
Binding Mechanism of CD47 with SIRPα Variants and Its Antibody: Elucidated by Molecular Dynamics Simulations
 ,
,
Abstract
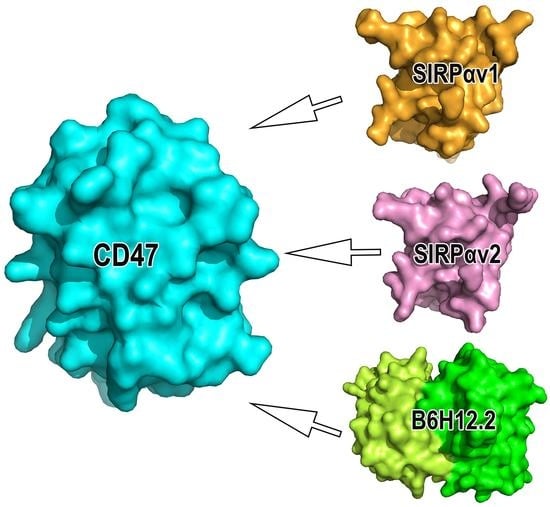
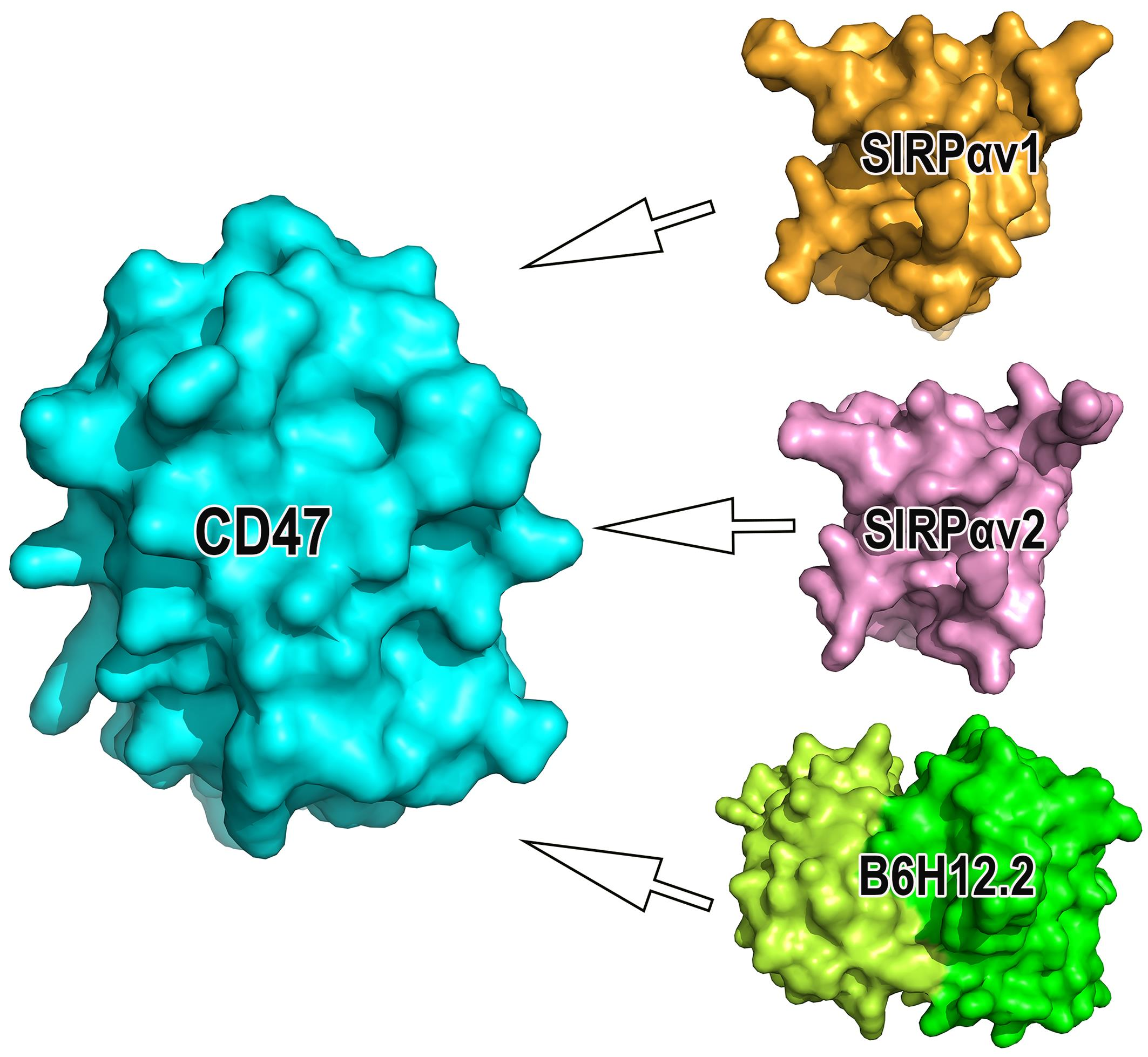
:
1. Introduction
2. Results
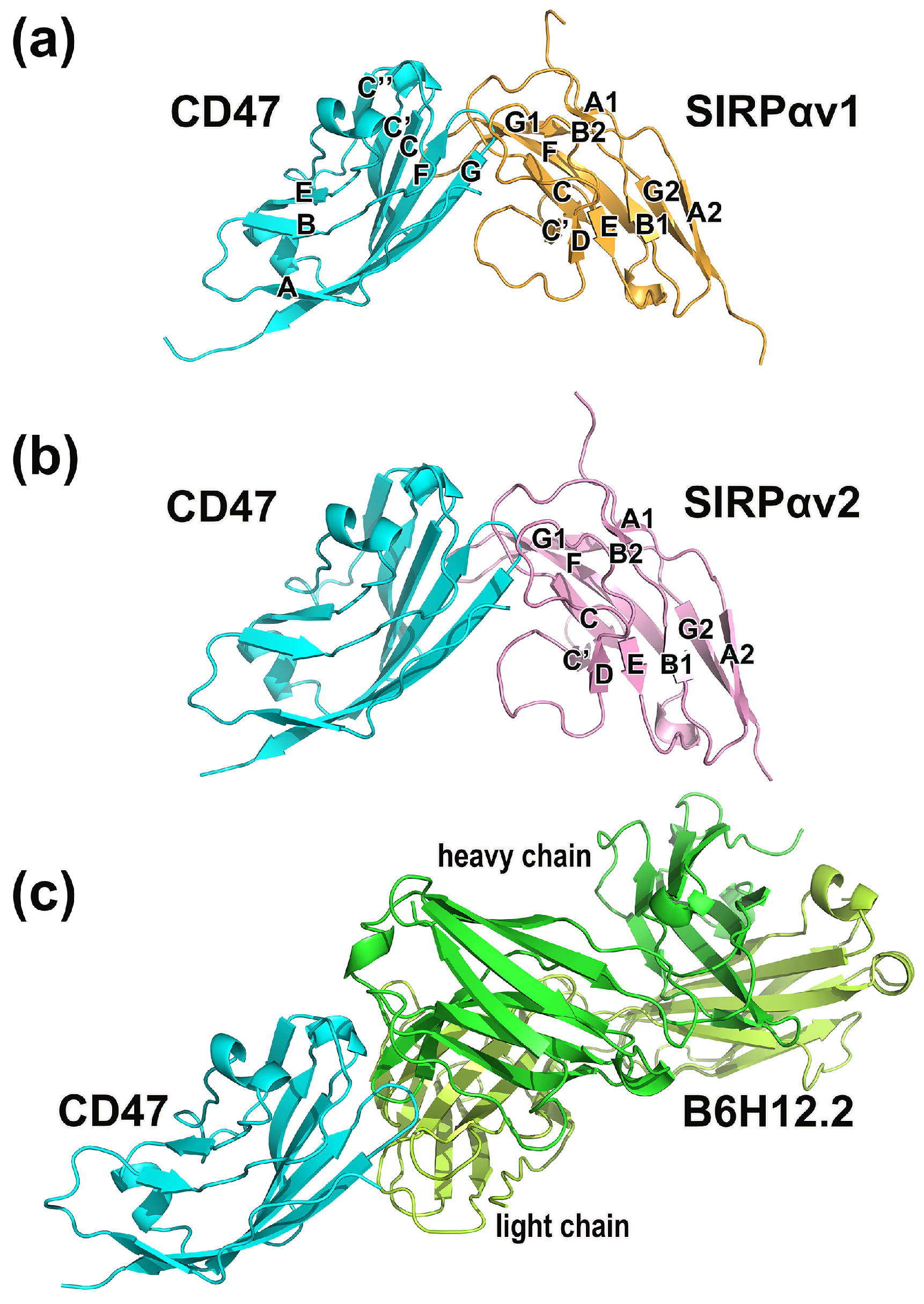
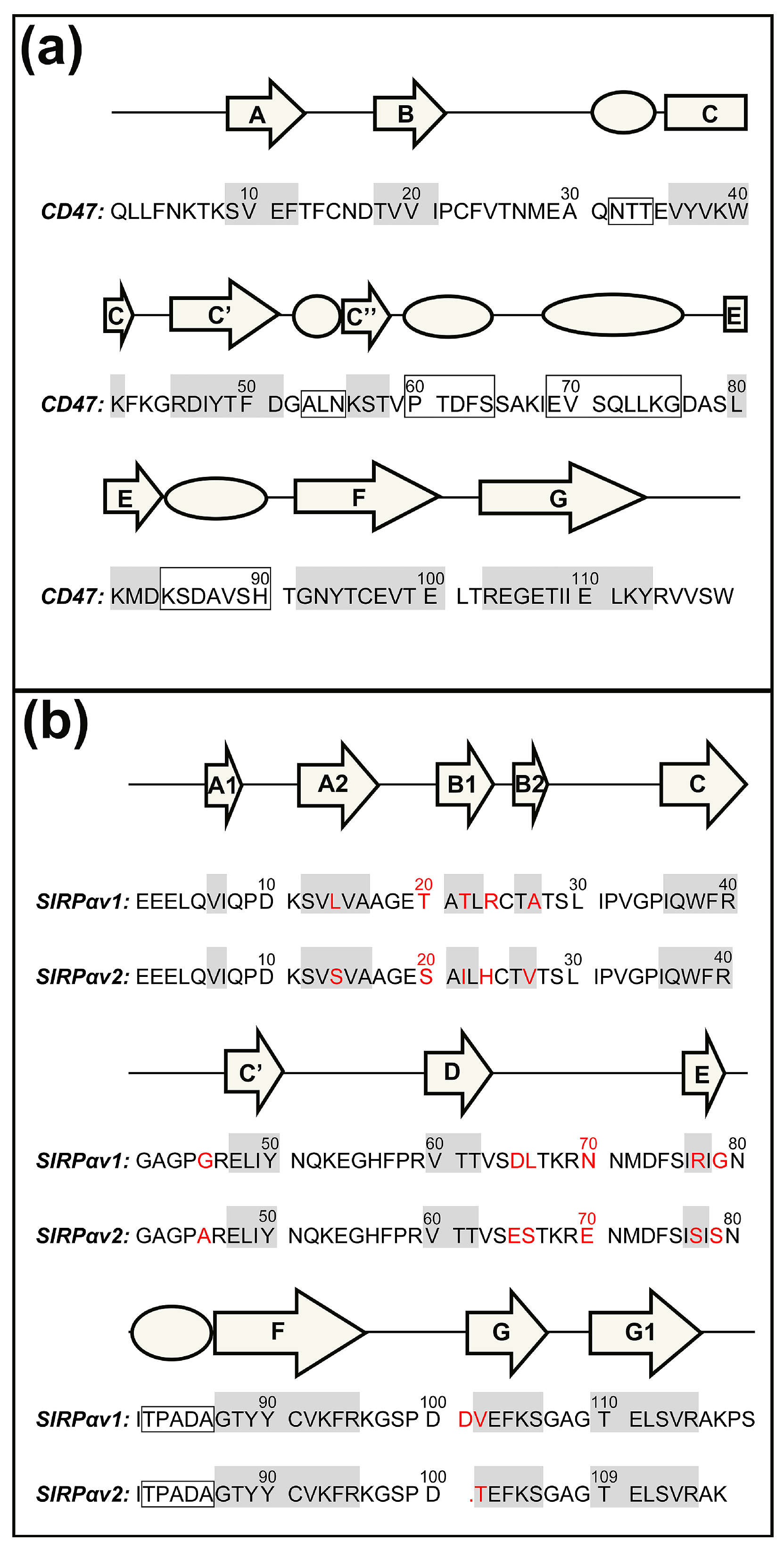
2.1. Static Structural Information Analysis
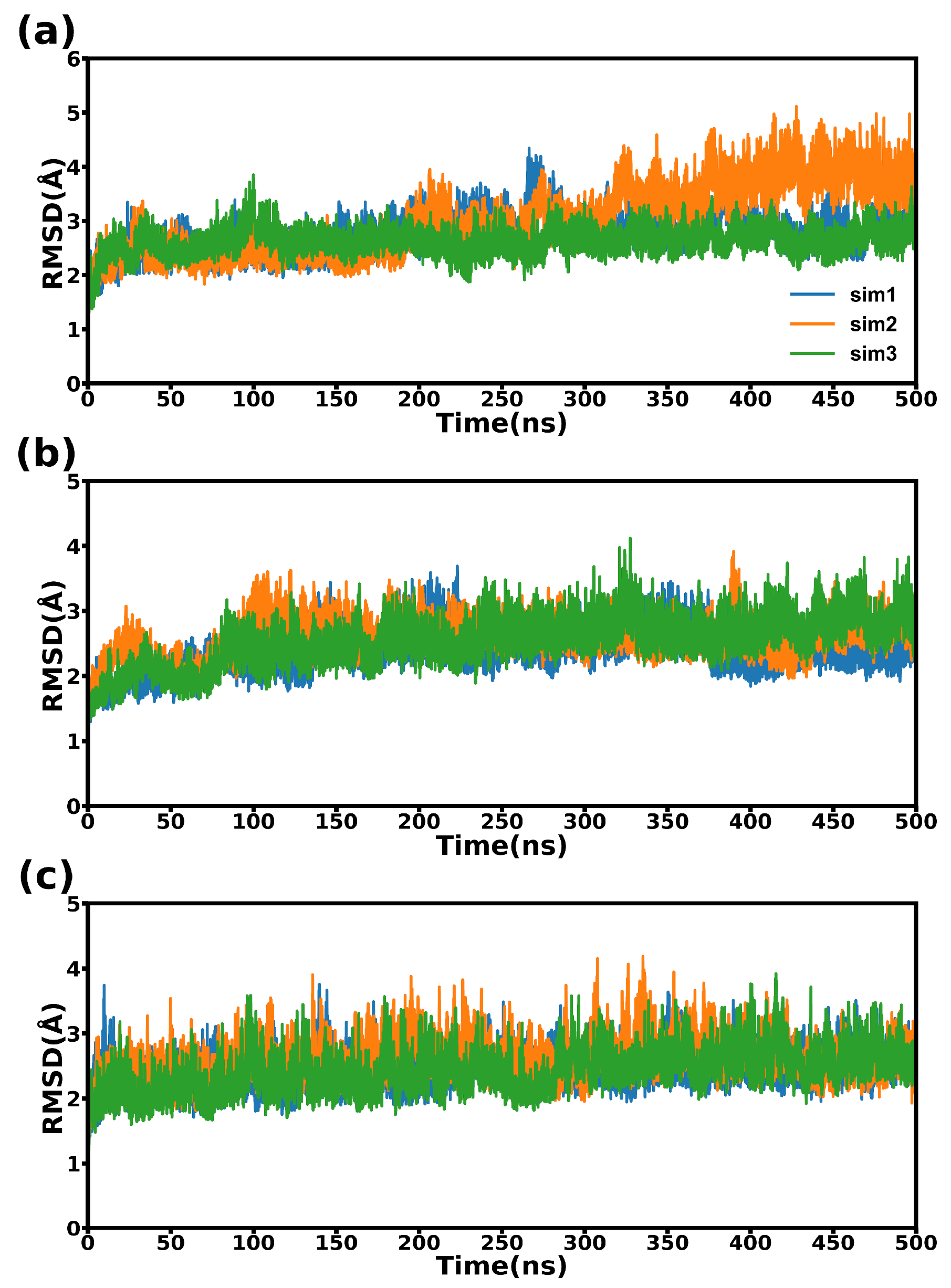
2.2. Structural Stability during Simulations
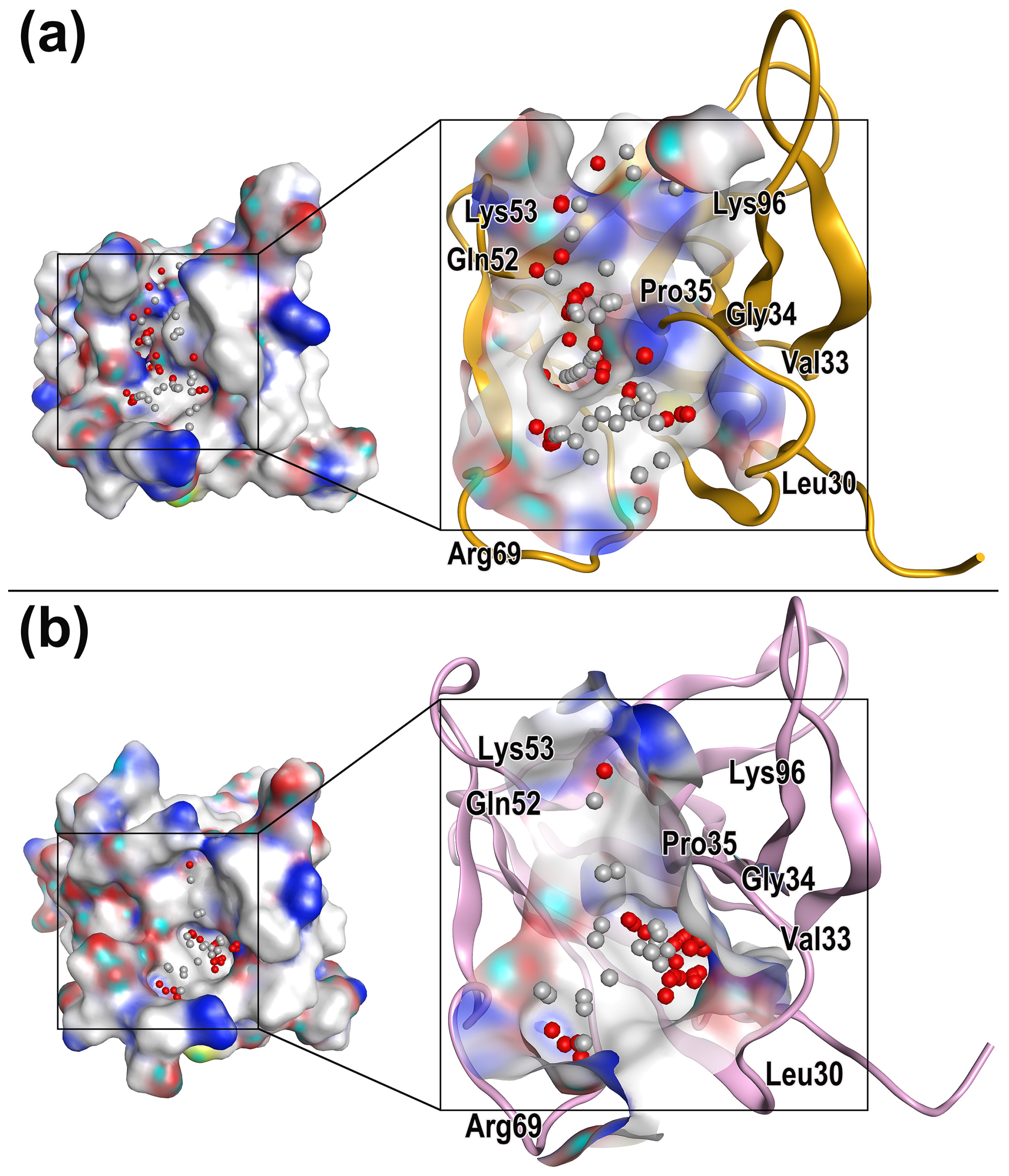
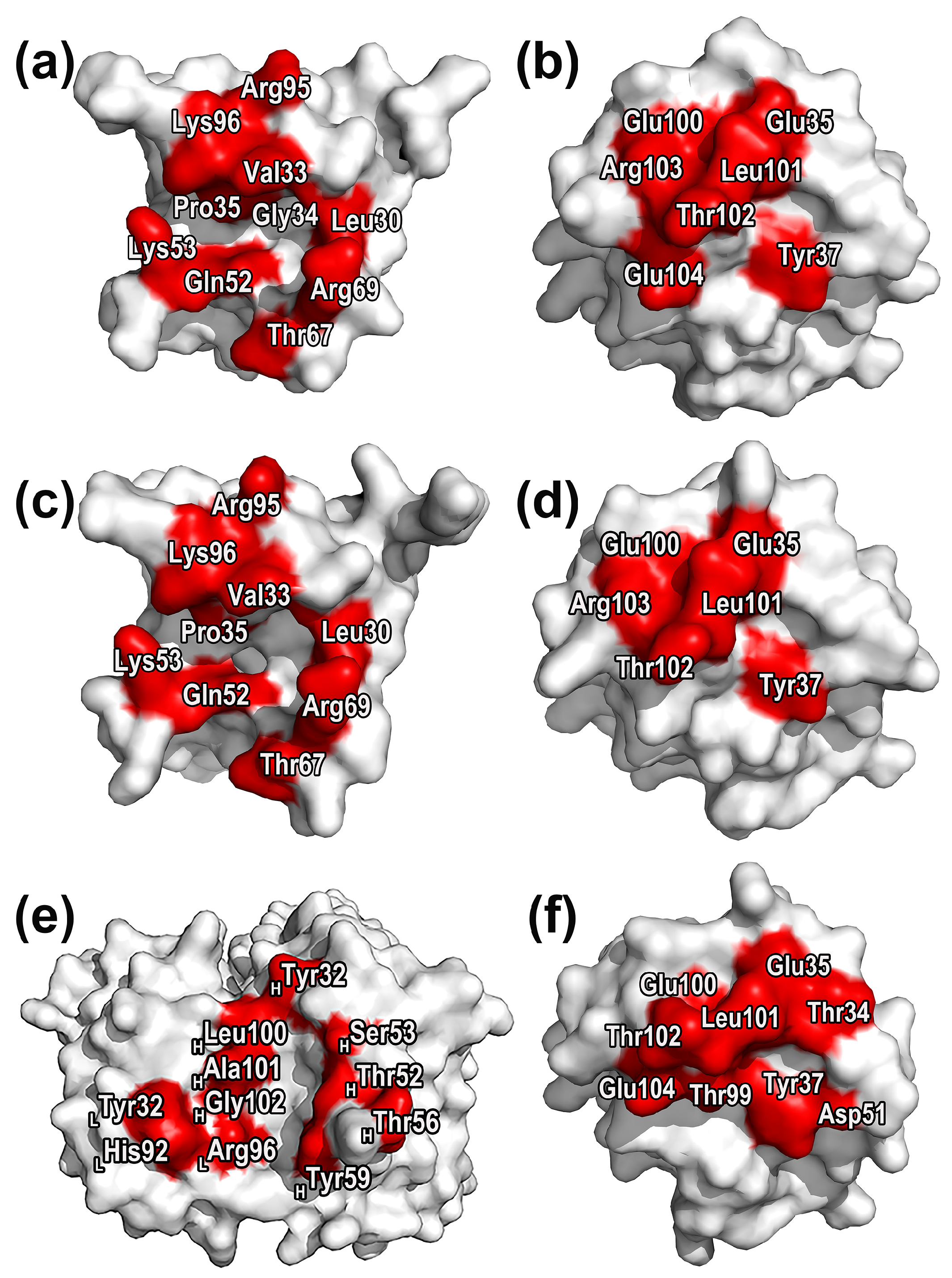
2.3. Characterization of Protein Druggable Sites
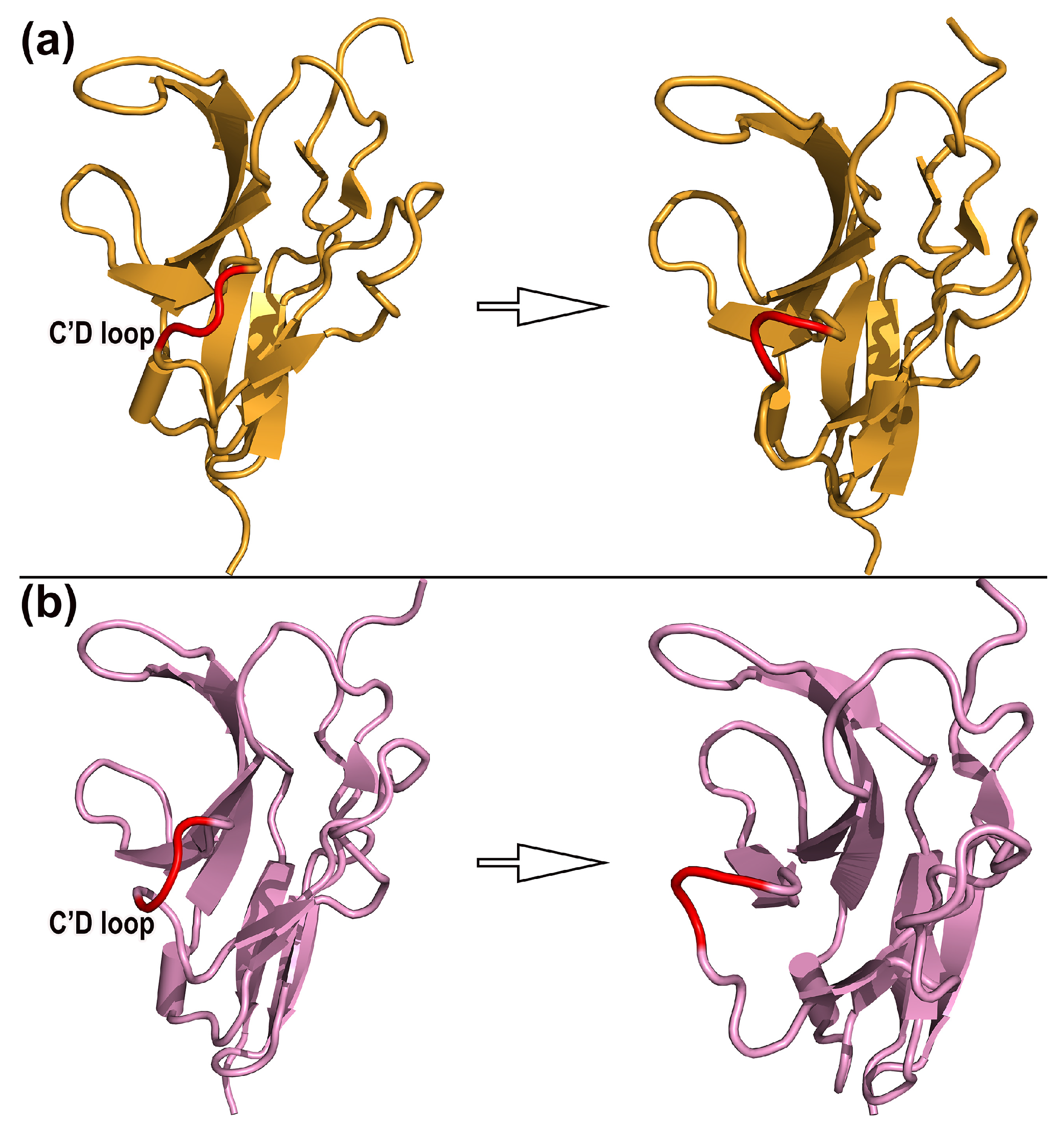
2.4. Dynamic Mechanism and Structural Change
2.5. Binging Free Energy Analysis
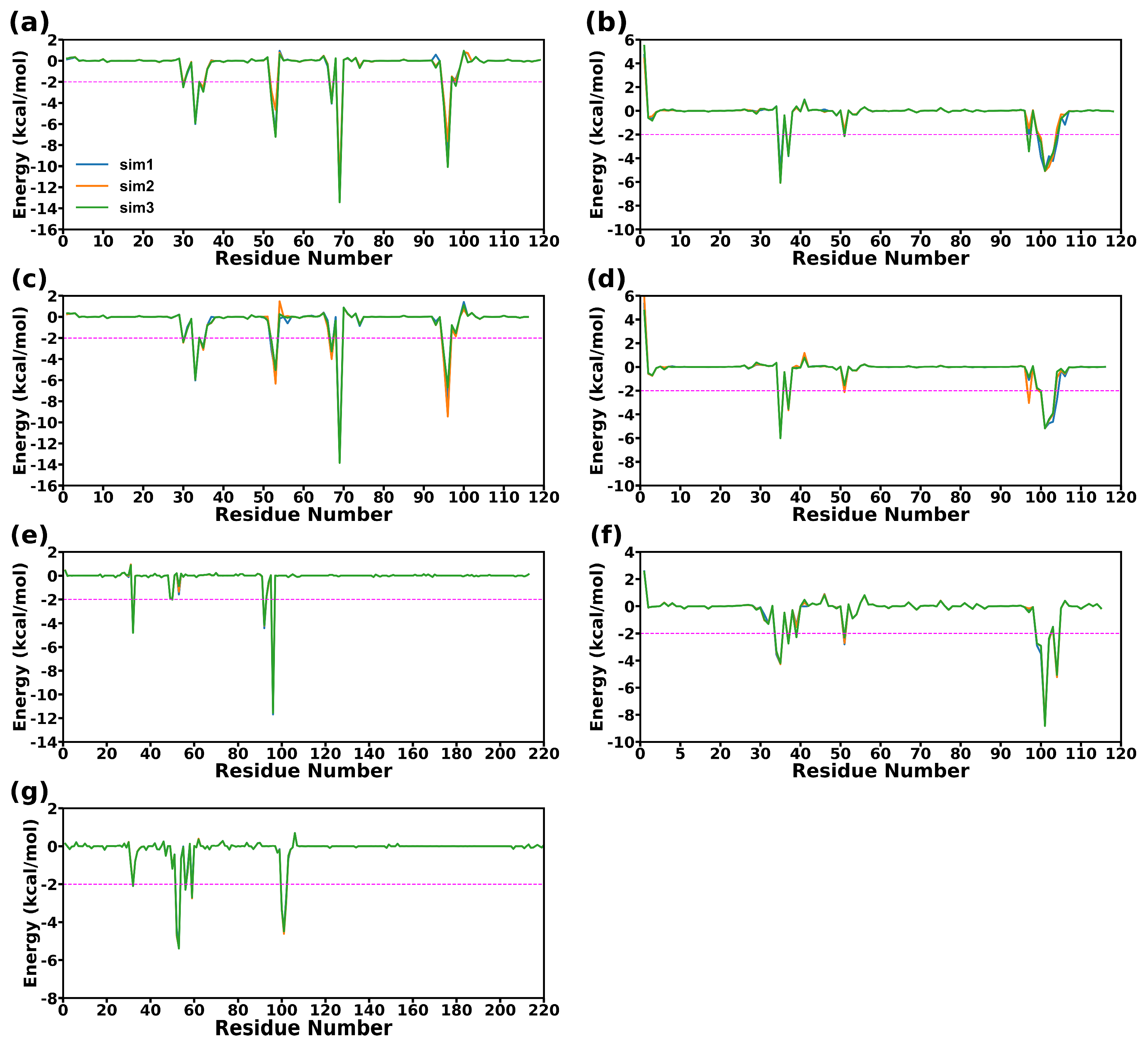
2.6. Energy Decomposition and Analysis of the Binding Hot Spots
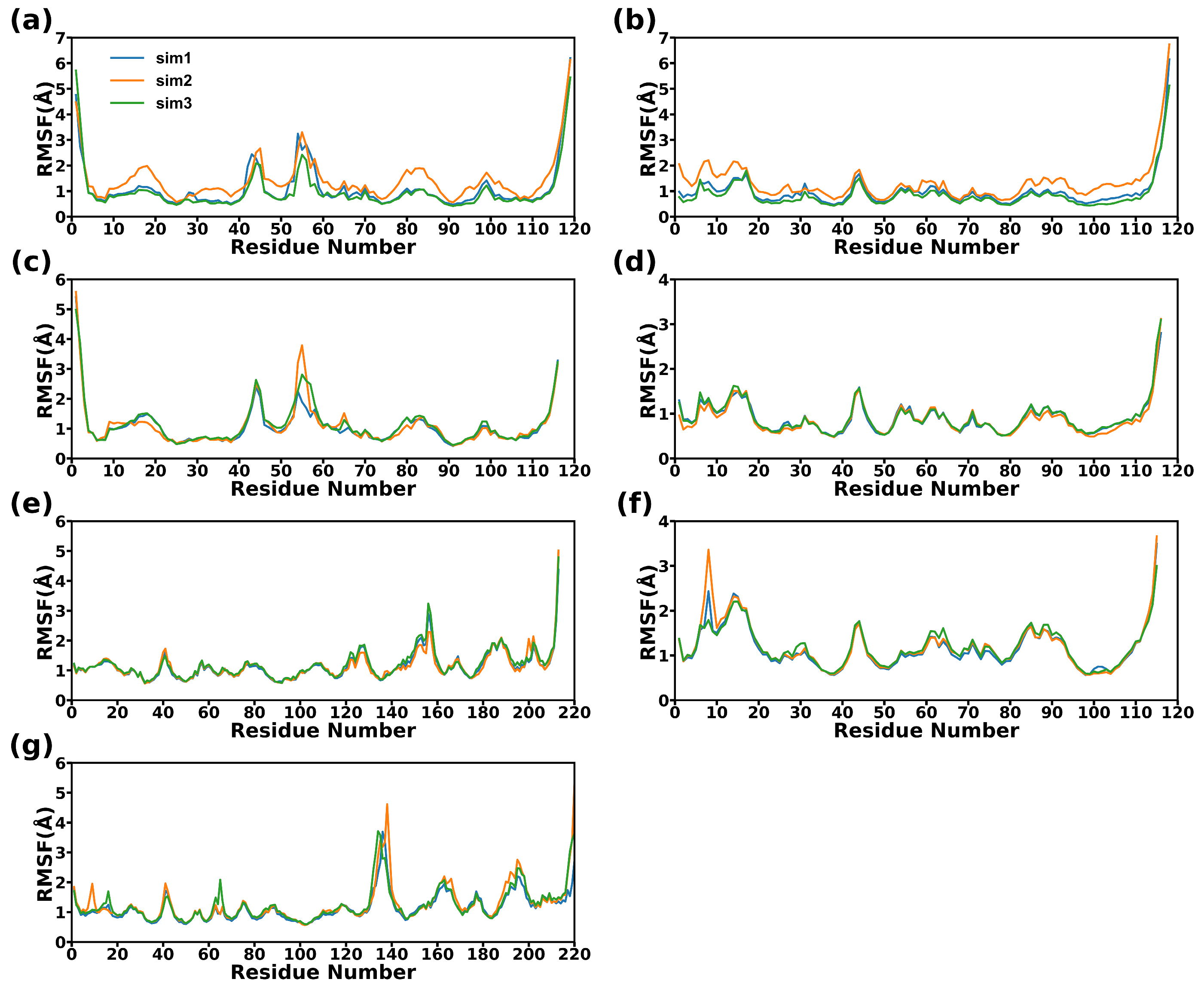
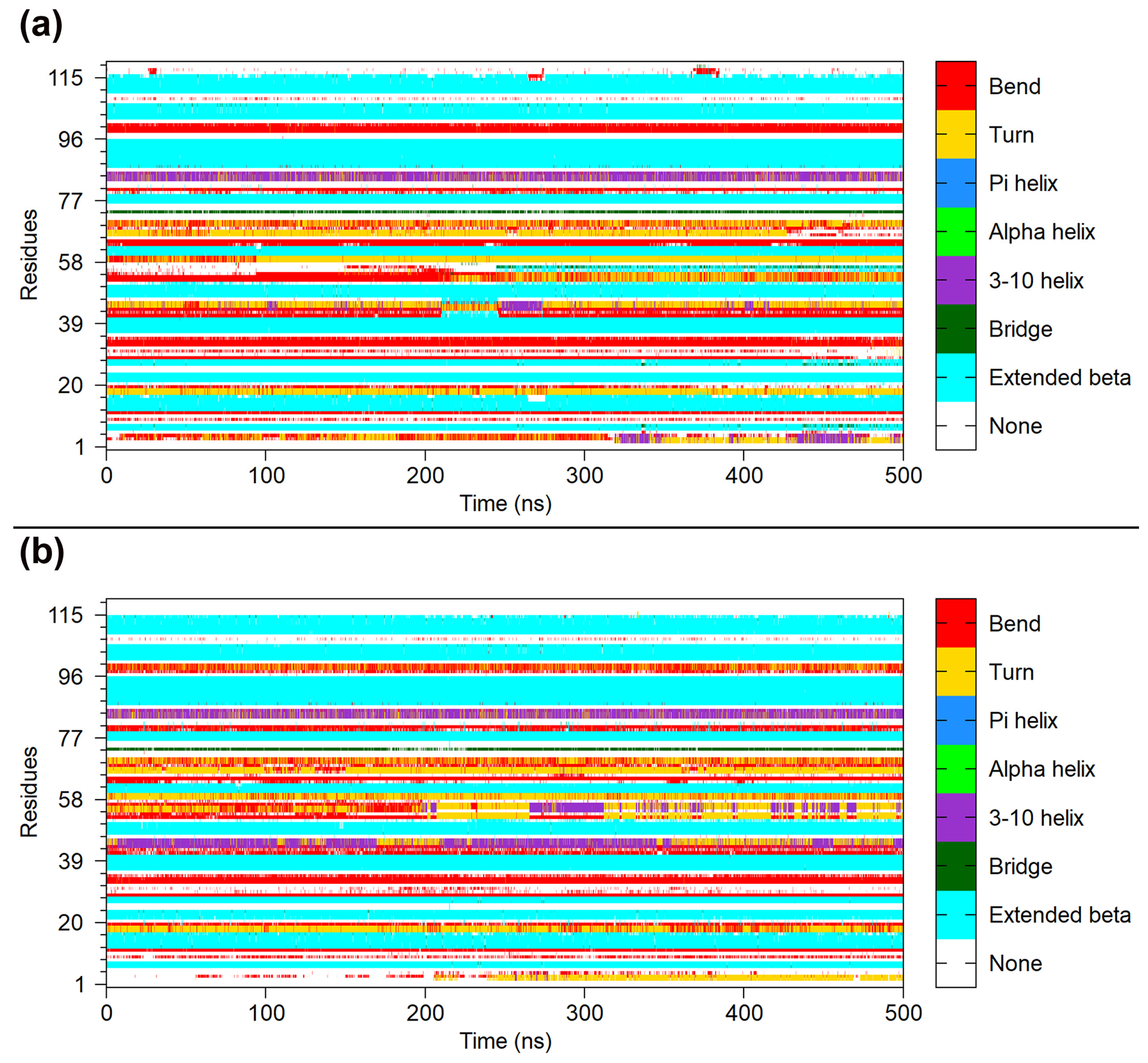
2.7. Dynamical Structure Analysis
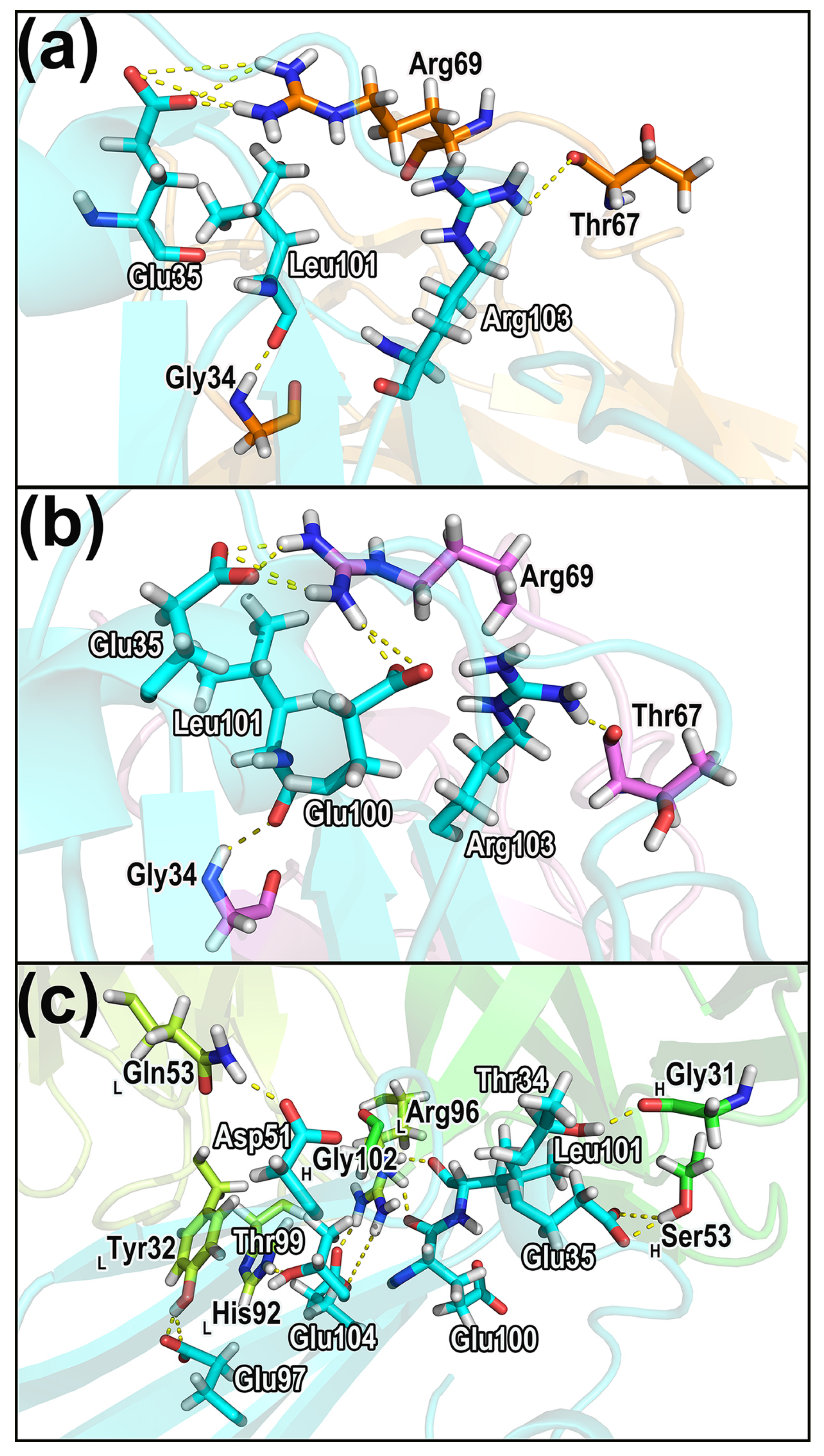
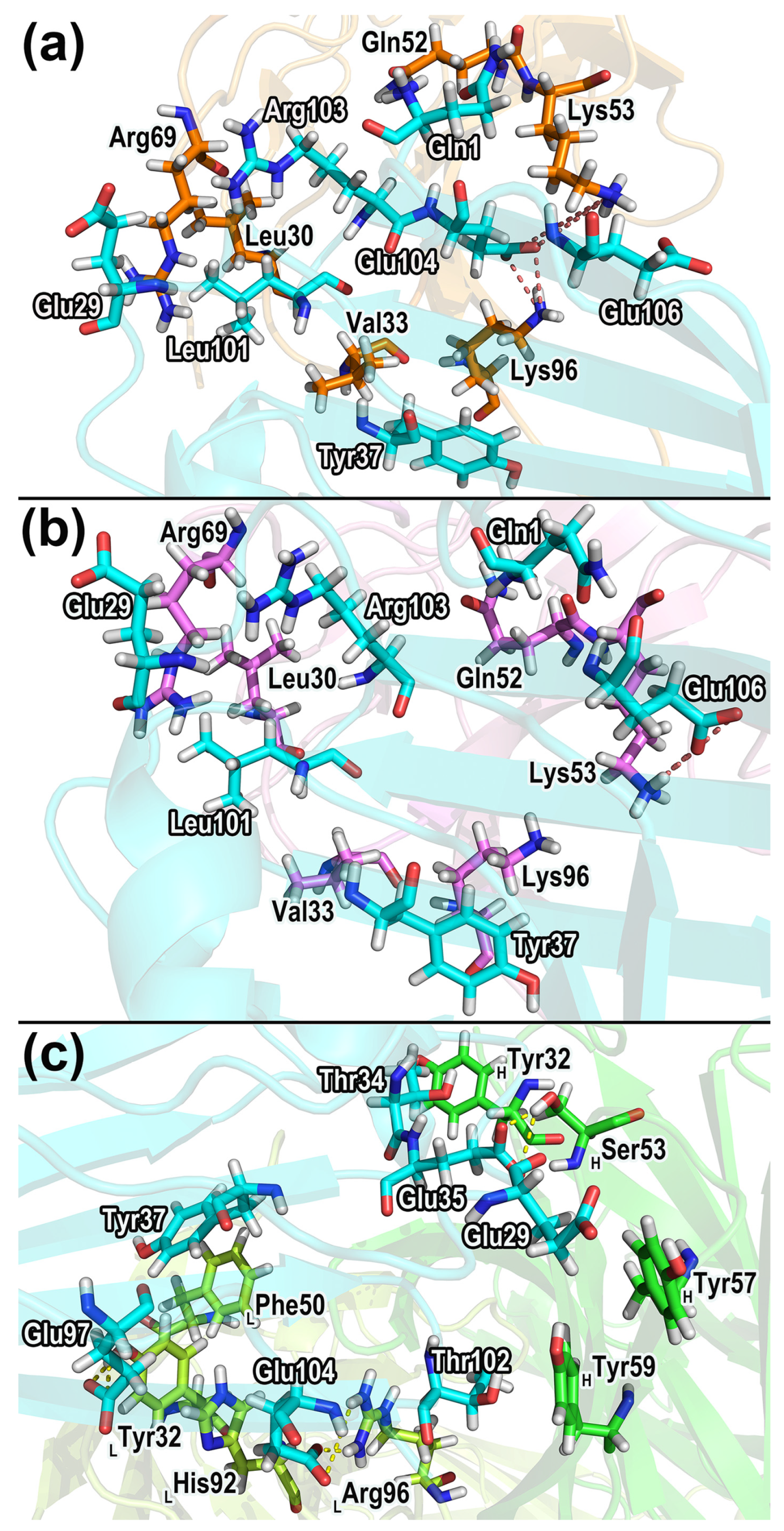
2.7.1. Hydrogen Bonds in the Complexes
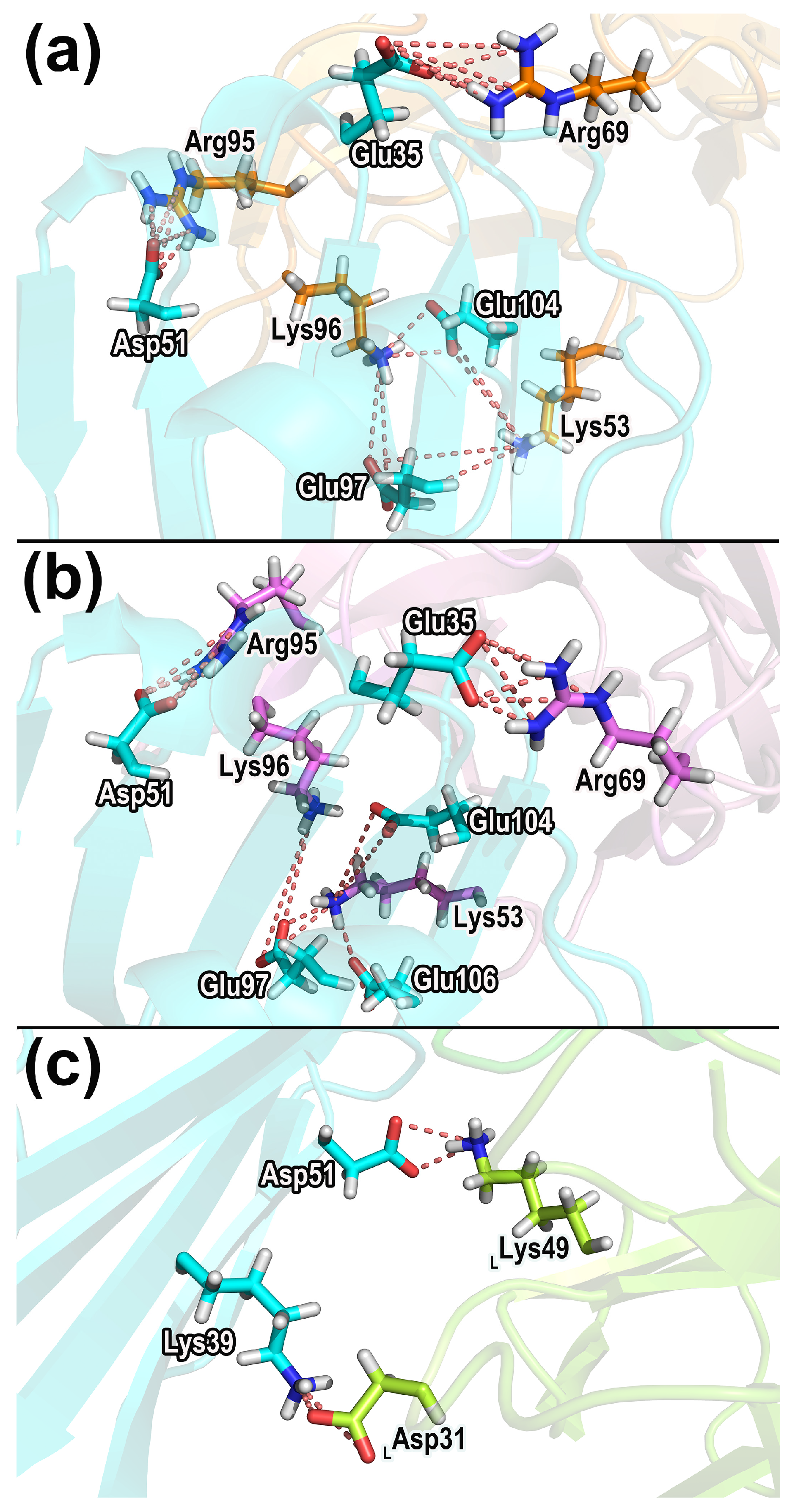
2.7.2. Salt Bridges
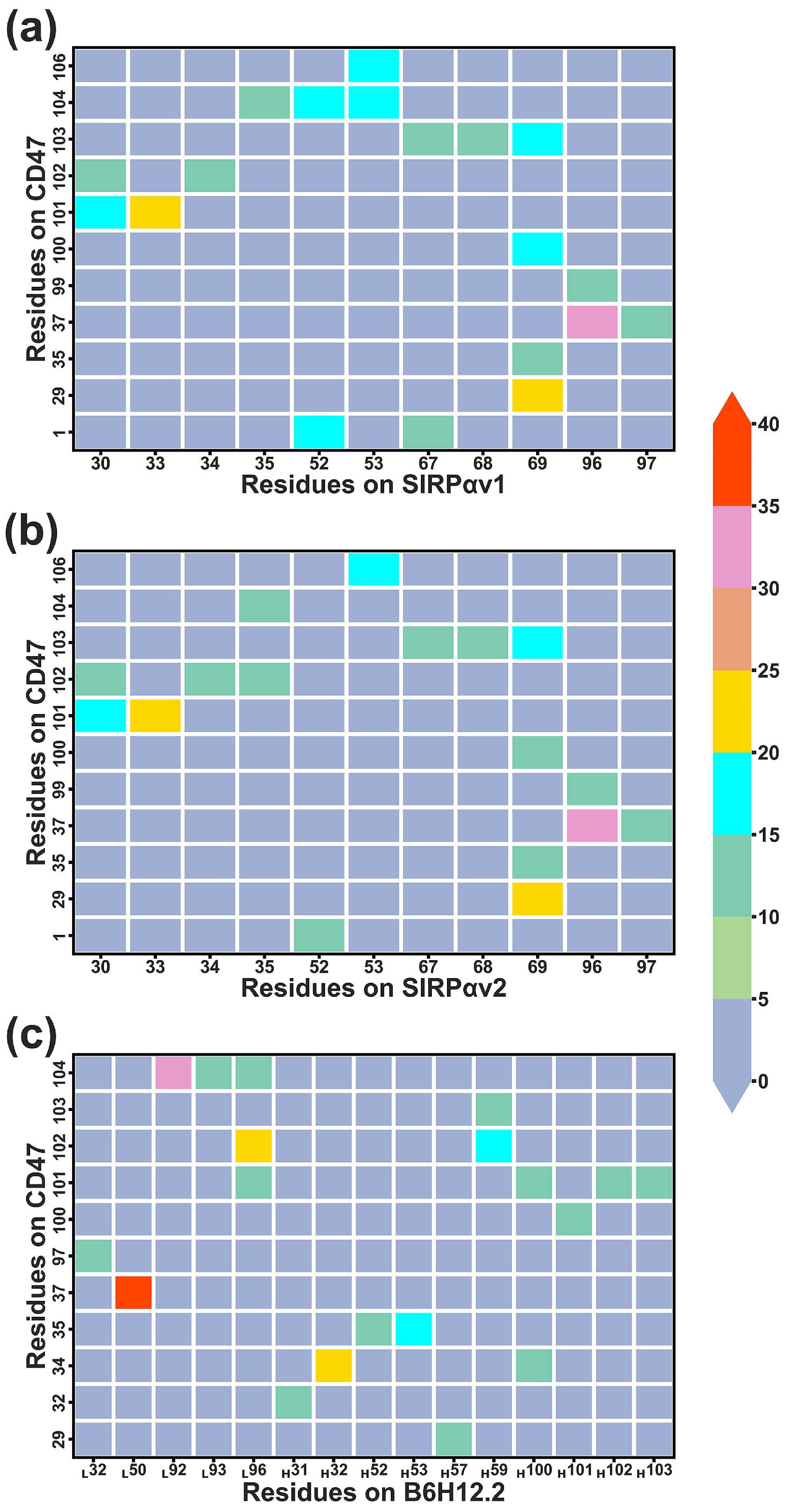
2.7.3. Dynamical Residue Contacts
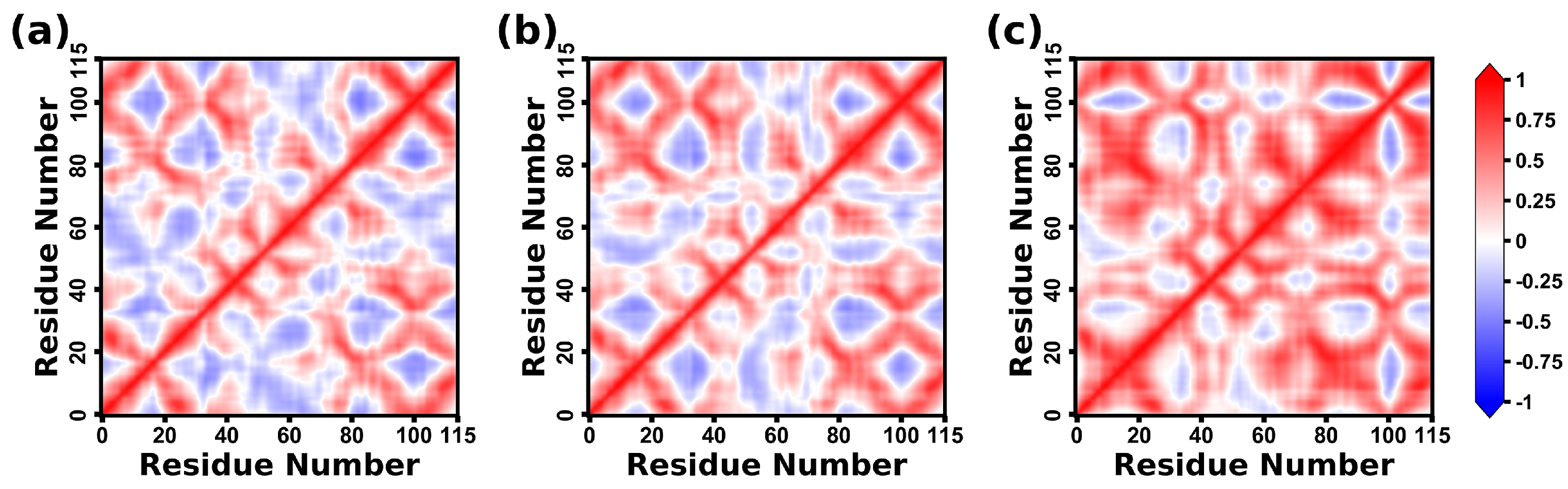
2.8. Dynamical Correlation Analysis of CD47 Proteins
3. Discussion
4. Materials and Methods
4.1. Structure Preparation
4.2. MD Simulations
4.3. Free Energy Calculations and Decomposition
4.4. Trajectory Analysis
4.5. Dynamical Residue Contacts Analysis
4.6. Correlation Analysis of Dynamical Motions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Sharpe, A.H. Introduction to checkpoint inhibitors and cancer immunotherapy. Immunol. Rev. 2017, 276, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Clin. Oncol. 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Brooke, G.; Holbrook, J.D.; Brown, M.H.; Barclay, A.N. Human lymphocytes interact directly with CD47 through a novel member of the signal regulatory protein (SIRP) family. J. Immunol. 2004, 173, 2562–2570. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.J.; Frazier, W.A. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001, 11, 130–135. [Google Scholar] [CrossRef]
- Oldenborg, P.A.; Zheleznyak, A.; Fang, Y.F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef]
- Matozaki, T.; Murata, Y.; Okazawa, H.; Ohnishi, H. Functions and molecular mechanisms of the CD47–SIRPα signalling pathway. Trends Cell Biol. 2009, 19, 72–80. [Google Scholar] [CrossRef]
- Barclay, A.N.; Berg, T.K.v.d. The Interaction Between Signal Regulatory Protein Alpha (SIRPα) and CD47: Structure, Function, and Therapeutic Target. Annu. Rev. Immunol. 2014, 32, 25–50. [Google Scholar] [CrossRef]
- Matlung, H.L.; Szilagyi, K.; Barclay, N.A.; van den Berg, T.K. The CD47-SIRPα signaling axis as an innate immune checkpoint in cancer. Immunol. Rev. 2017, 276, 145–164. [Google Scholar] [CrossRef]
- Weiskopf, K. Cancer immunotherapy targeting the CD47/SIRPα axis. Eur. J. Cancer 2017, 76, 100–109. [Google Scholar] [CrossRef]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, M.P.; Weissman, I.L.; Majeti, R. The CD47-SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr. Opin. Immunol. 2012, 24, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Ma, Y.; Gao, P.; Yao, Z. Targeting CD47: The achievements and concerns of current studies on cancer immunotherapy. J. Thorac. Dis. 2017, 9, E168–E174. [Google Scholar] [CrossRef] [Green Version]
- Logtenberg, M.E.W.; Scheeren, F.A.; Schumacher, T.N. The CD47-SIRPα Immune Checkpoint. Immunity 2020, 52, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Hatherley, D.; Graham, S.C.; Turner, J.; Harlos, K.; Stuart, D.I.; Barclay, A.N. Paired receptor specificity explained by structures of signal regulatory proteins alone and complexed with CD47. Mol. Cell 2008, 31, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Hatherley, D.; Lea, S.M.; Johnson, S.; Barclay, A.N. Polymorphisms in the human inhibitory signal-regulatory protein α do not affect binding to its ligand CD47. J. Biol. Chem. 2014, 289, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Pietsch, E.C.; Dong, J.; Cardoso, R.; Zhang, X.; Chin, D.; Hawkins, R.; Dinh, T.; Zhou, M.; Strake, B.; Feng, P.H.; et al. Anti-leukemic activity and tolerability of anti-human CD47 monoclonal antibodies. Blood Cancer J. 2017, 7, e536. [Google Scholar] [CrossRef]
- Sim, J.; Sockolosky, J.T.; Sangalang, E.; Izquierdo, S.; Pedersen, D.; Harriman, W.; Wibowo, A.S.; Carter, J.; Madan, A.; Doyle, L.; et al. Discovery of high affinity, pan-allelic, and pan-mammalian reactive antibodies against the myeloid checkpoint receptor SIRPα. MAbs 2019, 11, 1036–1052. [Google Scholar] [CrossRef] [Green Version]
- Takenaka, K.; Prasolava, T.K.; Wang, J.C.; Mortin-Toth, S.M.; Khalouei, S.; Gan, O.I.; Dick, J.E.; Danska, J.S. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat. Immunol. 2007, 8, 1313–1323. [Google Scholar] [CrossRef]
- Lascorz, J.; Bevier, M.; Schönfels, W.V.; Kalthoff, H.; Aselmann, H.; Beckmann, J.; Egberts, J.; Buch, S.; Becker, T.; Schreiber, S.; et al. Association study identifying polymorphisms in CD47 and other extracellular matrix pathway genes as putative prognostic markers for colorectal cancer. Int. J. Colorectal Dis. 2013, 28, 173–181. [Google Scholar] [CrossRef]
- Sano, S.; Ohnishi, H.; Kubota, M. Gene structure of mouse BIT/SHPS-1. Biochem. J. 1999, 344 Pt 3, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Veillette, A.; Chen, J. SIRPα-CD47 Immune Checkpoint Blockade in Anticancer Therapy. Trends Immunol. 2018, 39, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Zhao, H.; Xu, J.; Shen, C. CD47: The next checkpoint target for cancer immunotherapy. Crit. Rev. Oncol. Hematol. 2020, 152, 103014. [Google Scholar] [CrossRef] [PubMed]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [Green Version]
- Kuo, T.C.; Chen, A.; Harrabi, O.; Sockolosky, J.T.; Zhang, A.; Sangalang, E.; Doyle, L.V.; Kauder, S.E.; Fontaine, D.; Bollini, S.; et al. Targeting the myeloid checkpoint receptor SIRPα potentiates innate and adaptive immune responses to promote anti-tumor activity. J. Hematol. Oncol. 2020, 13, 160. [Google Scholar] [CrossRef]
- Wu, X.; Xu, L.Y.; Li, E.M.; Dong, G. Application of molecular dynamics simulation in biomedicine. Chem. Biol. Drug Des. 2022, 99, 789–800. [Google Scholar] [CrossRef]
- Hildebrand, P.W.; Rose, A.S.; Tiemann, J.K.S. Bringing Molecular Dynamics Simulation Data into View. Trends Biochem. Sci. 2019, 44, 902–913. [Google Scholar] [CrossRef] [Green Version]
- Feng, T.; Barakat, K. Molecular Dynamics Simulation and Prediction of Druggable Binding Sites. Methods Mol. Biol. 2018, 1762, 87–103. [Google Scholar]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Bera, I.; Payghan, P.V. Use of Molecular Dynamics Simulations in Structure-Based Drug Discovery. Curr. Pharm. Des. 2019, 25, 3339–3349. [Google Scholar] [CrossRef]
- Filipe, H.A.L.; Loura, L.M.S. Molecular Dynamics Simulations: Advances and Applications. Molecules 2022, 27, 2105. [Google Scholar] [CrossRef]
- Collier, T.A.; Piggot, T.J.; Allison, J.R. Molecular Dynamics Simulation of Proteins. Methods Mol. Biol. 2020, 2073, 311–327. [Google Scholar] [PubMed]
- St. West, S. 2022.02 Molecular Operating Environment (MOE); Chemical Computing Group ULC: Sherbooke, QC, Canada, 2023; p. 910. [Google Scholar]
- Delano, W.L. The PyMol Molecular Graphics System. Proteins Struct. Funct. Bioinform. 2002, 30, 442–454. [Google Scholar]
- Hatherley, D.; Harlos, K.; Dunlop, D.C.; Stuart, D.I.; Barclay, A.N. The Structure of the Macrophage Signal Regulatory Protein α (SIRPα) Inhibitory Receptor Reveals a Binding Face Reminiscent of That Used by T Cell Receptors. J. Biol. Chem. 2007, 282, 14567–14575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Do, R.K.; Sali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [Green Version]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E.; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Edelsbrunner, H. Alpha Shapes—A Survey. 2009. Available online: https://www.semanticscholar.org/paper/Alpha-Shapes-%E2%80%94-a-Survey-Edelsbrunner/0043d0b4317d685082f357b9edd9359969d59c2c (accessed on 3 June 2023).
- Edelsbrunner, H.; Facello, M.; Fu, P.; Liang, J. Measuring proteins and voids in proteins. In Proceedings of the 28th Hawaii International Conference on System Sciences, Wailea, HI, USA, 3–6 January 1995; p. 256. [Google Scholar]
- Volkamer, A.; Griewel, A.; Grombacher, T.; Rarey, M. Analyzing the topology of active sites: On the prediction of pockets and subpockets. J. Chem. Inf. Model. 2010, 50, 2041–2052. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tong, Q.; Zhou, Y.; Lee, H.-W.; Yang, J.J.; Bühring, H.-J.; Chen, Y.-T.; Ha, B.; Chen, C.X.J.; Yang, Y.; et al. Functional Elements on SIRPα IgV Domain Mediate Cell Surface Binding to CD47. J. Mol. Biol. 2007, 365, 680–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD47 Residues | Partner Residues a |
|---|---|
| CD47-SIRPαv1: | |
| Gln1, Leu2, Leu3, Lys6, Asn27, Glu29, Ala30, Gln31, Thr34, Glu35, Val36, Tyr37, Lys39, Asp46, Thr49, Glu97, Thr99, Glu100, Leu101, Thr102, Arg103, Glu104, Gly105, Glu106 | Ser29, Leu30, Ile31, Pro32, Val33, Gly34, Pro35, Ile36, Gln37, Tyr50, Asn51, Gln52, Lys53, Glu54, Leu66, Thr67, Lys68, Arg69, Phe74, Lys93, Lys96, Gly97, Ser98, Pro99, Asp100, Asp101 |
| CD47-SIRPαv2: | |
| Gln1, Leu2, Leu3, Lys6, Asn27, Glu29, Ala30,Gln31, Thr34, Glu35, Val36, Tyr37, Lys39, Lys41, Asp46, Thr49, Glu97, Thr99, Glu100, Leu101, Thr102, Arg103, Glu104, Gly105, Glu106 | Ser29, Leu30, Ile31, Pro32, Val33, Gly34, Pro35, Ile36, Gln37, Tyr50, Asn51, Gln52, Lys53, Glu54, His56, Ser66, Thr67, Lys68, Arg69, Glu70, Phe74, Lys93, Lys96, Gly97, Ser98, Pro99, Asp100 |
| CD47-B6H12.2 b: | |
| Met28, Glu29, Ala30, Gln31, Asn32, Thr34, Glu35, Val36, Tyr37, Lys39, Asp46, Asp51, Ala53, Leu54, Glu97, Thr99, Glu100, Leu101, Thr102, Arg103, Glu104 | Ser30LC, Asp31LC, Tyr32LC, Lys49LC, Phe50LC, Gln52LC, Gly91LC, His92LC, Gly93LC, Phe94LC, Arg96LC, Ser30HC, Gly31HC, Tyr32HC, Gly33HC, Trp47HC, Thr50HC, Ile51HC, Thr52HC, Ser53HC, Gly54HC, Gly55HC, Thr56HC, Tyr57HC, Tyr59HC, Ser99HC, Leu100HC, Ala101HC, Gly102HC, Asn103HC |
| Contribution | CD47-SIRPαv1 | CD47-SIRPαv2 | CD47-B6H12.2 |
|---|---|---|---|
| ΔEele | −557.87 ± 72.76 | −522.69 ± 69.19 | −549.61 ± 29.91 |
| ΔEvdw | −89.88 ± 6.72 | −86.09 ± 8.99 | −97.56 ± 6.01 |
| ΔEgas b | −647.74 ± 73.77 | −608.78 ± 72.85 | −647.17 ± 29.57 |
| ΔGsol-polar | 575.80 ± 68.36 | 540.53 ± 63.17 | 565.76 ± 26.28 |
| ΔGsol-np | −14.54 ± 0.79 | −13.76 ± 1.35 | −13.99 ± 0.58 |
| ΔGsol c | 561.27 ± 67.84 | 526.78 ± 62.13 | 551.77 ± 26.09 |
| ΔHtot d | −86.48 ± 9.72 | −82.00 ± 13.26 | −95.40 ± 7.85 |
| TΔS e | −53.75 ± 6.78 | −54.55 ± 5.78 | −51.76 ± 7.24 |
| ΔGbind f | −32.73 | −27.45 | −43.64 |
| Kd | 0.74 ± 0.07 μM | 0.64 ± 0.06 μM | 0.5 nM |
| Acceptor Residues | Acceptor Atoms | Donor Residues | Donor Atoms | Fraction 1 b | Fraction 2 | Fraction 3 |
|---|---|---|---|---|---|---|
| CD47-SIRPαv1: | ||||||
| SIRPα_Thr67 | O | CD47_Arg103 | NH1 | 88.39% | 84.93% | 91.16% |
| CD47_Leu101 | O | SIRPα_Gly34 | N | 79.75% | 73.11% | 80.38% |
| CD47_Glu35 | OE2 | SIRPα_Arg69 | NH2 | 49.75% | 49.16% | 52.81% |
| CD47_Glu35 | OE1 | SIRPα_Arg69 | NH2 | 49.22% | 50.90% | 48.36% |
| CD47_Glu35 | OE1 | SIRPα_Arg69 | NH1 | 34.85% | 45.86% | 43.36% |
| CD47_Glu35 | OE2 | SIRPα_Arg69 | NH1 | 27.67% | 41.40% | 47.19% |
| CD47-SIRPαv2: | ||||||
| SIRPα_Thr67 | O | CD47_Arg103 | NH1 | 86.75% | 90.19% | 85.80% |
| CD47_Leu101 | O | SIRPα_Gly34 | N | 85.18% | 83.24% | 83.27% |
| CD47_Glu35 | OE1 | SIRPα_Arg69 | NH2 | 50.61% | 48.62% | 52.99% |
| CD47_Glu35 | OE2 | SIRPα_Arg69 | NH2 | 51.46% | 52.00% | 47.72% |
| CD47_Glu35 | OE2 | SIRPα_Arg69 | NH1 | 51.63% | 46.76% | 49.83% |
| CD47_Glu100 | OE1 | SIRPα_Arg69 | NH1 | 49.31% | 45.71% | 41.79% |
| CD47_Glu100 | OE2 | SIRPα_Arg69 | NH1 | 37.97% | 41.74% | 45.84% |
| CD47_Glu35 | OE1 | SIRPα_Arg69 | NH1 | 39.36% | 44.43% | 41.80% |
| CD47-B6H12.2: | ||||||
| CD47_Thr99 | OG1 | LC_His92 | NE2 | 77.36% | 71.15% | 67.44% |
| CD47_Glu100 | O | HC_Gly102 | N | 76.87% | 80.10% | 80.90% |
| HC_Gly31 | O | CD47_Thr34 | OG1 | 70.27% | 63.52% | 68.58% |
| CD47_Asp51 | OD2 | LC_Gln53 | NE2 | 62.09% | 53.64% | 36.49% |
| CD47_Glu104 | OE1 | LC_Arg96 | NH1 | 55.32% | 53.87% | 41.68% |
| CD47_Leu101 | O | LC_Arg96 | NE | 54.75% | 55.88% | 49.23% |
| CD47_Glu35 | OE1 | HC_Ser53 | OG | 52.38% | 53.57% | 34.55% |
| CD47_Glu35 | OE2 | HC_Ser53 | OG | 46.38% | 45.29% | 63.99% |
| CD47_Glu97 | OE2 | LC_Tyr32 | OH | 50.82% | 41.96% | 48.33% |
| CD47_Glu97 | OE1 | LC_Tyr32 | OH | 42.93% | 50.97% | 46.96% |
| CD47_Glu104 | OE2 | LC_Arg96 | NH1 | 41.36% | 44.13% | 55.27% |
| Negative Residues b | Positive Residues | Fraction 1 c | Fraction 2 | Fraction 3 |
|---|---|---|---|---|
| CD47-SIRPαv1: | ||||
| CD47_Glu35 | SIRPαv1_Arg69 | 32.66% | 43.16% | 45.53% |
| CD47_Glu97 | SIRPαv1_Lys96 | 18.49% | 32.07% | 74.37% |
| CD47_Glu97 | SIRPαv1_Lys53 | 45.35% | 15.85% | 40.15% |
| CD47_Asp51 | SIRPαv1_Arg95 | 36.37% | 35.15% | 42.20% |
| CD47_Glu104 | SIRPαv1_Lys96 | 78.59% | 31.45% | 4.53% |
| CD47_Glu104 | SIRPαv1_Lys53 | 4.59% | 24.19% | 48.76% |
| CD47-SIRPαv2: | ||||
| CD47_Glu35 | SIRPαv2_Arg69 | 50.01% | 49.77% | 52.45% |
| CD47_Asp51 | SIRPαv2_Arg95 | 41.00% | 44.15% | 35.85% |
| CD47_Glu97 | SIRPαv2_Lys96 | 17.95% | 59.08% | 36.13% |
| CD47_Glu104 | SIRPαv2_Lys53 | 18.07% | 34.29% | 35.60% |
| CD47_Glu106 | SIRPαv2_Lys53 | 27.74% | 20.61% | 38.96% |
| CD47_Glu97 | SIRPαv2_Lys53 | 24.54% | 43.30% | 5.95% |
| CD47-B6H12.2: | ||||
| CD47_Asp51 | LC_Lys49 | 99.56% | 99.77% | 99.78% |
| LC_Asp31 | CD47_Lys39 | 28.65% | 35.78% | 60.68% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, K.; Liu, Y.; Wen, S.; Zhao, Y.; Ding, H.; Liu, H.; Kong, D.-X. Binding Mechanism of CD47 with SIRPα Variants and Its Antibody: Elucidated by Molecular Dynamics Simulations. Molecules 2023, 28, 4610. https://doi.org/10.3390/molecules28124610
Huang K, Liu Y, Wen S, Zhao Y, Ding H, Liu H, Kong D-X. Binding Mechanism of CD47 with SIRPα Variants and Its Antibody: Elucidated by Molecular Dynamics Simulations. Molecules. 2023; 28(12):4610. https://doi.org/10.3390/molecules28124610
Chicago/Turabian StyleHuang, Kaisheng, Yi Liu, Shuixiu Wen, Yuxin Zhao, Hanjing Ding, Hui Liu, and De-Xin Kong. 2023. "Binding Mechanism of CD47 with SIRPα Variants and Its Antibody: Elucidated by Molecular Dynamics Simulations" Molecules 28, no. 12: 4610. https://doi.org/10.3390/molecules28124610