | −8.4 | 0.400 |
| ZINC000214499182 |  | −8.0 | 0.333 |
| Characteristics | ZINC000252492504 (JAK1) | ZINC000072147089 (JAK2) | ZINC000072135158 (JAK3) |
|---|---|---|---|
| Formula | C22H27F3N6O2 | C19H19FN4O | C18H19N5O |
| Molecular weight | 464.48 g/mol | 338.38 g/mol | 321.38 g/mol |
| Num. rotatable bonds | 5 | 4 | 4 |
| Num. H-bond acceptors | 8 | 3 | 3 |
| Num. H-bond donors | 0 | 3 | 3 |
| Log P | 3.37 | 2.34 | 1.89 |
| Gastrointestinal absorption | High | High | High |
| CYP2C19 inhibitor | Yes | No | No |
| CYP2C9 inhibitor | Yes | No | No |
| CYP2D6 inhibitor | Yes | Yes | Yes |
| CYP3A4 inhibitor | Yes | Yes | Yes |
| Lipinski | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radu, A.-F.; Bungau, S.G.; Negru, A.P.; Uivaraseanu, B.; Bogdan, M.A. Novel Potential Janus Kinase Inhibitors with Therapeutic Prospects in Rheumatoid Arthritis Addressed by In Silico Studies. Molecules 2023, 28, 4699. https://doi.org/10.3390/molecules28124699
Radu A-F, Bungau SG, Negru AP, Uivaraseanu B, Bogdan MA. Novel Potential Janus Kinase Inhibitors with Therapeutic Prospects in Rheumatoid Arthritis Addressed by In Silico Studies. Molecules. 2023; 28(12):4699. https://doi.org/10.3390/molecules28124699
Chicago/Turabian StyleRadu, Andrei-Flavius, Simona Gabriela Bungau, Andrei Paul Negru, Bogdan Uivaraseanu, and Mihaela Alexandra Bogdan. 2023. "Novel Potential Janus Kinase Inhibitors with Therapeutic Prospects in Rheumatoid Arthritis Addressed by In Silico Studies" Molecules 28, no. 12: 4699. https://doi.org/10.3390/molecules28124699