
Synthesis, In Vitro Biological Evaluation and In Silico Molecular Docking Studies of Indole Based Thiadiazole Derivatives as Dual Inhibitor of Acetylcholinesterase and Butyrylchloinesterase

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Result and Discussion
2.1. Chemistry
2.2. Spectral Analysis
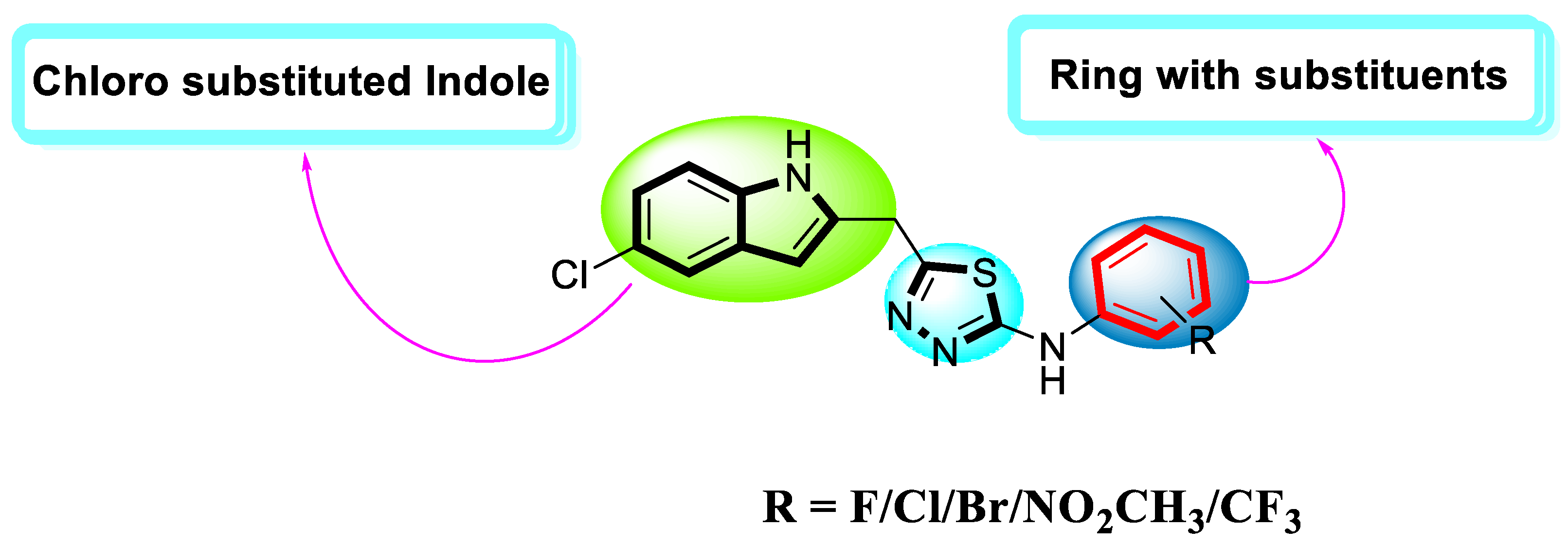
2.3. An Illustration of a Molecule
3. Bio-Activity of Indole Based Thiadiazole Derivatives
Inhibitory Profile of Synthesized Compound against AchE and BuChE
4. Molecular Docking
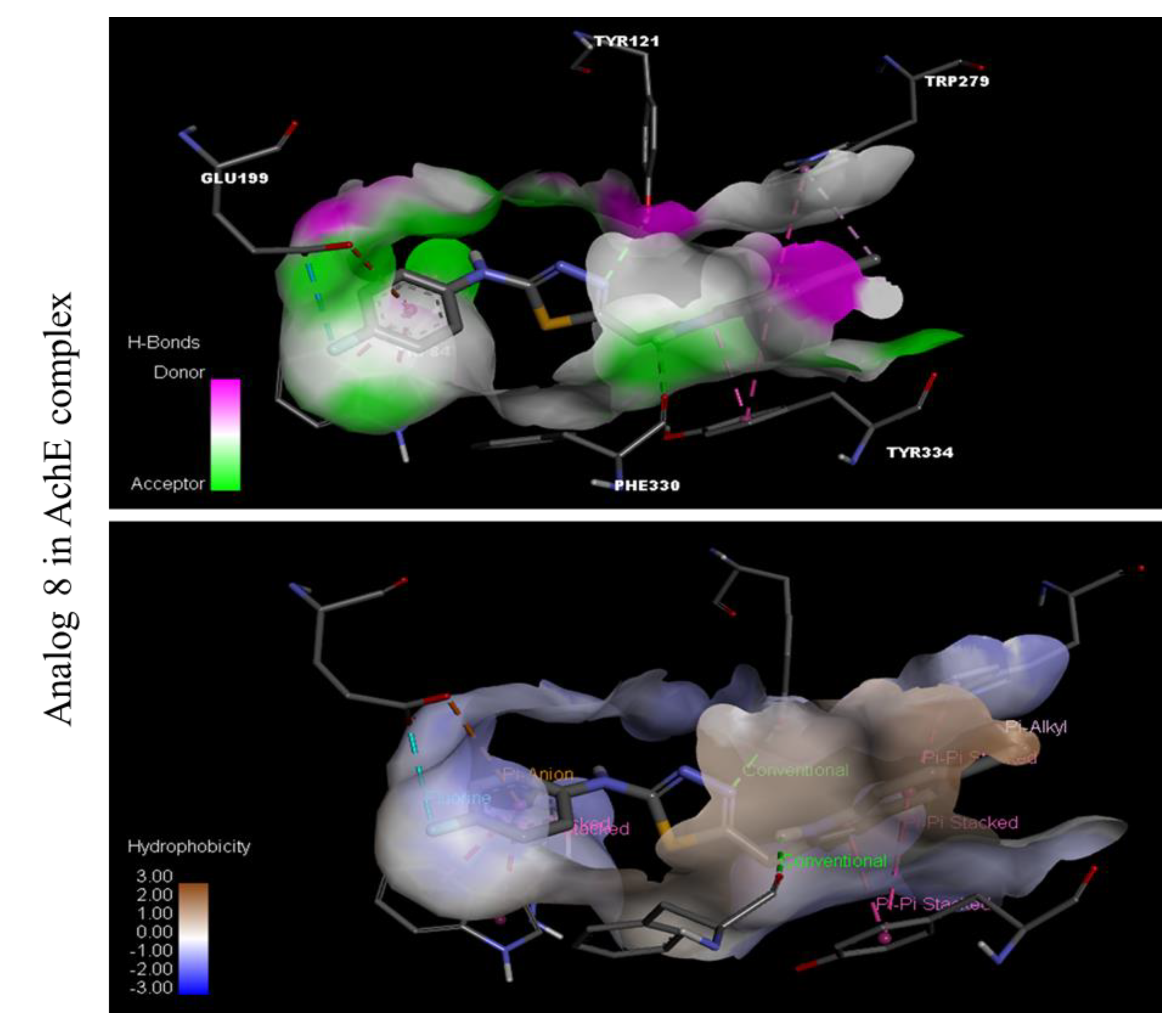
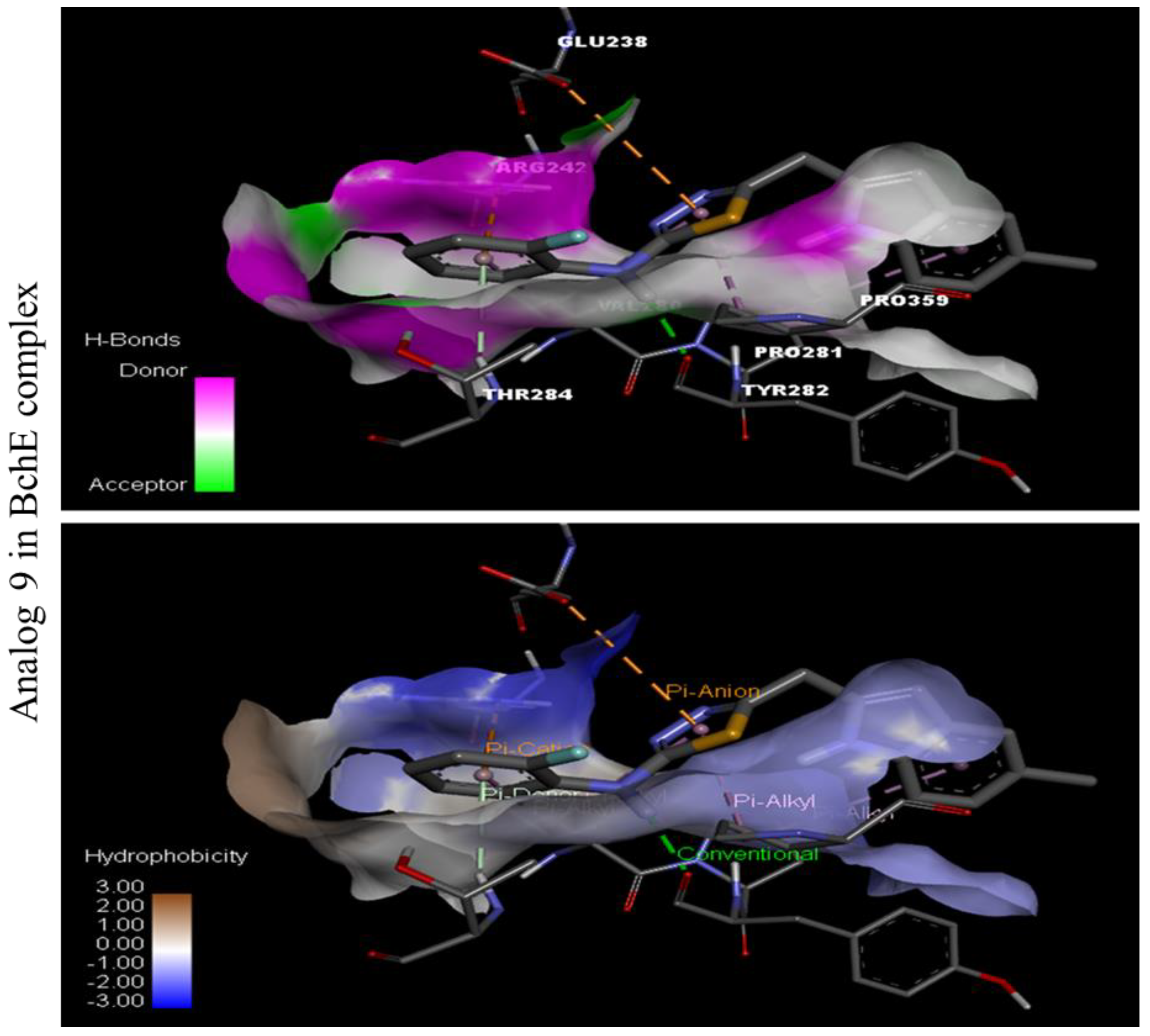
Docking Results
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ahmad, S.; Iftikhar, F.; Ullah, F.; Sadiq, A.; Rashid, U. Rational design and synthesis of dihydropyrimidine based dual binding site acetylcholinesterase inhibitors. Bioorg. Chem. 2016, 69, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Auld, D.S.; Kornecook, T.J.; Bastianetto, S.; Quirion, R. Alzheimer’s disease and the basal forebrain cholinergic system: Relations to β-amyloid peptides, cognition, and treatment strategies. Prog. Neurobiol. 2002, 68, 209–245. [Google Scholar] [CrossRef]
- Adams, R.L.; Craig, P.L.; Parsons, O.A. Neuropsychology of dementia. Neurol. Clin. 1986, 4, 387–404. [Google Scholar] [CrossRef]
- Aisen, P.S.; Davis, K.L. The search for disease-modifying treatment for Alzheimer’s disease. Neurology 1997, 48, 35–41. [Google Scholar] [CrossRef]
- Jann, M.W. Preclinical pharmacology of metrifonate. Pharmacotherapy 1998, 18, 55–67. [Google Scholar] [PubMed]
- Massoulié, J.; Pezzementi, L.; Bon, S.; Krejci, E.; Vallette, F.M. Molecular and cellular biology of cholinesterases. Prog. Neurobiol. 1993, 41, 31–91. [Google Scholar] [CrossRef]
- Mushtaq, G.; Greig, N.H.; AKhan, J.; Kamal, M.A. Status of acetylcholinesterase and butyrylcholinesterase in Alzheimer’s disease and type 2 diabetes mellitus. CNS Neurol. Disord. Drug Targets 2014, 13, 1432–1439. [Google Scholar] [CrossRef] [PubMed]
- Ecobichon, D.J.; Comeau, A.M. Pseudocholinesterases of mammalian plasma: Physicochemical properties and organophosphate inhibition in eleven species. Toxicol. Appl. Pharmacol. 1973, 24, 92–100. [Google Scholar] [CrossRef]
- Rahim, F.; Ullah, H.; Taha, M.; Wadood, A.; Javed, M.T.; Rehman, W.; Nawaz, M.; Ashraf, M.; Ali, M.; Sajid, M.; et al. Synthesis and in vitro acetylcholinesterase and butyrylcholinesterase inhibitory potential of hydrazide-based Schiff bases. Bioorg. Chem. 2016, 68, 30–40. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef] [PubMed]
- Rockwood, K.; Mintzer, J.; Truyen, L.; Wessel, T.; Wilkinson, D. Effects of a flexible galantamine dose in Alzheimer’s disease: A randomised, controlled trial. J. Neurol. Neurosurg. Psychiatry 2001, 71, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesulam, M.; Guillozet, A.; Shaw, P.; Quinn, B. Widely spread butyrylcholinesterase can hydrolyze acetylcholine in the normal and Alzheimer brain. Neurobiol. Dis. 2002, 9, 88–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greig, N.H.; Utsuki, T.; Yu, Q.S.; Zhu, X.; Holloway, H.W.; Perry, T.; Lee, B.; Ingram, D.K.; Lahiri, D.K. A new therapeutic target in Alzheimer’s disease treatment: Attention to butyrylcholinesterase. Curr. Med. Res. Opin. 2001, 17, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Gabr, M.T.; Abdel-Raziq, M.S. Design and synthesis of donepezil analogues as dual AChE and BACE-1 inhibitors. Bioorg. Chem. 2018, 80, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Small, G.W.; Rabins, P.V.; Barry, P.P.; Buckholtz, N.S.; DeKosky, S.T.; Ferris, S.H.; Finkel, S.I.; Gwyther, L.P.; Khachaturian, Z.S.; Lebowitz, B.D.; et al. Diagnosis and treatment of Alzheimer disease and related disorders: Consensus statement of the American Association for Geriatric Psychiatry, the Alzheimer’s Association, and the American Geriatrics Society. J. Am. Med. Assoc. 1997, 278, 1363–1371. [Google Scholar] [CrossRef]
- Melzer, D. New drug treatment for Alzheimer’s disease: Lessons for healthcare policy. Brit. Med. J. 1998, 316, 762–764. [Google Scholar] [CrossRef]
- Sharma, V.; Kumar, P.; Pathak, D. Biological importance of the indole nucleus in recent years: A comprehensive review. J. Heter. Chem. 2010, 47, 491–502. [Google Scholar] [CrossRef]
- MacDonough, M.T.; Strecker, T.E.; Hamel, E.; Hall, J.J.; Chaplin, D.J.; Trawick, M.L.; Pinney, K.G. Synthesis and biological evaluation of indole-based, anti-cancer agents inspired by the vascular disrupting agent 2-(3′-hydroxy-4′-methoxyphenyl)-3- (3″,4″,5″-trimethoxybenzoyl)-6-methoxyindole (OXi8006). Bioorg. Med. Chem. 2013, 21, 6831–6843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, T.; Inoue, H.; Horigome, M.; Momose, K.; Sugita, M.; Katsuyama, K.; Suzuki, C.; Nagai, S.; Nagase, M.; Nakamaru, K. Nisshin Seifun Group Inc. Indole derivatives and anti-ulcer compositions thereof. U.S. Patent 5,252,580, 26 May 1993. [Google Scholar]
- Agarwal, A.; Srivastava, K.; Puri, S.K.; Chauhan, P.M. Synthesis of substituted indole derivatives as a new class of antimalarial agents. Bioorg. Med. Chem. Lett. 2005, 15, 3133–3136. [Google Scholar] [CrossRef]
- Park, M.K.; Rhee, Y.H.; Lee, H.J.; Lee, E.O.; Kim, K.H.; Park, M.J.; Jeon, B.H.; Shim, B.S.; Jung, C.H.; Choi, S.H.; et al. Antiplatelet and antithrombotic activity of indole-3-carbinol in vitro and in vivo. Phytother. Res. 2008, 22, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Delorenzi, J.C.; Attias, M.; Gattass, C.R.; Andrade, M.; Rezende, C.; da Cunha Pinto, Â.; Henriques, A.T.; Bou-Habib, D.C.; Saraiva, E.M. Antileishmanial activity of an indole alkaloid Frompeschiera australis, Antimicrob. Agents Chemother. 2001, 45, 1349–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Süzen, S. Antioxidant activities of synthetic indole derivatives and possible activity mechanisms. In Bioactive Heterocycles; Springer: Berlin/Heidelberg, Germany, 2007; pp. 145–178. [Google Scholar]
- Berger, L.; Corraz, A.J. Hoffmann La Roche Inc, Cyclopenta [b] indole-2-carboxylic acids and derivatives thereof. U.S. Patent 4,009,181, 22 February 1977. [Google Scholar]
- Yamamoto, Y.; Kurazono, M. A new class of anti-MRSA and anti-VRE agents: Preparation and antibacterial activities of indole-containing compounds. Bioorg. Med. Chem. Lett. 2007, 17, 1626–1628. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Suzuki, M.; Xie, L.; Morris-Natschke, S.L.; Lee, K.H. Recent progress in the development of coumarin derivatives as potent anti-HIV agents. Med. Res. Rev. 2003, 23, 322–345. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.P.; Khanna, R.; Karmarkar, R.; Anwer, M.K.; Khar, R.K. Shilajit: A review. Phytother. Res. An Int. J. Devoted Pharmacol. Toxicol. Evalu. Natur. Prod.Derivat. 2007, 21, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Büyükbingöl, E.; Süzen, S.; Klopman, G. Studies on the synthesis and structure-activity relationships of 5-(3′-indolal)-2-thiohydantoin derivatives as aldose reductase enzyme inhibitors. Il Farmaco. 1994, 49, 443–447. [Google Scholar] [PubMed]
- Kharb, M.; Jat, R.K.; Parjapati, G.; Gupta, A. Introduction to molecular docking software technique in medicinal chemistry. Int. J. Drug Res. Technol. 2012, 2, 89–197. [Google Scholar]
- Li, Z.; Gu, J.; Zhuang, H.; Kang, L.; Zhao, X.; Guo, Q. Adaptive molecular docking method based on information entropy genetic algorithm. Appl. Soft Comput. 2015, 26, 299–302. [Google Scholar] [CrossRef]
- Rao, C.M.M.P.; Naidu, N.; Priya, J.; Rao, K.P.C.; Ranjith, K.; Shobha, S.; Chowdary, B.S.; Siddiraju, S. Molecular docking and dynamic simulations of benzimidazoles with beta-tubulins. Bioinformation 2021, 17, 404. [Google Scholar] [PubMed]
- Rahim, F.; Tariq, S.; Taha, M.; Ullah, H.; Zaman, K.; Zafar, S.; Shah, S.A.A. New triazinoindole bearing thiazole/oxazole analogues: Synthesis, α-amylase inhibitory potential and molecular docking study. Bioorg. Chem. 2019, 92, 103284. [Google Scholar] [CrossRef]
- Khan, S.; Ullah, H.; Rahim, F.; Nawaz, M.; Hussain, R.; Rasheed, L. Synthesis, in vitro α-amylase, α-glucosidase activities and molecular docking study of new benzimidazole bearing thiazolidinone derivatives. J. mol. Struct. 2022, 1269, 133812. [Google Scholar] [CrossRef]
- Zada, H.; Ullah, H.; Hayat, S.; Rahim, F.; Khan, F.; Wadood, A. Synthesis of triazinoindole bearing sulfonamide derivatives, in vitro α-amylase activity and their molecular docking study. Chem. Data Collect. 2022, 39, 100875. [Google Scholar] [CrossRef]
- Ghose, A.K.; Crippen, G.M. Atomic physicochemical parameters for three-dimensional structure-directed quantitative structure-activity relationships. 2. Modeling dispersive and hydrophobic interactions. J. Chem. Inf. Comput. Sci. 1987, 27, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Chigurupati, S.; Selvaraj, M.; Mani, V.; Selvarajan, K.K.; Mohammad, J.I.; Kaveti, B.; Bera, H.; Palaniuthu, V.R.; Teh, L.K.; Salleh, M.Z. Identification of novel acetylcholinesterase inhibitors: Indolopyrazoline derivatives and molecular docking studies. Bioorganic Chem. 2016, 67, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Chigurupati, S.; Selvaraj, M.; Mani, V.; Mohammad, J.I.; Selvarajan, K.K.; Akhtar, S.S.; Marikannan, M.; Raj, S.; Teh, L.K.; Salleh, M.Z. Syn thesis of azomethines derived from cinnamal dehyde and vanillin: In vitro acetylcholinesterase inhibitory, antioxidant and in silico molecular docking studies. Med. Chem. Res. 2018, 27, 807–816. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | R | AChE IC50 (µM ± SEM) | BuChE IC50 (µM ± SEM) |
|---|---|---|---|
| 1 |  | 2.10 ± 0.10 | 2.80 ± 0.20 |
| 2 |  | 1.90 ± 0.10 | 4.30 ± 0.10 |
| 3 |  | 2.90 ± 0.10 | 4.60 ± 0.10 |
| 4 |  | 2.80 ± 0.10 | 5.40 ± 0.10 |
| 5 |  | 5.80 ± 0.20 | 9.30 ± 0.10 |
| 6 |  | 8.70 ± 0.20 | 10.90 ± 0.10 |
| 7 |  | 9.60 ± 0.20 | 13.60 ± 0.30 |
| 8 |  | 0.15 ± 0.010 | 0.20 ± 0.10 |
| 9 |  | 0.35 ± 0.050 | 0.50 ± 0.050 |
| 10 |  | 1.10 ± 0.10 | 2.70 ± 0.10 |
| 11 |  | 12.30 ± 0.20 | 18.40 ± 0.30 |
| 12 |  | 14.70 ± 0.30 | 19.20 ± 0.30 |
| 13 |  | 19.10 ± 0.30 | 25.30 ± 0.40 |
| 14 |  | 0.40 ± 0.050 | 2.60 ± 0.10 |
| 15 |  | 22.20 ± 0.40 | 32.10 ± 0.10 |
| 16 |  | 36.160 ± 0.50 | 34.30 ± 0.60 |
| Standard Donepezil | 0.21 ± 0.12 | 0.30 ± 0.32 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, S.; Iqbal, S.; Taha, M.; Rahim, F.; Shah, M.; Ullah, H.; Bahadur, A.; Alrbyawi, H.; Dera, A.A.; Alahmdi, M.I.; et al. Synthesis, In Vitro Biological Evaluation and In Silico Molecular Docking Studies of Indole Based Thiadiazole Derivatives as Dual Inhibitor of Acetylcholinesterase and Butyrylchloinesterase. Molecules 2022, 27, 7368. https://doi.org/10.3390/molecules27217368
Khan S, Iqbal S, Taha M, Rahim F, Shah M, Ullah H, Bahadur A, Alrbyawi H, Dera AA, Alahmdi MI, et al. Synthesis, In Vitro Biological Evaluation and In Silico Molecular Docking Studies of Indole Based Thiadiazole Derivatives as Dual Inhibitor of Acetylcholinesterase and Butyrylchloinesterase. Molecules. 2022; 27(21):7368. https://doi.org/10.3390/molecules27217368
Chicago/Turabian StyleKhan, Shoaib, Shahid Iqbal, Muhammad Taha, Fazal Rahim, Mazloom Shah, Hayat Ullah, Ali Bahadur, Hamad Alrbyawi, Ayed A. Dera, Mohammed Issa Alahmdi, and et al. 2022. "Synthesis, In Vitro Biological Evaluation and In Silico Molecular Docking Studies of Indole Based Thiadiazole Derivatives as Dual Inhibitor of Acetylcholinesterase and Butyrylchloinesterase" Molecules 27, no. 21: 7368. https://doi.org/10.3390/molecules27217368