
Matrix Isolation FTIR and Theoretical Study of Weakly Bound Complexes of Isocyanic Acid with Nitrogen


Abstract
:1. Introduction
2. Results and Discussion
2.1. Computational Results
2.1.1. Structure and Energetics of the 1:1 HNCO⋯N2 Complexes
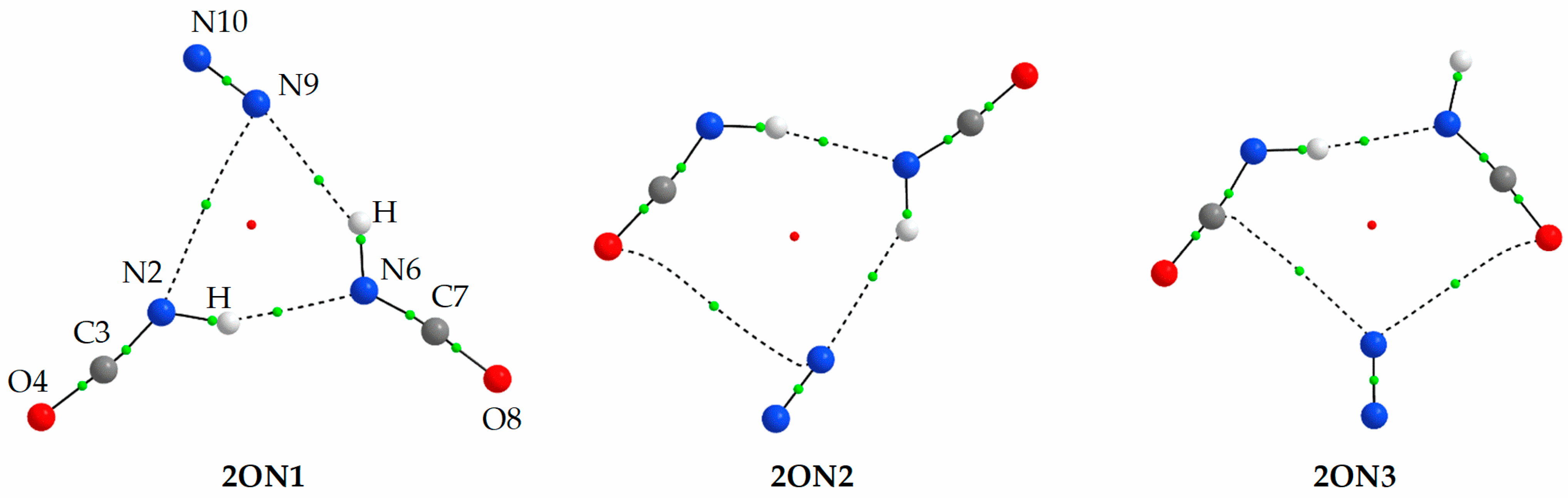
2.1.2. Structure and Energetics of the 1:2 and 2:1 HNCO⋯N2 Complexes
2.2. Matrix Isolation Infrared Spectra
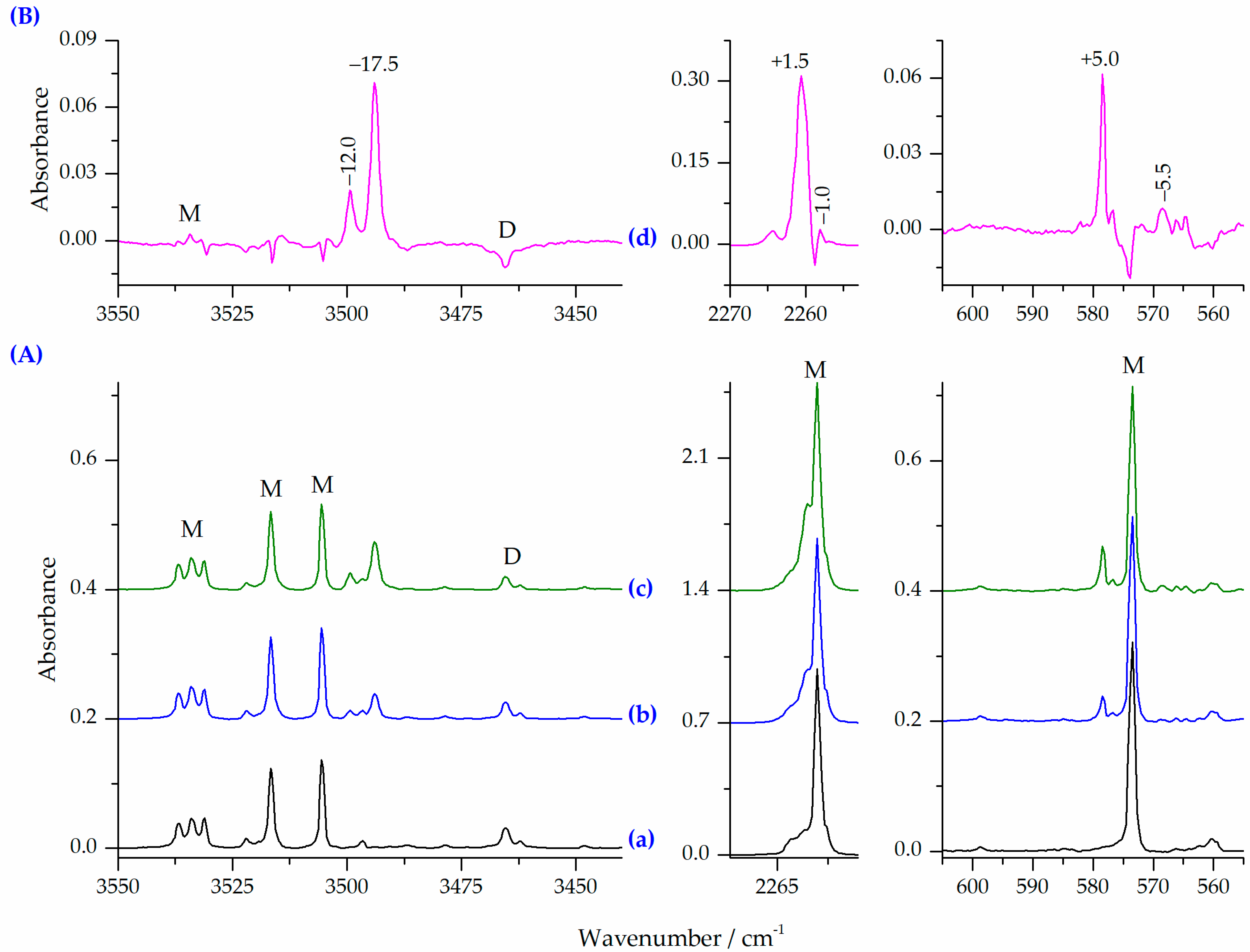
2.2.1. HNCO⋯N2 Complexes of the 1:1 Stoichiometry
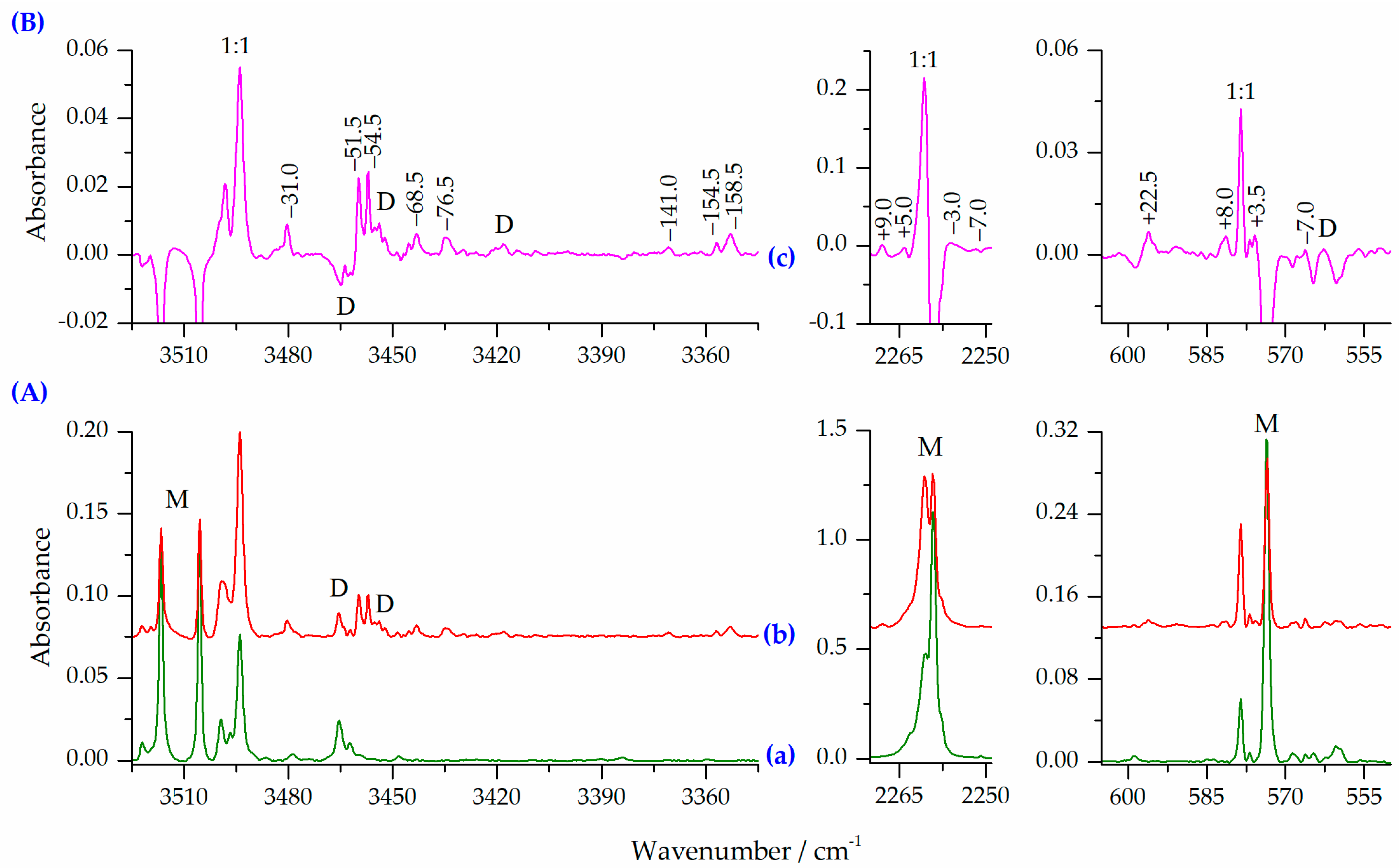
2.2.2. HNCO⋯N2 Complexes of the 1:2 and 2:1 Stoichiometry
3. Experimental and Computational Details
3.1. Matrix Isolation FTIR Studies
3.2. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Vigasin, A.A.; Slanina, Z. Molecular Complexes in Earth’s, Planetary, Cometary, and Interstellar Atmospheres; World Scientific Publishing: Singapore, 1998. [Google Scholar]
- Hobza, P.; Muller-Dethlefs, K. Non-Covalent Interactions: Theory and Experiment; The Royal Society of Chemistry: Cambridge, UK, 2009. [Google Scholar]
- Ritchie, G.L.D.; Watson, J.N.; Keir, R.I. Temperature dependence of electric field-gradient induced birefringence (Buckingham effect) and molecular quadrupole moment of N2. Comparison of experiment and theory. Chem. Phys. Lett. 2003, 370, 376–380. [Google Scholar] [CrossRef]
- Andrews, L.; Kelsall, B.J.; Arlinghaus, R.T. FTIR observation of the N2—HF complex in solid argon. J. Chem. Phys. 1983, 79, 2488–2490. [Google Scholar] [CrossRef]
- Andrews, L.; Davis, S.R. FTIR observation of N≡N stretching fundamentals in hydrogen-bonded complexes in solid argon. J. Chem. Phys. 1985, 83, 4983–4989. [Google Scholar] [CrossRef]
- Barnes, A.J.; Hallam, H.E.; Scrimshaw, G.F. Infra-red cryogenic studies. Part 3.—Hydrogen halides in doped argon matrices. Trans. Faraday Soc. 1969, 65, 3172–3178. [Google Scholar] [CrossRef]
- Coussan, S.; Loutellier, A.; Perchard, J.P.; Racine, S.; Bouteiller, Y. Matrix isolation infrared spectroscopy and DFT calculations of complexes between water and nitrogen. J. Mol. Struct. 1998, 471, 37–47. [Google Scholar] [CrossRef]
- Hirabayashi, S.; Ohno, K.; Abe, H.; Yamada, K.M.T. Infrared spectra of the water-nitrogen complexes (H2O)2−(N2)n(n = 1–4) in argon matrices. J. Chem. Phys. 2005, 122, 194506. [Google Scholar] [CrossRef] [PubMed]
- Murto, J.; Ovaska, M. Matrix infrared study of the specific interactions between methanol and nitrogen, and methanol and carbon monoxide. Spectrochim. Acta Part A Mol. Spectrosc. 1983, 39, 149–152. [Google Scholar] [CrossRef]
- Oswald, S.; Wallrabe, M.; Suhm, M.A. Cooperativity in Alcohol–Nitrogen Complexes: Understanding Cryomatrices through Slit Jet Expansions. J. Phys. Chem. A 2017, 121, 3411–3422. [Google Scholar] [CrossRef] [Green Version]
- Lundell, J.; Räsänen, M.; Latajka, Z. Matrix isolation FTIR and ab initio study of complexes between formic acid and nitrogen. Chem. Phys. 1994, 189, 245–260. [Google Scholar] [CrossRef]
- Marushkevich, K.; Räsänen, M.; Khriachtchev, L. Interaction of Formic Acid with Nitrogen: Stabilization of the Higher-Energy Conformer. J. Phys. Chem. A 2010, 114, 10584–10589. [Google Scholar] [CrossRef]
- Mielke, Z.; Latajka, Z.; Kołodziej, J.; Tokhadze, K.G. Matrix Infrared Spectra and ab Initio Calculations of the Nitrous Acid Complexes with N2 and CO. J. Phys. Chem. 1996, 100, 11610–11615. [Google Scholar] [CrossRef]
- Barnes, A.J.; Lasson, E.; Nielsen, C.J. Molecular complexes of nitric acid with N2, CO and NO studied by matrix isolation IR spectroscopy. J. Chem. Soc. Faraday Trans. 1995, 91, 3111–3116. [Google Scholar] [CrossRef]
- Wierzejewska-Hnat, M.; Latajka, Z.; Mielke, Z.; Ratajczak, H. Theoretical and infrared matrix isolation studies of the CF3COOH—N2 system. J. Mol. Struct. 1985, 129, 229–235. [Google Scholar] [CrossRef]
- Givan, A.; Loewenschuss, A.; Nielsen, C.J. IR spectrum of molecular complexes of sulfuric acid with N2 and NO trapped in solid argon. Phys. Chem. Chem. Phys. 1999, 1, 37–43. [Google Scholar] [CrossRef]
- Wierzejewska, M.; Wieczorek, R. Infrared matrix isolation and ab initio studies on isothiocyanic acid HNCS and its complexes with nitrogen and xenon. Chem. Phys. 2003, 287, 169–181. [Google Scholar] [CrossRef]
- Golec, B.; Grzegorzek, J.; Mielke, Z. Complexation of formaldoxime and acetaldoxime with nitrogen. Chem. Phys. 2008, 353, 13–18. [Google Scholar] [CrossRef]
- Sałdyka, M.; Mielke, Z. The interaction of formohydroxamic acid with nitrogen: FTIR matrix isolation and ab initio studies. J. Mol. Struct. 2004, 708, 183–188. [Google Scholar] [CrossRef]
- Sałdyka, M.; Mielke, Z.; Mierzwicki, K.; Coussan, S.; Roubin, P. CH stretching vibration of N-methylformamide as a sensitive probe of its complexation: Infrared matrix isolation and computational study. Phys. Chem. Chem. Phys. 2011, 13, 13992–14002. [Google Scholar] [CrossRef]
- Cao, Q.; Andrijchenko, N.; Ahola, A.-E.; Domanskaya, A.; Räsänen, M.; Ermilov, A.; Nemukhin, A.; Khriachtchev, L. Interaction of phenol with xenon and nitrogen: Spectroscopic and computational characterization. J. Chem. Phys. 2012, 137, 134305. [Google Scholar] [CrossRef]
- Oswald, S.; Suhm, M.A.; Coussan, S. Incremental NH stretching downshift through stepwise nitrogen complexation of pyrrole: A combined jet expansion and matrix isolation study. Phys. Chem. Chem. Phys. 2019, 21, 1277–1284. [Google Scholar] [CrossRef] [Green Version]
- Sałdyka, M.; Mielke, Z.; Haupa, K. Structural and spectroscopic characterization of DMF complexes with nitrogen, carbon dioxide, ammonia and water. Infrared matrix isolation and theoretical studies. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 190, 423–432. [Google Scholar] [CrossRef]
- Oswald, S.; Coussan, S. Chloroform–nitrogen aggregates: Upshifted CH and downshifted CCl stretching vibrations observed by matrix isolation and jet expansion infrared spectroscopy. Low Temp. Phys. 2019, 45, 639–648. [Google Scholar] [CrossRef]
- Banerjee, P.; Chakraborty, T. Confinement effects on C–H and C–F stretching vibrational frequencies of difluoromethane in cold inert gas matrixes: A combined infrared spectroscopy and electronic structure theory study. Eur. Phys. J. D 2021, 75, 131. [Google Scholar] [CrossRef]
- Khriachtchev, L.; Tapio, S.; Räsänen, M.; Domanskaya, A.; Lignell, A. HY⋯N2 and HXeY⋯N2 complexes in solid xenon (Y = Cl and Br): Unexpected suppression of the complex formation for deposition at higher temperature. J. Chem. Phys. 2010, 133, 084309. [Google Scholar] [CrossRef]
- Lignell, A.; Khriachtchev, L.; Pettersson, M.; Räsänen, M. Interaction of rare-gas-containing molecules with nitrogen: Matrix-isolation and ab initio study of HArF⋯N2, HKrF⋯N2, and HKrCl⋯N2 complexes. J. Chem. Phys. 2003, 118, 11120. [Google Scholar] [CrossRef]
- Lignell, A.; Khriachtchev, L.; Lignell, H.; Räsänen, M. Protons solvated in noble-gas matrices: Interaction with nitrogen. Phys. Chem. Chem. Phys. 2006, 8, 2457–2463. [Google Scholar] [CrossRef] [PubMed]
- Lalov, A.V.; Boganov, S.E.; Faustov, V.I.; Egorov, M.P.; Nefedov, O.M. Experimental and quantum-chemical study of complexation of carbene analogs with dinitrogen. Direct IR-spectroscopic observation of Cl2Si·N2 complexes in low-temperature argon-nitrogen matrices. Russ. Chem. Bull. 2003, 52, 526–538. [Google Scholar] [CrossRef]
- Maier, G.; Reisenauer, H.P.; Glatthaar, J.; Zetzmann, R. Complex of Silylene with Nitrogen: A Combined Matrix-Spectroscopic and Density Functional Theory Study. Chem. Asian J. 2006, 1, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Castaño, J.A.G.; Fantoni, A.; Romano, R.M. Matrix-isolation FTIR study of carbon dioxide: Reinvestigation of the CO2 dimer and CO2⋯N2 complex. J. Mol. Struct. 2008, 881, 68–75. [Google Scholar] [CrossRef]
- Leslie, M.D.; Ridoli, M.; Murphy, J.G.; Borduas-Dedekind, N. Isocyanic acid (HNCO) and its fate in the atmosphere: A review. Environ. Sci. Process. Impacts 2019, 21, 793–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borduas, N.; Murphy, J.G.; Wang, C.; da Silva, G.; Abbatt, J.P.D. Gas Phase Oxidation of Nicotine by OH Radicals: Kinetics, Mechanisms, and Formation of HNCO. Environ. Sci. Technol. Lett. 2016, 3, 327–331. [Google Scholar] [CrossRef]
- Roberts, J.M.; Veres, P.R.; Cochran, A.K.; Warneke, C.; Burling, I.R.; Yokelson, R.J.; Lerner, B.; Gilman, J.B.; Kuster, W.C.; Fall, R.; et al. Isocyanic acid in the atmosphere and its possible link to smoke-related health effects. Proc. Natl. Acad. Sci. USA 2011, 108, 8966–8971. [Google Scholar] [CrossRef] [Green Version]
- Jathar, S.H.; Heppding, C.; Link, M.F.; Farmer, D.K.; Akherati, A.; Kleeman, M.J.; de Gouw, J.A.; Veres, P.R.; Roberts, J.M. Investigating diesel engines as an atmospheric source of isocyanic acid in urban areas. Atmos. Chem. Phys. 2017, 17, 8959–8970. [Google Scholar] [CrossRef] [Green Version]
- Link, M.F.; Friedman, B.; Fulgham, R.; Brophy, P.; Galang, A.; Jathar, S.H.; Veres, P.; Roberts, J.M.; Farmer, D.K. Photochemical processing of diesel fuel emissions as a large secondary source of isocyanic acid (HNCO). Geophys. Res. Lett. 2016, 43, 4033–4041. [Google Scholar] [CrossRef]
- Suarez-Bertoa, R.; Astorga, C. Isocyanic acid and ammonia in vehicle emissions. Transp. Res. Part D Transp. Environ. 2016, 49, 259–270. [Google Scholar] [CrossRef]
- Barnes, I.; Solignac, G.; Mellouki, A.; Becker, K.H. Aspects of the Atmospheric Chemistry of Amides. Chem. Phys. Chem. 2010, 11, 3844–3857. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yuan, B.; Ye, C.; Roberts, J.M.; Wisthaler, A.; Lin, Y.; Li, T.; Wu, C.; Peng, Y.; Wang, C.; et al. High Concentrations of Atmospheric Isocyanic Acid (HNCO) Produced from Secondary Sources in China. Environ. Sci. Technol. 2020, 54, 11818–11826. [Google Scholar] [CrossRef]
- Kewley, R.; Sastry, K.V.L.N.; Winnewisser, M. The millimeter wave spectra of isocyanic and isothiocyanic acids. J. Mol. Spectrosc. 1963, 10, 418–441. [Google Scholar] [CrossRef]
- Jacox, M.E.; Milligan, D.E. Low-Temperature Infrared Study of Intermediates in the Photolysis of HNCO and DNCO. J. Chem. Phys. 1964, 40, 2457–2460. [Google Scholar] [CrossRef]
- Steiner, D.A.; Wishah, K.A.; Polo, S.R.; McCubbin, T.K. Infrared spectrum of isocyanic acid between 465 and 1100 cm−1. J. Mol. Spectrosc. 1979, 76, 341–373. [Google Scholar] [CrossRef]
- Drozdoski, W.S.; Baronavski, A.P.; McDonald, J.R. Photodissociation of HNCO at 193 nm. NH(a1Δ) internal energy distribution and reaction rate with O2. Chem. Phys. Lett. 1979, 64, 421–425. [Google Scholar] [CrossRef]
- Yamada, K. Molecular structure and centrifugal distortion constants of isocyanic acid from the microwave, millimeter wave, and far-infrared spectra. J. Mol. Spectrosc. 1980, 79, 323–344. [Google Scholar] [CrossRef]
- Bondybey, V.E.; English, J.H.; Mathews, C.W.; Contolini, R.J. Infrared spectra and isomerization of CHNO species in rare gas matrices. J. Mol. Spectrosc. 1982, 92, 431–442. [Google Scholar] [CrossRef]
- Fujimoto, G.T.; Umstead, M.E.; Lin, M.C. Dynamics of CO formation in the photodissociation of HNCO and CH2CO at 193nm. Chem. Phys. 1982, 65, 197–203. [Google Scholar] [CrossRef]
- Teles, J.H.; Maier, G.; Hess, B.A., Jr.; Schaad, L.J.; Winnewisser, M.; Winnewisser, B.P. The CHNO Isomers. Chem. Ber. 1989, 122, 753–766. [Google Scholar] [CrossRef]
- Ruscic, B.; Berkowitz, J. The H–NCO bond energy and ΔH0f (NCO) from photoionization mass spectrometric studies of HNCO and NCO. J. Chem. Phys. 1994, 100, 4498. [Google Scholar] [CrossRef]
- Droz-Georget, T.; Zyrianov, M.; Sanov, A.; Reisler, H. Photodissociation of HNCO: Three competing pathways. Ber. Der Bunsenges. Phys. Chem. 1997, 101, 469–477. [Google Scholar] [CrossRef]
- Pettersson, M.; Khriachtchev, L.; Jolkkonen, S.; Räsänen, M. Photochemistry of HNCO in Solid Xe: Channels of UV Photolysis and Creation of H2NCO Radicals. J. Phys. Chem. A 1999, 103, 9154–9162. [Google Scholar] [CrossRef]
- Pettersson, M.; Khriachtchev, L.; Lundell, J.; Jolkkonen, A.S.; Räsänen, M. Photochemistry of HNCO in Solid Xenon: Photoinduced and Thermally Activated Formation of HXeNCO. J. Phys. Chem. A 2000, 104, 3579–3583. [Google Scholar] [CrossRef]
- Yu, S.; Su, S.; Dai, D.; Yuan, K.; Yang, X. Vacuum Ultraviolet Photodissociation Dynamics of Isocyanic Acid: The Hydrogen Elimination Channel. J. Phys. Chem. A 2013, 117, 13564–13571. [Google Scholar] [CrossRef]
- Yu, S.; Su, S.; Dorenkamp, Y.; Wodtke, A.M.; Dai, N.; Yuan, K.; Yang, X. Competition between Direct and Indirect Dissociation Pathways in Ultraviolet Photodissociation of HNCO. J. Phys. Chem. A 2013, 117, 11673–11678. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, Z.; Huang, C.; Chen, Y.; Dai, N.; Parker, D.H.; Yang, X. Imaging the Pair-Correlated HNCO Photodissociation: The NH(a1Δ) + CO(X1Σ+) Channel. J. Phys. Chem. A 2014, 118, 2413–2418. [Google Scholar] [CrossRef]
- Holzmeier, F.; Wolf, T.J.A.; Gienger, C.; Wagner, I.; Bozek, J.; Nandi, S.; Nicolas, C.; Fischer, I.; Gühr, M.; Fink, R.F. Normal and resonant Auger spectroscopy of isocyanic acid, HNCO. J. Chem. Phys. 2018, 149, 034308. [Google Scholar] [CrossRef]
- Shapley, W.A.; Bacskay, G.B. A Gaussian-2 Quantum Chemical Study of CHNO: Isomerization and Molecular Dissociation Reactions. J. Phys. Chem. A 1999, 103, 6624–6631. [Google Scholar] [CrossRef]
- Mladenović, M.; Elhiyani, M.; Lewerenz, M. Electric and magnetic properties of the four most stable CHNO isomers fromab initioCCSD(T) studies. J. Chem. Phys. 2009, 131, 034302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keresztes, B.; Csonka, I.P.; Lajgút, G.G.; Bazsó, G.; Tarczay, G. Matrix isolation study on the 1:1 hydrogen-bonded complexes [H, C, N]⋯[H, C, N, O]. J. Mol. Struct. 2020, 1219, 128535. [Google Scholar] [CrossRef]
- Boyce, C.W.; Gillies, C.W.; Warner, H.; Gillies, J.Z.; Lovas, F.J.; Suenram, R.D. Microwave Spectra, Structure, and Electric Dipole Moment of the Ar-Isocyanic Acid van der Waals Complex. J. Mol. Spectrosc. 1995, 171, 533–545. [Google Scholar] [CrossRef]
- Sałdyka, M.; Mielke, Z. Photodecomposition of formohydroxamic acid. Matrix isolation FTIR and DFT studies. Phys. Chem. Chem. Phys. 2003, 5, 4790–4797. [Google Scholar] [CrossRef]
- Mullaney, J.C.; Medcraft, C.; Tew, D.P.; Lewis-Borrell, L.; Golding, B.T.; Walker, N.R.; Legon, A.C. Cooperative hydrogen bonds form a pseudocycle stabilizing an isolated complex of isocyanic acid with urea. Phys. Chem. Chem. Phys. 2017, 19, 25080–25085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sałdyka, M.; Mielke, Z. Photochemistry of Acetohydroxamic Acid in Solid Argon. FTIR and Theoretical Studies. J. Phys. Chem. A 2018, 122, 60–71. [Google Scholar] [CrossRef]
- Krupa, J.; Wierzejewska, M.; Lundell, J. Structure and IR Spectroscopic Properties of HNCO Complexes with SO2 Isolated in Solid Argon. Molecules 2021, 26, 6441. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Head-Gordon, T. Analytic MP2 frequencies without fifth-order storage. Theory and application to bifurcated hydrogen bonds in the water hexamer. Chem. Phys. Lett. 1994, 220, 122–128. [Google Scholar] [CrossRef]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. Semi-direct algorithms for the MP2 energy and gradient. Chem. Phys. Lett. 1990, 166, 281–289. [Google Scholar] [CrossRef]
- Sæbø, S.; Almlöf, J. Avoiding the integral storage bottleneck in LCAO calculations of electron correlation. Chem. Phys. Lett. 1989, 154, 83–89. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, T.; Grimme, S. Double-hybrid density functionals with long-range dispersion corrections: Higher accuracy and extended applicability. Phys. Chem. Chem. Phys. 2007, 9, 3397–3406. [Google Scholar] [CrossRef]
- Biczysko, M.; Panek, P.; Scalmani, G.; Bloino, J.; Barone, V. Harmonic and Anharmonic Vibrational Frequency Calculations with the Double-Hybrid B2PLYP Method: Analytic Second Derivatives and Benchmark Studies. J. Chem. Theory Comput. 2010, 6, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Davidson, E.R. Comment on “Comment on Dunning’s correlation-consistent basis sets”. Chem. Phys. Lett. 1996, 260, 514–518. [Google Scholar] [CrossRef]
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Grabowski, S.J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef] [PubMed]
- Cugley, J.A.; Pullin, A.D.E. Matrix isolated spectra of HNCO and HNCS in the far infrared. Chem. Phys. Lett. 1973, 19, 203–208. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Boys, S.F.; Bernardi, F. The Calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Simon, S.; Duran, M.; Dannenberg, J.J. How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers? J. Chem. Phys. 1996, 105, 11024–11031. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Keith, T.A. AIMAll, version 19.10.12; TK Gristmill Software; Overland Park, KS, USA, 2019. Available online: aim.tkgristmill.com (accessed on 8 January 2022).
- Barnes, A.J. Molecular complexes of the hydrogen halides studied by matrix isolation infrared spectroscopy. J. Mol. Struct. 1983, 100, 259–280. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Intermolecular Parameters 1 | AIM Parameters | ||||
|---|---|---|---|---|---|---|
| Interatomic Distances | Angle | BCP | ρ(r) | ∇2ρ(r) | ||
| H⋯Y | X⋯Y | X–H⋯Y | ||||
| ON1 | 4.372 | 6.258 | 170.7 | H1⋯N5 | 0.0106 | 0.0419 |
| ON2 | 6.187 | N2⋯N5 | 0.0048 | 0.0197 | ||
| ON3 | 5.992 | C3⋯N5 | 0.0051 | 0.0215 | ||
| Complex | MP2 | B2PLYPD3 | B3LYPD3 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Eint | ΔE | ΔG | A | TΔS 1 | Eint | ΔE | ΔG | A | TΔS | Eint | ΔE | ΔG | A | TΔS | |
| ON1 | −6.53 | 0.00 | 4.72 | 9 | −7.79 | −6.49 | 0.00 | 5.40 | 7 | −8.90 | −6.40 | 0.00 | 4.51 | 14 | −7.91 |
| ON2 | −3.10 | 3.40 | 0.00 | 61 | 0.00 | −2.59 | 3.86 | 0.00 | 63 | 0.00 | −2.68 | 3.72 | 0.00 | 86 | 0.00 |
| ON3 | −4.52 | 2.00 | 1.76 | 30 | −3.11 | −4.31 | 2.21 | 1.87 | 30 | −3.44 | |||||
| Complex | MP2 | B2PLYPD3 | B3LYPD3 | |||
|---|---|---|---|---|---|---|
| Eint | ΔE | Eint | ΔE | Eint | ΔE | |
| O2N1 | −11.67 | 0.00 | −11.13 | 0.00 | −11.34 | 0.00 |
| O2N2 | −11.00 | 0.69 | −10.17 | 0.98 | −10.04 | 1.26 |
| O2N3 | −9.58 | 2.10 | −9.04 | 2.08 | −9.04 | 2.27 |
| O2N4 | −9.25 | 2.47 | ||||
| Complex | MP2 | B2PLYPD3 | B3LYPD3 | |||
|---|---|---|---|---|---|---|
| Eint | ΔE | Eint | ΔE | Eint | ΔE | |
| 2ON1 | −24.77 | 0.00 | −24.60 | 0.00 | −24.85 | 0.00 |
| 2ON2 | −23.81 | 1.09 | −23.97 | 0.71 | −24.81 | 0.19 |
| 2ON3 | −22.89 | 2.02 | −22.64 | 2.07 | −23.56 | 1.43 |
| 2ON4 | −22.13 | 2.62 | −22.72 | 1.86 | −24.31 | 0.63 |
| 2ON5 | −21.17 | 3.88 | −22.13 | 2.70 | −22.84 | 2.26 |
| 2ON6 | −20.38 | 4.31 | −20.67 | 3.83 | −21.80 | 2.96 |
| 2ON7 | −19.50 | 5.19 | −19.66 | 4.84 | −20.88 | 3.89 |
| 2ON8 | −18.37 | 6.32 | −18.70 | 5.79 | −20.84 | 3.97 |
| 2ON9 | −18.28 | 6.43 | −18.62 | 5.90 | −19.75 | 5.04 |
| 2ON10 | −16.48 | 8.18 | −17.03 | 7.44 | −18.74 | 6.06 |
| MP2 | B2PLYPD3 | B3LYPD3 | Mode | Exp.1 | |||||
|---|---|---|---|---|---|---|---|---|---|
| ON1 | ON2 | ON3 | ON1 | ON2 | ON3 | ON1 | ON2 | ||
| −25 (405) | −6 (169) | −9 (163) | −25 (401) | −5 (155) | −8 (149) | −23 (392) | −4 (155) | νNH | −17.5, −12.0 |
| +2 (706) | −2 (631) | −2 (636) | +2 (732) | −1 (654) | −2 (654) | +2 (798) | 0 (712) | νasNCO | +1.5, −1.0 |
| +21 (76) | −3 (83) | −6 (97) | +22 (63) | −3 (72) | −4 (81) | +20 (67) | −3 (75) | δNCO | +5.0, −5.5 |
| MP2 | B2PLYPD3 | B3LYPD3 | Mode | Exp.1 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| O2N1 | O2N2 | O2N3 | O2N4 | O2N1 | O2N2 | O2N3 | O2N1 | O2N2 | O2N3 | ||
| −35 (380) | −33 (398) | −31 (397) | −11 (159) | −32 (376) | −31 (391) | −28 (394) | −29 (364) | −23 (345) | −26 (386) | νNH | −31.0 |
| 0 (662) | 0 (740) | 0 (661) | −1 (604) | 0 (687) | 0 (769) | 0 (688) | 0 (748) | 0 (837) | +1 (751) | νasNCO | |
| +16 (73) | +22 (81) | +19 (69) | −6 (96) | +17 (61) | +21 (68) | +19 (58) | +16 (66) | +16 (78) | +17 (62) | δNCO | +22.5 |
| MP2 | B2PLYPD3 | B3LYPD3 | Mode | Exp.1 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2ON1 | 2ON2 | 2ON3 | 2ON1 | 2ON2 | 2ON3 | 2ON1 | 2ON2 | 2ON3 | ||
| −64 (361) | −61 (340) | −39 (177) | −64 (367) | −60 (339) | −36 (163) | −60 (344) | −53 (315) | −34 (163) | νNH | −51.5, −54.5, −68.5, −76,5 |
| −145 (769) | −150 (770) | −153 (784) | −154 (780) | −157 (762) | −158 (780) | −170 (830) | −174 (810) | −171 (808) | −141.0, −154.5, −158.5 | |
| +5 (486) | +5 (21) | +5 (697) | +7 (532) | +6 (32) | +7 (724) | +9 (635) | +8 (43) | +9 (799) | νasNCO | +9.0, +5.0 |
| −6 (1090) | −7 (1553) | −9 (650) | −7 (1103) | −8 (1599) | −10 (679) | −7 (1145) | −8 (1738) | −9 (734) | −3.0, −7.0 | |
| +66 (0) | +63 (37) | +61 (22) | +61 (1) | +62 (2) | +57 (2) | +64 (2) | +65 (3) | +61 (4) | δNCO | n.o. |
| +4 (187) | +11 (26) | −5 (72) | +9 (145) | +13 (20) | −3 (60) | +10 (137) | +14 (22) | −1 (60) | +8.0, +2.5, −7.0 | |
| Proton Donor HA | Relative Shifts (%) 1 | Reference | Proton Donor HA | Relative Shifts (%) | Reference |
|---|---|---|---|---|---|
| HF | 0.99 | [4] | HNCO | 0.50 | This work |
| HNO3 | 0.97 | [14] | HNCS | 0.46 | [17] |
| CF3COOH | 0.91 | [15] | HONO-trans | 0.34 | [13] |
| H2SO4 | 0.56 | [16] | HCl | 0.27 | [88] |
| HCOOH | 0.53 | [11] | CH3OH | 0.19 | [9] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krupa, J.; Wierzejewska, M.; Lundell, J. Matrix Isolation FTIR and Theoretical Study of Weakly Bound Complexes of Isocyanic Acid with Nitrogen. Molecules 2022, 27, 495. https://doi.org/10.3390/molecules27020495
Krupa J, Wierzejewska M, Lundell J. Matrix Isolation FTIR and Theoretical Study of Weakly Bound Complexes of Isocyanic Acid with Nitrogen. Molecules. 2022; 27(2):495. https://doi.org/10.3390/molecules27020495
Chicago/Turabian StyleKrupa, Justyna, Maria Wierzejewska, and Jan Lundell. 2022. "Matrix Isolation FTIR and Theoretical Study of Weakly Bound Complexes of Isocyanic Acid with Nitrogen" Molecules 27, no. 2: 495. https://doi.org/10.3390/molecules27020495