
Virtual Screening of Artemisia annua Phytochemicals as Potential Inhibitors of SARS-CoV-2 Main Protease Enzyme
 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Active Site of the SARS-CoV-2 Mpro
2.2. Molecular Docking of the Retrieved Phytochemical with SARS-CoV Mpro
2.3. Selection of Top Compounds for Simulation
2.4. Physiochemical Properties of the Selected Compounds
2.5. Molecular Dynamic Simulation Assay for the Selected Compounds
2.6. Binding Free Energy Calculation
3. Discussion
4. Materials and Methods
4.1. Retrieval of Antiviral Drugs
4.2. Receptor and Ligand Preparation
4.3. Molecular Docking Study
4.4. Physiochemical and Pharmacokinetic Properties of the Selected Compounds
4.5. Molecular Dynamic Simulation
4.6. Binding Free Energy Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Perlman, S. Another decade, another coronavirus. N. Engl. J. Med. 2020, 382, 760–762. [Google Scholar] [CrossRef] [PubMed]
- Zu, Z.Y.; Di Jiang, M.; Xu, P.P.; Chen, W.; Ni, Q.Q.; Lu, G.M.; Zhang, L.J. Coronavirus disease 2019 (COVID-19): A perspective from China. Radiology 2020, 296, E15–E25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): An update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Tiwari, S.; Deb, M.K.; Marty, J.L. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): A global pandemic and treatment strategies. Int. J. Antimicrob. Agents 2020, 56, 106054. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xiao, Z.; Ye, K.; He, X.; Sun, B.; Qin, Z.; Yu, J.; Yao, J.; Wu, Q.; Bao, Z. SARS-CoV-2: Characteristics and current advances in research. Virol. J. 2020, 17, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Li, R.; Pan, W.; Huang, W.; Liu, B.; Xie, Y.; Wang, Z.; Li, C.; Jiang, H.; Huang, J. Phillyrin (KD-1) exerts anti-viral and anti-inflammatory activities against novel coronavirus (SARS-CoV-2) and human coronavirus 229E (HCoV-229E) by suppressing the nuclear factor kappa B (NF-κB) signaling pathway. Phytomedicine 2020, 78, 153296. [Google Scholar] [CrossRef] [PubMed]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural basis of potential inhibitors targeting SARS-CoV-2 main protease. Front. Chem. 2021, 9, 622898. [Google Scholar] [CrossRef]
- Agost-Beltrán, L.; de la Hoz-Rodriguez, S.; Bou-Iserte, L.; Rodriguez, S.; Fernández-de-la-Pradilla, A.; González, F.V. Advances in the Development of SARS-CoV-2 Mpro Inhibitors. Molecules 2022, 27, 2523. [Google Scholar] [CrossRef]
- Narayanan, A.; Narwal, M.; Majowicz, S.A.; Varricchio, C.; Toner, S.A.; Ballatore, C.; Brancale, A.; Murakami, K.S.; Jose, J. Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun. Biol. 2022, 5, 1–17. [Google Scholar] [CrossRef]
- Qiao, J.; Li, Y.-S.; Zeng, R.; Liu, F.-L.; Luo, R.-H.; Huang, C.; Wang, Y.F.; Zhang, J.; Quan, B.; Shen, C.; et al. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science 2021, 371, 1374–1378. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan Baig, M.; Ahmad, K.; Roy, S.; Mohammad Ashraf, J.; Adil, M.; Haris Siddiqui, M.; Khan, S.; Amjad Kamal, M.; Provazník, I.; Choi, I. Computer aided drug design: Success and limitations. Curr. Pharm. Des. 2016, 22, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.V.; Balamuralikrishnan, B.; Kaviya, M.; Bharathi, K.; Parithathvi, A.; Arun, M.; Senthilkumar, N.; Velayuthaprabhu, S.; Saradhadevi, M.; Al-Dhabi, N.A. Medicinal plants, phytochemicals, and herbs to combat viral pathogens including SARS-CoV-2. Molecules 2021, 26, 1775. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.H. Health-promoting components of fruits and vegetables in the diet. Adv. Nutr. 2013, 4, 384S–392S. [Google Scholar] [CrossRef] [Green Version]
- Forni, C.; Facchiano, F.; Bartoli, M.; Pieretti, S.; Facchiano, A.; D’Arcangelo, D.; Norelli, S.; Valle, G.; Nisini, R.; Beninati, S. Beneficial role of phytochemicals on oxidative stress and age-related diseases. Biomed. Res. Int. 2019, 2019, 8748253. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, K.; Yazdanpanah, N.; Saghazadeh, A.; Rezaei, N. Computational drug discovery and repurposing for the treatment of COVID-19: A systematic review. Bioorg. Chem. 2021, 106, 104490. [Google Scholar] [CrossRef]
- Mahdian, S.; Ebrahim-Habibi, A.; Zarrabi, M. Drug repurposing using computational methods to identify therapeutic options for COVID-19. J. Diabetes Metab. Disord. 2020, 19, 691–699. [Google Scholar] [CrossRef]
- Sharma, P.P.; Bansal, M.; Sethi, A.; Pena, L.; Goel, V.K.; Grishina, M.; Chaturvedi, S.; Kumar, D.; Rathi, B. Computational methods directed towards drug repurposing for COVID-19: Advantages and limitations. RSC Adv. 2021, 11, 36181–36198. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the SC’06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H. The FF14SB force field. Amber 2014, 14, 29–31. [Google Scholar]
- Siva Kumar, B.; Anuragh, S.; Kammala, A.K.; Ilango, K. Computer Aided Drug Design Approach to Screen Phytoconstituents of Adhatoda vasica as Potential Inhibitors of SARS-CoV-2 Main Protease Enzyme. Life 2022, 12, 315. [Google Scholar] [CrossRef]
- Lu, J.; Chen, S.A.; Khan, M.B.; Brassard, R.; Arutyunova, E.; Lamer, T.; Vuong, W.; Fischer, C.; Young, H.S.; Vederas, J.C.; et al. Crystallization of Feline Coronavirus Mpro With GC376 Reveals Mechanism of Inhibition. Front. Chem. 2022, 10, 852210. [Google Scholar] [CrossRef]
- Bhardwaj, V.K.; Singh, R.; Sharma, J.; Rajendran, V.; Purohit, R.; Kumar, S. Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2020, 39, 3449–3458. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, J.; Ikram, S.; Ahmad, F.; Rehman, I.U.; Mushtaq, M. SARS-CoV-2 RNA dependent RNA polymerase (RdRp)–a drug repurposing study. Heliyon 2020, 6, e04502. [Google Scholar] [CrossRef] [PubMed]
- Shamsi, A.; Mohammad, T.; Anwar, S.; Amani, S.; Khan, M.S.; Husain, F.M.; Rehman, M.T.; Islam, A.; Hassan, M.I. Potential drug targets of SARS-CoV-2: From genomics to therapeutics. Int. J. Biol. Macromol. 2021, 177, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Pradhan, A.; Maurya, V.K.; Kumar, S.; Theengh, A.; Puri, B.; Saxena, S.K. Therapeutic approaches for SARS-CoV-2 infection. Methods 2021, 195, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Swargiary, A.; Mahmud, S.; Saleh, M.A. Screening of phytochemicals as potent inhibitor of 3-chymotrypsin and papain-like proteases of SARS-CoV2: An in silico approach to combat COVID-19. J. Biomol. Struct. Dyn. 2022, 40, 2067–2081. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Kollar, J.; Frecer, V. How accurate is the description of ligand–protein interactions by a hybrid QM/MM approach? J. Mol. Model. 2018, 24, 1–20. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. In Chemical Biology; Springer: Berlin/Heidelberg, Germany, 2015; pp. 243–250. [Google Scholar]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 1078–1084. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Fu, A.; Zhang, L. Progress in molecular docking. Quant. Biol. 2019, 7, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Lim-Wilby, M. Molecular docking. In Molecular Modeling of Proteins; Springer: Berlin/Heidelberg, Germany, 2008; pp. 365–382. [Google Scholar]
- Chen, G.; Seukep, A.J.; Guo, M. Recent advances in molecular docking for the research and discovery of potential marine drugs. Mar. Drugs 2020, 18, 545. [Google Scholar] [CrossRef]
- Pawar, R.P.; Rohane, S.H. Role of autodock vina in PyRx molecular docking. Asian J. Res. Chem. 2021, 14, 132–134. [Google Scholar]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.; Cheatham, T.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. Amber. 2020. Available online: https://ambermd.org/doc12/Amber20.pdf (accessed on 16 November 2022).
- Naqvi, A.A.T.; Mohammad, T.; Hasan, G.M.; Hassan, M. Advancements in docking and molecular dynamics simulations towards ligand-receptor interactions and structure-function relationships. Curr. Top. Med. Chem. 2018, 18, 1755–1768. [Google Scholar] [CrossRef]
- Rafi, S.; Yasmin, S.; Uddin, R. A molecular dynamic simulation approach: Development of dengue virus vaccine by affinity improvement techniques. J. Biomol. Struct. Dyn. 2020, 40, 61–76. [Google Scholar] [CrossRef]
- Ahmad, S.; Raza, S.; Uddin, R.; Azam, S.S. Binding mode analysis, dynamic simulation and binding free energy calculations of the MurF ligase from Acinetobacter baumannii. J. Mol. Graph. Model. 2017, 77, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, K.G.; Jaeger, V.W.; Pfaendtner, J. The general AMBER force field (GAFF) can accurately predict thermodynamic and transport properties of many ionic liquids. J. Phys. Chem. B 2015, 119, 5882–5895. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Kuhn, O.; Mikulskis, P.; Hoffmann, D.; Ryde, U. The normal-mode entropy in the MM/GBSA method: Effect of system truncation, buffer region, and dielectric constant. J. Chem. Inf. Model. 2012, 52, 2079–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Compound | Interactive Amino Acids |
|---|---|---|
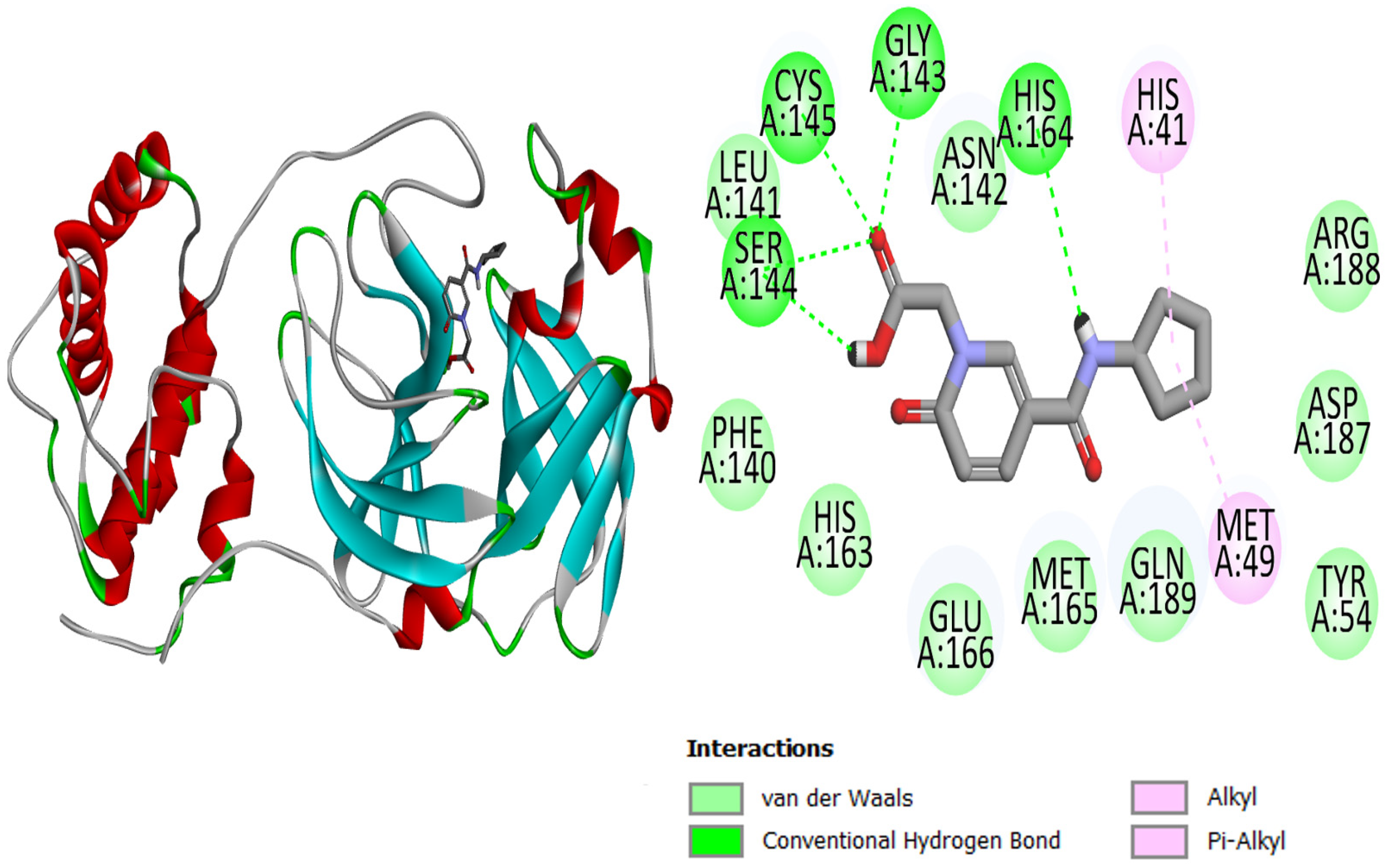
| 1 | Top-1 | His41, Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, Met149, His163, Met165, Glu166, Asp187, Arg188, Gln189 |
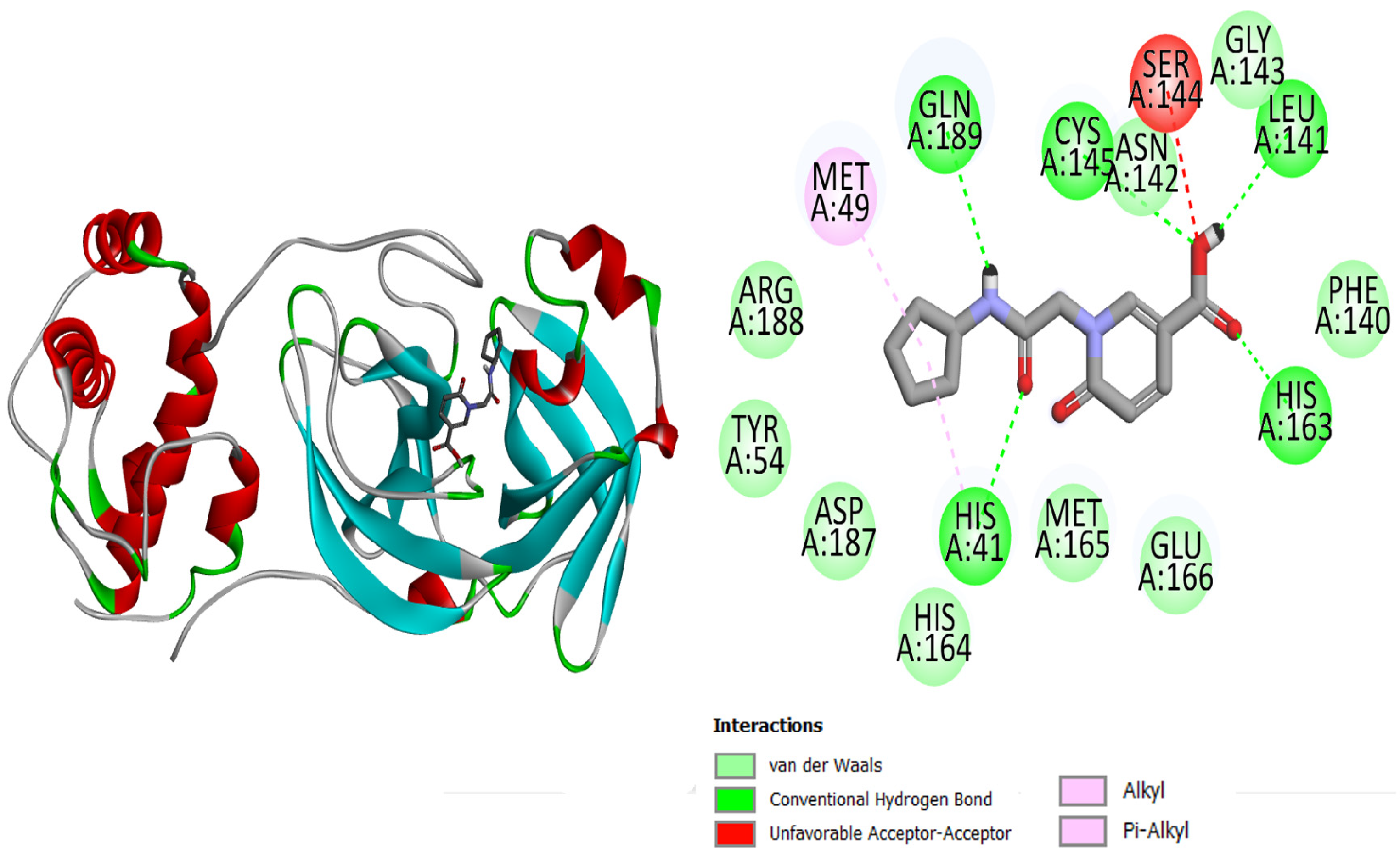
| 2 | Top-2 | His41, Tyr54, Leu141, Asn142, Gly143, Ser144, Cys145, His163, His164, Met165, Glu166, Asp187, Arg188 |
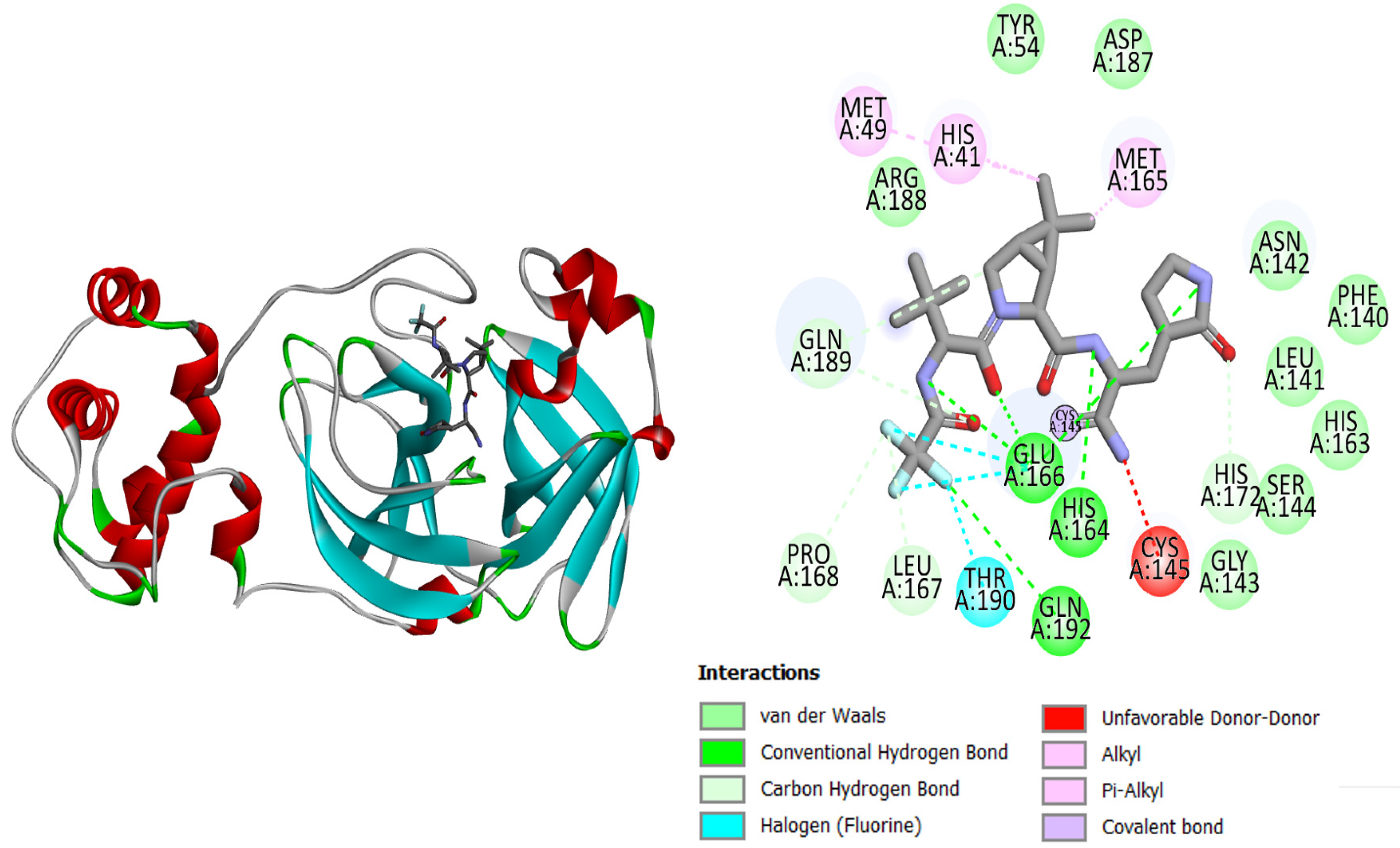
| 3 | Top-3 | Met49, Tyr54, Phe140, Gly143, Ser144, Cys145, Met165, His164, His172, Asp187, Gln189 |
| 4 | Control (N3) | Leu14, Thr24, Thr26, Tyr54, Phe140, Asn142, Gly143, Ser144, Cys145, His163, His164, Glu166, His172, Gln189, Thr190, Gln192 |
| S. No | Compounds | Structure | Binding Energy Score (kcal/mol) |
|---|---|---|---|
| 1 | BBB_26580140 (top-1) |  | −7.2 |
| 2 | BBB_26580153 (top-2) |  | −7 |
| 3 | BBB_26580155 (top-3) |  | −7 |
| 4 | BBB_26580162 (top-4) |  | −6.9 |
| 5 | BBB_265801565 (top-5) |  | −6.8 |
| 6 | BBB_26580166 (top-6) |  | −6.3 |
| 7 | BBB_26580172 (top-7) |  | −6 |
| 8 | BBB_26580189 (top-8) |  | −5.6 |
| 9 | BBB_26580191 (top-9) |  | −5.2 |
| 10 | BBB_29843330 (top-10) |  | −5.1 |
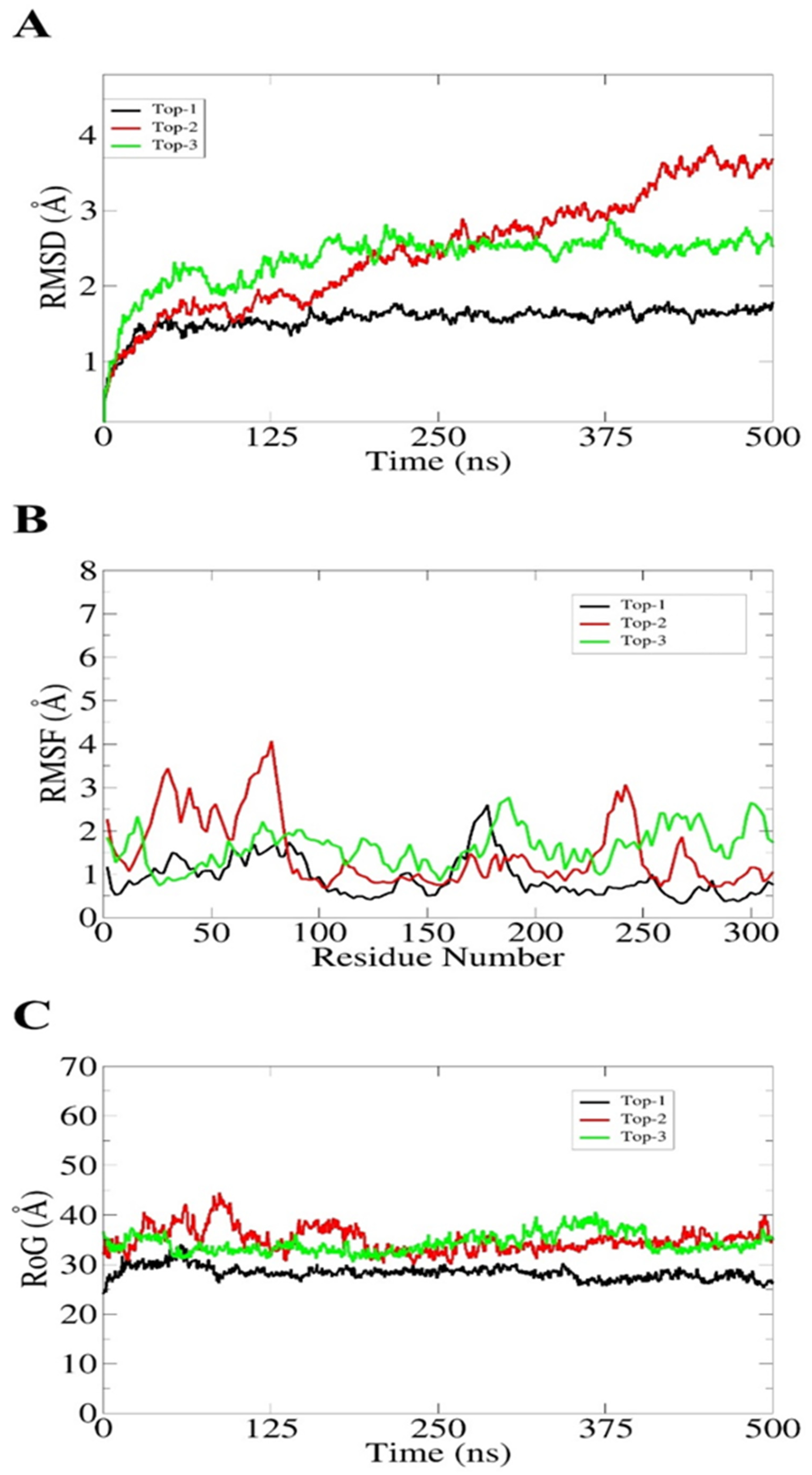
| Energy Parameter | Top-1 Complex | Top-2 Complex | Top-3 Complex |
|---|---|---|---|
| MM-GBSA | |||
| VDWAALS | −27.11 | −22.66 | −24.69 |
| EEL | −15.90 | −15.85 | −12.97 |
| EGB | 22.77 | 15.62 | 18.64 |
| ESURF | −3.99 | −3.49 | −5.98 |
| Delta G gas | −43.01 | −38.51 | −37.66 |
| Delta G solv | 18.78 | 12.13 | 12.66 |
| Delta total | −24.23 | −26.38 | −25 |
| MM-PBSA | |||
| VDWAALS | −27.11 | −22.66 | −24.69 |
| EEL | −15.90 | −15.85 | −12.97 |
| EPB | 29.11 | 18.12 | 16.44 |
| ENPOLAR | −3.33 | −4.36 | −3.64 |
| Delta G gas | −43.01 | −38.51 | −37.66 |
| Delta G solv | 25.78 | 13.76 | 12.8 |
| Delta total | −17.23 | −24.75 | −24.86 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miandad, K.; Ullah, A.; Bashir, K.; Khan, S.; Abideen, S.A.; Shaker, B.; Alharbi, M.; Alshammari, A.; Ali, M.; Haleem, A.; et al. Virtual Screening of Artemisia annua Phytochemicals as Potential Inhibitors of SARS-CoV-2 Main Protease Enzyme. Molecules 2022, 27, 8103. https://doi.org/10.3390/molecules27228103
Miandad K, Ullah A, Bashir K, Khan S, Abideen SA, Shaker B, Alharbi M, Alshammari A, Ali M, Haleem A, et al. Virtual Screening of Artemisia annua Phytochemicals as Potential Inhibitors of SARS-CoV-2 Main Protease Enzyme. Molecules. 2022; 27(22):8103. https://doi.org/10.3390/molecules27228103
Chicago/Turabian StyleMiandad, Khalid, Asad Ullah, Kashif Bashir, Saifullah Khan, Syed Ainul Abideen, Bilal Shaker, Metab Alharbi, Abdulrahman Alshammari, Mahwish Ali, Abdul Haleem, and et al. 2022. "Virtual Screening of Artemisia annua Phytochemicals as Potential Inhibitors of SARS-CoV-2 Main Protease Enzyme" Molecules 27, no. 22: 8103. https://doi.org/10.3390/molecules27228103