
The Inhibitory Potential of 2′-dihalo Ribonucleotides against HCV: Molecular Docking, Molecular Simulations, MM-BPSA, and DFT Studies



Abstract
:1. Introduction
2. Results and Discussion
2.1. Docking Studies
2.1.1. Docking Protocol Validation
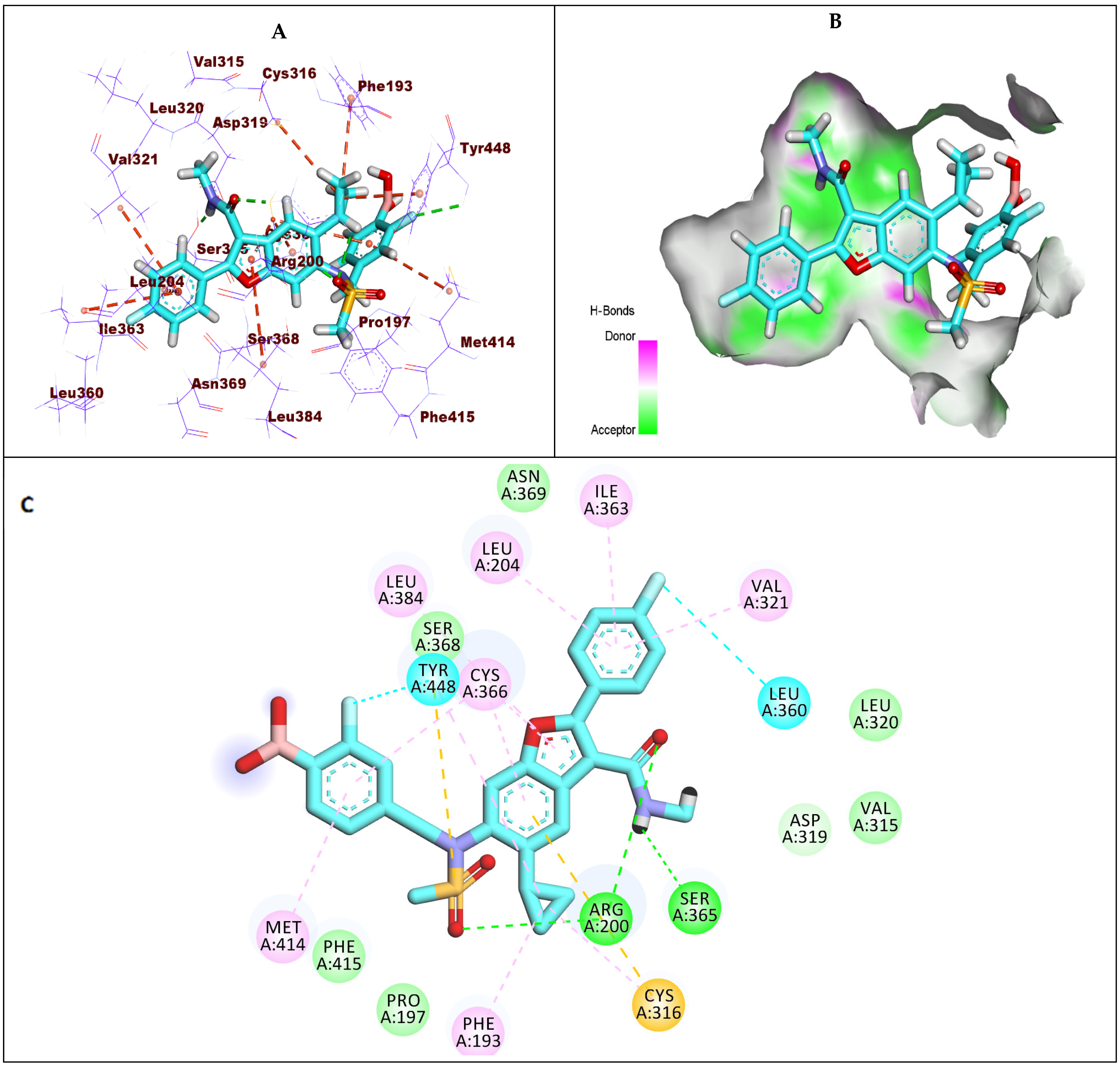
2.1.2. Docking Studies against GT1a
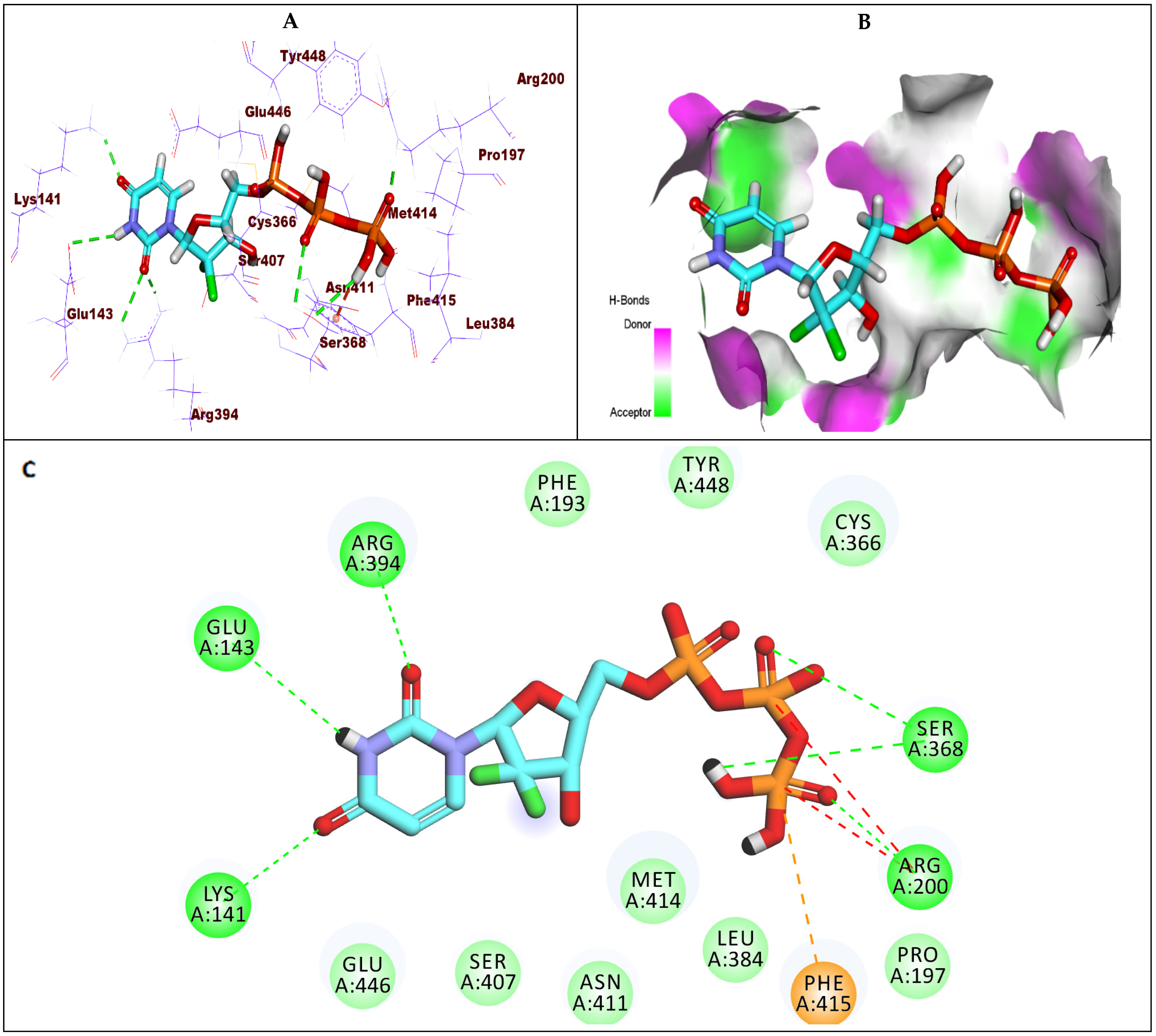
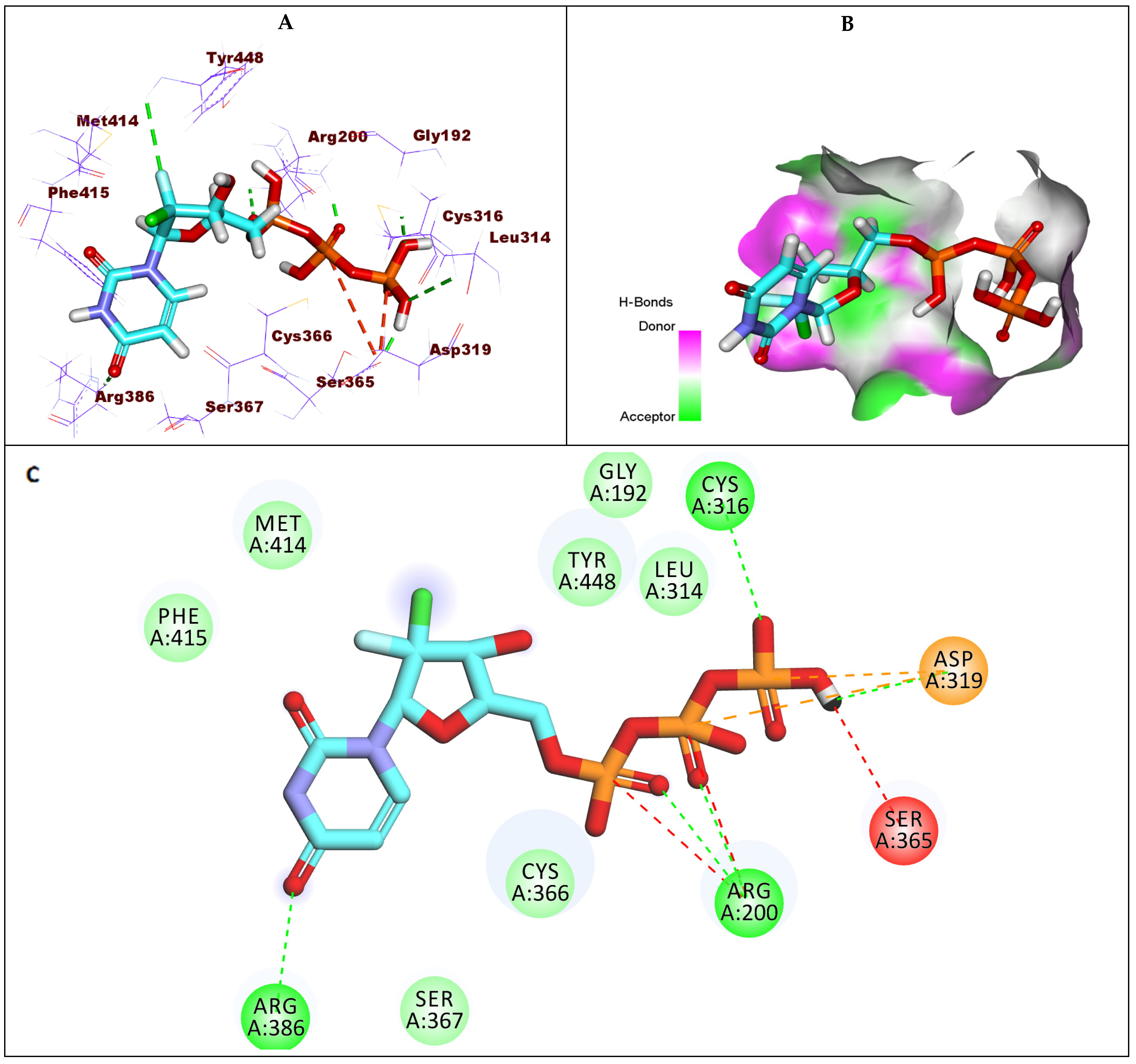
2.1.3. Docking Studies against GT1b
2.2. In Silico ADMET Analysis
2.3. Toxicity Studies
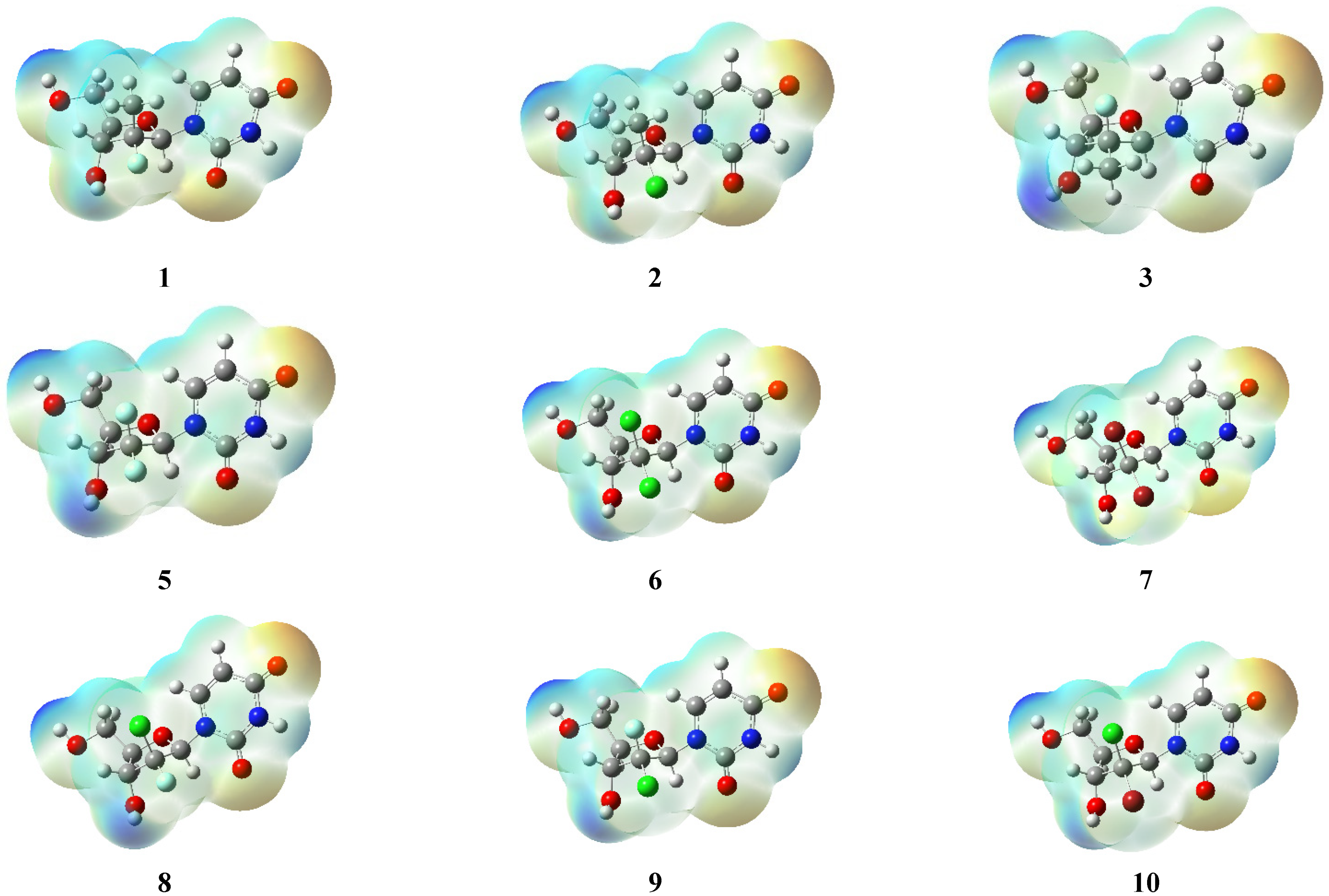
2.4. DFT Studies
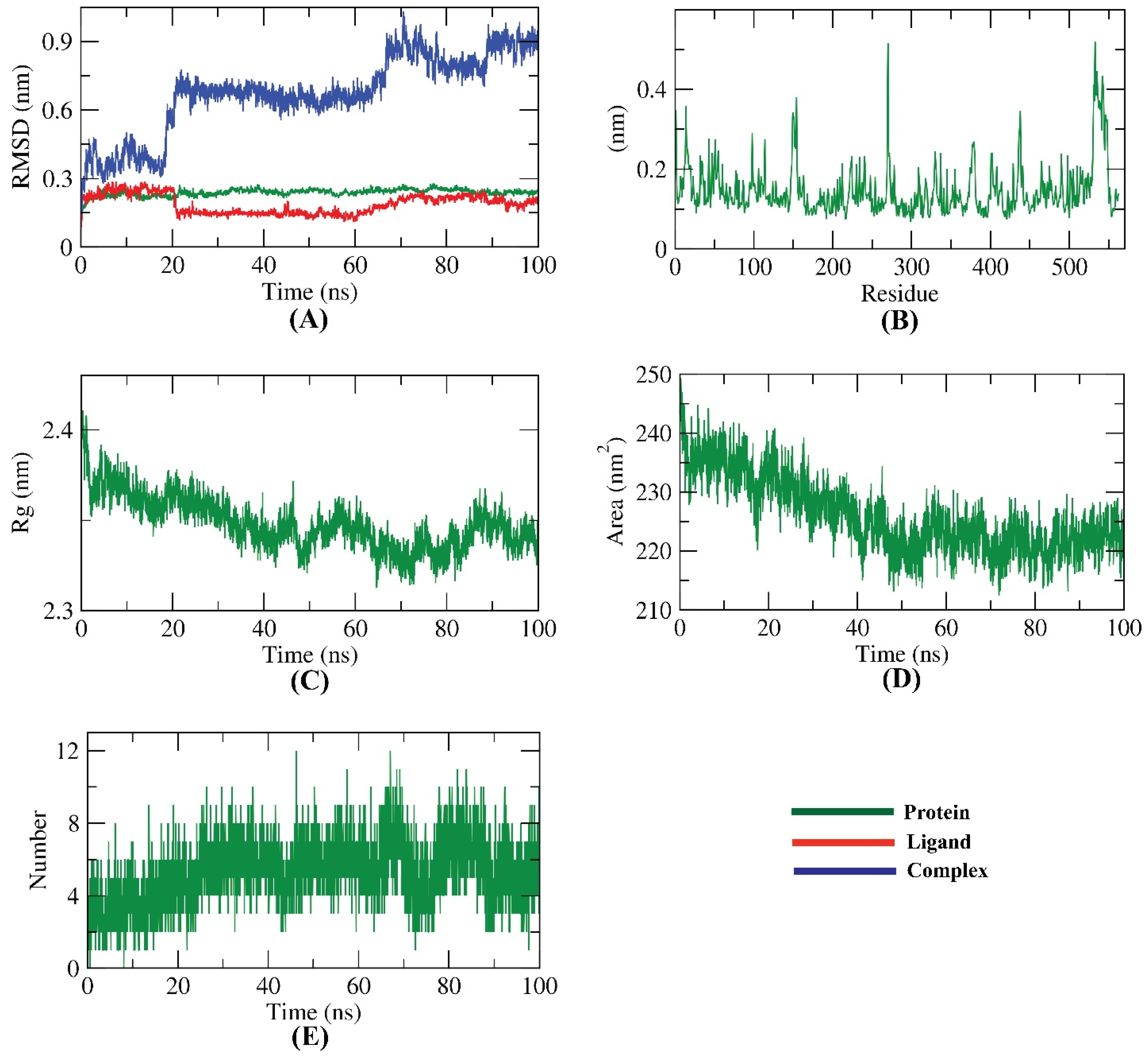
2.5. Molecular Dynamics Simulation Studies
2.6. MM-PBSA Studies
3. Experimental
3.1. Docking Studies
3.2. ADMET
3.3. Toxicity
3.4. DFT Studies
3.5. MD Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, J.; Nguyen, D.; Hu, K.-Q. Chronic Hepatitis C Virus Infection: A Review of Current Direct-Acting Antiviral Treatment Strategies. Am. Chin. J. Med. Sci. 2016, 9, 47–54. [Google Scholar]
- Chopp, S.; Vanderwall, R.; Hult, A.; Klepser, M. Simeprevir and sofosbuvir for treatment of hepatitis C infection. Am. J. Health Pharm. 2015, 72, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.; Lu, X.; Hollenbaugh, J.A.; Cho, J.H.; Amblard, F.; Schinazi, R.F. Metabolism, Biochemical Actions, and Chemical Synthesis of Anticancer Nucleosides, Nucleotides, and Base Analogs. Chem. Rev. 2016, 116, 14379–14455. [Google Scholar] [CrossRef]
- Pradere, U.; Garnier-Amblard, E.C.; Coats, S.J.; Amblard, F.; Schinazi, R.F. Synthesis of Nucleoside Phosphate and Phosphonate Prodrugs. Chem. Rev. 2014, 114, 9154–9218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuigan, C.; Bourdin, C.; Derudas, M.; Hamon, N.; Hinsinger, K.; Kandil, S.; Madela, K.; Meneghesso, S.; Pertusati, F.; Serpi, M.; et al. Design, synthesis and biological evaluation of phosphorodiamidate prodrugs of antiviral and anticancer nucleosides. Eur. J. Med. Chem. 2013, 70, 326–340. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, C.; Perrone, P.; Madela, K.; Neyts, J. The phosphoramidate ProTide approach greatly enhances the activity of beta-2′-C-methylguanosine against hepatitis C virus. Bioorg. Med. Chem. Lett. 2009, 19, 4316–4320. [Google Scholar] [CrossRef]
- Sofia, M.J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P.G.; Ross, B.S.; Wang, P.; Zhang, H.R.; et al. Discovery of a beta-d-2′-deoxy-2′-alpha-fluoro-2′-beta-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J. Med. Chem. 2010, 53, 7202–7218. [Google Scholar] [CrossRef]
- Lam, A.M.; Espiritu, C.; Bansal, S.; Steuer, H.M.M.; Niu, C.; Zennou, V.; Keilman, M.; Zhu, Y.; Lan, S.; Otto, M.J.; et al. Genotype and Subtype Profiling of PSI-7977 as a Nucleotide Inhibitor of Hepatitis C Virus. Antimicrob. Agents Chemother. 2012, 56, 3359–3368. [Google Scholar] [CrossRef] [Green Version]
- Denning, J.; Cornpropst, M.; Flach, S.D.; Berrey, M.M.; Symonds, W.T. Pharmacokinetics, Safety, and Tolerability of GS-9851, a Nucleotide Analog Polymerase Inhibitor for Hepatitis C Virus, following Single Ascending Doses in Healthy Subjects. Antimicrob. Agents Chemother. 2013, 57, 1201–1208. [Google Scholar] [CrossRef] [Green Version]
- Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, UK, 2009. [Google Scholar]
- Brown, N. Bioisosterism in Medicinal Chemistry. In Bioisosteres in Medical Chemistry; 2012; Wiley: Hoboken, NJ, USA; pp. 1–14. [Google Scholar] [CrossRef] [Green Version]
- Krueger, A.C.; Chen, H.-J.; Randolph, J.T.; Brown, B.S.; Halvorsen, G.T.; Heyman, H.R.; Li, T.; Marvin, C.; Shanley, J.P.; Voight, E.A.; et al. Synthesis and evaluation of 2′-dihalo ribonucleotide prodrugs with activity against hepatitis C virus. Bioorg. Med. Chem. 2019, 28, 115208. [Google Scholar] [CrossRef]
- Randolph, J.T.; Li, T.; Krueger, A.C.; Heyman, H.R.; Chen, H.-J.; Bow, D.A.; Van Handel, C.; Peterkin, V.; Carr, R.A.; Stolarik, D.; et al. Discovery of 2-aminoisobutyric acid ethyl ester (AIBEE) phosphoramidate prodrugs for delivering nucleoside HCV NS5B polymerase inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 126986. [Google Scholar] [CrossRef] [PubMed]
- Mengshetti, S.; Zhou, L.; Sari, O.; de Schutter, C.; Zhang, H.; Cho, J.H.; Tao, S.; Bassit, L.C.; Verma, K.; Domaoal, R.A.; et al. Discovery of a Series of 2′-alpha-Fluoro,2′-beta-bromo-ribonucleosides and Their Phosphoramidate Prodrugs as Potent Pan-Genotypic Inhibitors of Hepatitis C Virus. J. Med. Chem. 2019, 62, 1859–1874. [Google Scholar] [CrossRef] [PubMed]
- Ovadia, R.; Khalil, A.; Li, H.; de Schutter, C.; Mengshetti, S.; Zhou, S.; Bassit, L.; Coats, S.J.; Amblard, F.; Schinazi, R.F. Synthesis and anti-HCV activity of beta-d-2′-deoxy-2′-alpha-chloro-2′-beta-fluoro and beta-d-2′-deoxy-2′-alpha-bromo-2′-beta-fluoro nucleosides and their phosphoramidate prodrugs. Bioorg. Med. Chem. 2019, 27, 664–676. [Google Scholar] [CrossRef]
- Zhou, S.; Mahmoud, S.; Liu, P.; Zhou, L.; Ehteshami, M.; Bassit, L.; Tao, S.; Domaoal, R.A.; Sari, O.; De Schutter, C.; et al. 2′-Chloro,2′-fluoro Ribonucleotide Prodrugs with Potent Pan-genotypic Activity against Hepatitis C Virus Replication in Culture. J. Med. Chem. 2017, 60, 5424–5437. [Google Scholar] [CrossRef] [PubMed]
- Pinho, P.; Kalayanov, G.; Westerlind, H.; Rosenquist, A.; Wahling, H.; Sund, C.; Almeida, M.; Ayesa, S.; Tejbrant, J.; Targett-Adams, P.; et al. Discovery of beta-d-2′-deoxy-2′-dichlorouridine nucleotide prodrugs as potent inhibitors of hepatitis C virus replication. Bioorg. Med. Chem. Lett. 2017, 27, 3468–3471. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Cox, B.D.; Garnier-Amblard, E.C.; McBrayer, T.R.; Coats, S.J.; Schinazi, R.F.; Amblard, F. Synthesis and anti-HCV activity of a series of beta-d-2′-deoxy-2′-dibromo nucleosides and their corresponding phosphoramidate prodrugs. Bioorg. Med. Chem. Lett. 2017, 27, 5296–5299. [Google Scholar] [CrossRef]
- Appleby, T.C.; Perry, J.K.; Murakami, E.; Barauskas, O.; Feng, J.; Cho, A.; Fox, D., 3rd; Wetmore, D.R.; McGrath, M.E.; Ray, A.S.; et al. Viral replication. Structural basis for RNA replication by the hepatitis C virus polymerase. Science 2015, 347, 771–775. [Google Scholar] [CrossRef]
- Barakat, K.H.; Law, J.; Prunotto, A.; Magee, W.C.; Evans, D.H.; Tyrrell, D.L.; Tuszynski, J.; Houghton, M. Detailed Computational Study of the Active Site of the Hepatitis C Viral RNA Polymerase to Aid Novel Drug Design. J. Chem. Inf. Model. 2013, 53, 3031–3043. [Google Scholar] [CrossRef]
- Kramer, M.; Halleran, D.; Rahman, M.; Iqbal, M.; Anwar, M.I.; Sabet, S.; Ackad, E.; Yousef, M.S. Comparative molecular dynamics simulation of Hepatitis C Virus NS3/4A protease (Genotypes 1b, 3a and 4b) predicts conformational instability of the catalytic triad in drug resistant strains. PLoS ONE 2014, 9, e104425. [Google Scholar] [CrossRef]
- Maynard, A.; Crosby, R.M.; Ellis, B.; Hamatake, R.; Hong, Z.; Johns, B.A.; Kahler, K.M.; Koble, C.; Leivers, A.L.; Leivers, M.R.; et al. Discovery of a Potent Boronic Acid Derived Inhibitor of the HCV RNA-Dependent RNA Polymerase. J. Med. Chem. 2013, 57, 1902–1913. [Google Scholar] [CrossRef]
- Van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug. Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Mannhold, H.K.; Folkers, G.; Smith, D.A.; Allerton, C.; Kalgutkar, A.S.; van de Waterbeemd, H.; Walker, D.K. Pharmacokinetics and Metabolism in Drug Design, 3rd ed.; Smith, D.A., Allerton, C., Kalgutkar, A.S., Walker, D.K., Eds.; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Klopman, G.; Stefan, L.R.; Saiakhov, R.D. ADME evaluation: 2. A computer model for the prediction of intestinal absorption in humans. Eur. J. Pharm. Sci. 2002, 17, 253–263. [Google Scholar] [CrossRef]
- Roy, P.P.; Roy, K. QSAR Studies of CYP2D6 Inhibitor Aryloxypropanolamines Using 2D and 3D Descriptors. Chem. Biol. Drug Des. 2009, 73, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Amin, Z.; Ghafourian, T. QSAR Models for the Prediction of Plasma Protein Binding. BioImpacts 2013, 3, 21–27. [Google Scholar] [CrossRef]
- Xia, X.; Maliski, E.G.; Gallant, A.P.; Rogers, D. Classification of Kinase Inhibitors Using a Bayesian Model. J. Med. Chem. 2004, 47, 4463–4470. [Google Scholar] [CrossRef] [Green Version]
- BIOVIA. QSAR, Admet and Predictive Toxicology. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-discovery-studio/qsar-admet-and-predictive-toxicology/ (accessed on 11 May 2022).
- Venkatapathy, R.; Wang, N.C.Y.; Martin, T.M.; Harten, P.F.; Young, D. Structure-Activity Relationships for Carcinogenic Potential. In General Applied and System Toxicology; Wiley: Hoboken, NJ, USA, 2009; Volume 1. [Google Scholar] [CrossRef]
- Goodman, G.; Wilson, R. Comparison of the dependence of the TD50 on maximum tolerated dose for mutagens and nonmutagens. Risk Anal. 1992, 12, 525–533. [Google Scholar]
- Krewski, D.; Gaylor, D.; Soms, A.P.; Szyszkowicz, M. An Overview of the Report: Correlation Between Carcinogenic Potency and the Maximum Tolerated Dose: Implications for Risk Assessment. Risk Anal. 1993, 13, 383–398. [Google Scholar] [CrossRef]
- Pizzo, F.; Benfenati, E. In Silico Models for Repeated-Dose Toxicity (RDT): Prediction of the No Observed Adverse Effect Level (NOAEL) and Lowest Observed Adverse Effect Level (LOAEL) for Drugs. Methods Mol. Biol. 2016, 1425, 163–176. [Google Scholar] [CrossRef]
- Venkatapathy, R.; Moudgal, C.J.; Bruce, R.M. Assessment of the Oral Rat Chronic Lowest Observed Adverse Effect Level Model in TOPKAT, a QSAR Software Package for Toxicity Prediction. J. Chem. Inf. Comput. Sci. 2004, 44, 1623–1629. [Google Scholar] [CrossRef]
- Saliner, A.G.; Patlewicz, G.; Worth, A. A Review of (Q)SAR Models for Skin and Eye Irritation and Corrosion. QSAR Comb. Sci. 2007, 27, 49–59. [Google Scholar] [CrossRef]
- Chakraborty, A.; Chattaraj, P.K.; Pan, S. Biological Activity and Toxicity: A Conceptual DFT Approach. In Applications of Density Functional Theory to Biological and Bioinorganic Chemistry; Putz, M.M.D., Ed.; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Badenhoop, J.K.; Weinhold, F. Natural steric analysis: Ab initio van der Waals radii of atoms and ions. J. Chem. Phys. 1997, 107, 5422–5432. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Abraham, M.H.; Zissimos, A.M. Fast Calculation of van der Waals Volume as a Sum of Atomic and Bond Contributions and Its Application to Drug Compounds. J. Org. Chem. 2003, 68, 7368–7373. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 2017, 13, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Hansson, T.; Oostenbrink, C.; van Gunsteren, W. Molecular dynamics simulations. Curr. Opin. Struct. Biol. 2002, 12, 190–196. [Google Scholar] [CrossRef]
- McCammon, J.A.; Gelin, B.R.; Karplus, M. Dynamics of folded proteins. Nature 1977, 267, 585–590. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Malathi, K.; Ramaiah, S.; Anbarasu, A. Comparative Molecular Field Analysis and Molecular Docking Studies on Quinolinone Derivatives Indicate Potential Hepatitis C Virus Inhibitors. Cell Biophys. 2019, 77, 139–156. [Google Scholar] [CrossRef]
- Liu, P.; Lu, J.; Yu, H.; Ren, N.; Lockwood, F.E.; Wang, Q.J. Lubricant shear thinning behavior correlated with variation of radius of gyration via molecular dynamics simulations. J. Chem. Phys. 2017, 147, 084904. [Google Scholar] [CrossRef]
- Kumar, K.M.; Anbarasu, A.; Ramaiah, S. Molecular docking and molecular dynamics studies on beta-lactamases and penicillin binding proteins. Mol. Biosyst. 2014, 10, 891–900. [Google Scholar] [CrossRef]
- Ren, J.; Yuan, X.; Li, J.; Lin, S.; Yang, B.; Chen, C.; Zhao, J.; Zheng, W.; Liao, H.; Yang, Z.; et al. Assessing the performance of the g_mmpbsa tools to simulate the inhibition of oseltamivir to influenza virus neuraminidase by molecular mechanics Poisson–Boltzmann surface area methods. J. Chin. Chem. Soc. 2019, 67, 46–53. [Google Scholar] [CrossRef]
- Li, N.; Wang, Y.; Li, W.; Li, H.; Yang, L.; Wang, J.; Mahdy, H.A.; Mehany, A.B.M.; Jaiash, D.A.; Santali, E.Y.; et al. Screening of Some Sulfonamide and Sulfonylurea Derivatives as Anti-Alzheimer’s Agents Targeting BACE1 and PPARγ. J. Chem. 2020, 2020, 1631243. [Google Scholar] [CrossRef]
- El-Gamal, K.M.; El-Morsy, A.M.; Saad, A.M.; Eissa, I.H.; Alswah, M. Synthesis, docking, QSAR, ADMET and antimicrobial evaluation of new quinoline-3-carbonitrile derivatives as potential DNA-gyrase inhibitors. J. Mol. Struct. 2018, 1166, 15–33. [Google Scholar] [CrossRef]
- Sapundzhi, F.; Prodanova, K.; Lazarova, M. Survey of the scoring functions for protein-ligand docking. AIP Conf. Proc. 2019, 2172, 100008. [Google Scholar] [CrossRef]
- Frisch, M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | X | Y | EC50 GT1a | EC50 GT1b | CC50 |
|---|---|---|---|---|---|
| 1; Sofosbuvir | CH3 | F | 0.155 | 0.230 | >100 |
| 2 | CH3 | Cl | 0.139 | 0.167 | >100 |
| 3 | F | CH3 | >10 | >10 | >32 |
| 4 | CH3 | CH3 | 5.63 | 4.75 | 31.6 |
| 5 | F | F | 0.226 | 0.286 | 0.384 |
| 6 | Cl | Cl | 0.043 | 0.048 | >100 |
| 7 | Br | Br | 0.408 | 0.522 | >100 |
| 8 | Cl | F | 0.053 | 0.061 | 11 |
| 9 | F | Cl | 0.9 | NR | 1.3 |
| 10 | Cl | Br | 0.114 | 0.147 | 80 |
| 11 | Br | Cl | 0.200 | 0.258 | >100 |
| 12 | Br | F | 0.056 | 0.087 | >100 |
| 13 | F | Br | 3.03 | 2.50 | 26.5 |
| Comp. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | Ligand |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GTIa | −7.19 | −6.65 | −6.44 | −6.73 | −6.53 | −6.21 | −6.87 | −6.79 | −6.74 | −7.27 | −6.48 | −7.21 | −7.57 | −9.16 |
| GTIb | −6.30 | −6.87 | −6.79 | −6.95 | −6.45 | −6.63 | −7.04 | −6.39 | −7.09 | −7.34 | −6.37 | −6.57 | −6.68 | −7.24 |
| Comp. | BBB Level a | Solubility Level b | Absorption Level c | CYP2D6 Prediction d | PPB Prediction e | |||
|---|---|---|---|---|---|---|---|---|
| 1 (Sofosbuvir) | 4 | 3 | −3.72 | 1 | −7.28 | FALSE | 1.30 | FALSE |
| 2 | 4 | 4 | −0.66 | 0 | −3.34 | FALSE | −0.85 | FALSE |
| 3 | 4 | 4 | −0.45 | 1 | −2.51 | FALSE | −1.11 | FALSE |
| 4 | 4 | 4 | −0.41 | 0 | −3.19 | FALSE | −0.83 | FALSE |
| 5 | 4 | 4 | −0.60 | 1 | −2.90 | FALSE | −1.25 | FALSE |
| 6 | 3 | 4 | −1.09 | 0 | −3.09 | FALSE | −0.66 | FALSE |
| 7 | 3 | 4 | −1.45 | 0 | −5.45 | FALSE | −0.24 | FALSE |
| 8 | 4 | 4 | −0.84 | 0 | −2.66 | FALSE | −0.96 | FALSE |
| 9 | 4 | 4 | −0.84 | 0 | −2.66 | FALSE | −0.96 | FALSE |
| 10 | 3 | 4 | −1.27 | 0 | −5.49 | FALSE | −0.45 | FALSE |
| 11 | 3 | 4 | −1.27 | 0 | −5.49 | FALSE | −0.45 | FALSE |
| 12 | 3 | 4 | −1.02 | 0 | −2.65 | FALSE | −0.75 | FALSE |
| 13 | 3 | 4 | −1.02 | 0 | −2.65 | FALSE | −0.75 | FALSE |
| Comp. | FDA Rodent Carcinogenicity (Rat- Male) | Carcinogenic Potency TD50 Rat a | Rat Maximum Tolerated Dose (Feed) b | Rat Chronic LOAEL b | Skin Irritancy | Ocular Irritancy |
|---|---|---|---|---|---|---|
| 1 (Sofosbuvir) | Non-Carcinogen | 4.929 | 0.028 | 0.0011 | None | Moderate |
| 2 | Non-Carcinogen | 4.059 | 0.051 | 0.0013 | Mild | Moderate |
| 3 | Non-Carcinogen | 4.101 | 0.056 | 0.0013 | Mild | Moderate |
| 4 | Non-Carcinogen | 5.463 | 0.063 | 0.0026 | Mild | Moderate |
| 5 | Non-Carcinogen | 4.998 | 0.050 | 0.0011 | Mild | Moderate |
| 6 | Non-Carcinogen | 4.810 | 0.042 | 0.0011 | Mild | Moderate |
| 7 | Non-Carcinogen | 5.159 | 0.021 | 0.0013 | Mild | Moderate |
| 8 | Non-Carcinogen | 4.911 | 0.046 | 0.0011 | Mild | Moderate |
| 9 | Non-Carcinogen | 4.911 | 0.046 | 0.0011 | Mild | Moderate |
| 10 | Non-Carcinogen | 5.024 | 0.030 | 0.0012 | Mild | Moderate |
| 11 | Non-Carcinogen | 5.024 | 0.030 | 0.0012 | Mild | Moderate |
| 12 | Non-Carcinogen | 5.169 | 0.033 | 0.0012 | Mild | Moderate |
| 13 | Non-Carcinogen | 5.169 | 0.033 | 0.0012 | Mild | Moderate |
| B3LYP/6-311+g(d,p) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Comp. | X | Y | G | μ (D) | εLUMO | εHOMO | Egap | IP | EA | χ | η | S | ω |
| 1 | CH3 | F | −611,483.98 | 6.44 | −1.64409 | −7.14044 | 5.50 | 7.14 | 1.64 | 4.39 | 5.50 | 0.18 | 1.75 |
| 2 | CH3 | Cl | −837,608.55 | 6.47 | −1.64082 | −7.15921 | 5.52 | 7.16 | 1.64 | 4.40 | 5.52 | 0.18 | 1.75 |
| 3 | F | CH3 | −611,488.44 | 5.77 | −1.47538 | −7.03921 | 5.56 | 7.04 | 1.48 | 4.26 | 5.56 | 0.18 | 1.63 |
| 5 | F | F | −649,124.45 | 5.83 | −1.65171 | −7.24656 | 5.59 | 7.25 | 1.65 | 4.45 | 5.59 | 0.18 | 1.77 |
| 6 | Cl | Cl | −1,101,368.36 | 5.78 | −1.60926 | −7.20493 | 5.60 | 7.20 | 1.61 | 4.41 | 5.60 | 0.18 | 1.74 |
| 7 | Br | Br | −37,54,376.37 | 5.68 | −1.98205 | −7.18942 | 5.21 | 7.19 | 1.98 | 4.59 | 5.21 | 0.19 | 2.02 |
| 8 | Cl | F | −875,244.85 | 5.97 | −1.61878 | −7.19813 | 5.58 | 7.20 | 1.62 | 4.41 | 5.58 | 0.18 | 1.74 |
| 9 | F | Cl | −875,246.68 | 5.66 | −1.63973 | −7.2452 | 5.61 | 7.25 | 1.64 | 4.44 | 5.61 | 0.18 | 1.76 |
| 10 | Cl | Br | −2,427,872.55 | 5.62 | −1.68926 | −7.20493 | 5.52 | 7.20 | 1.69 | 4.45 | 5.52 | 0.18 | 1.79 |
| 11 | Br | Cl | −2,427,872.17 | 5.82 | −1.82695 | −7.1867 | 5.36 | 7.19 | 1.83 | 4.51 | 5.36 | 0.19 | 1.89 |
| 12 | Br | F | −2,201,748.52 | 5.99 | −1.60871 | −7.17935 | 5.57 | 7.18 | 1.61 | 4.39 | 5.57 | 0.18 | 1.73 |
| 13 | F | Br | −2,201,750.54 | 5.52 | −1.64518 | −7.25146 | 5.61 | 7.25 | 1.65 | 4.45 | 5.61 | 0.18 | 1.76 |
| Mulliken Charges | Bond Length (Å) | Bondi Radii of Atoms and Their Volumes | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| X | Y | ||||||||||
| Comp. | X | Y | C-2′ | X | Y | C-X | C-Y | RBondi (Å) | VvdW (Å3) | RvdW (Å) | VvdW (Å3) |
| 1 | CH3 | F | 0.509 | C (−0.413) H (0.193, 0.188, 0.154) | −0.092 | 1.515 | 1.407 | C (1.70) H (1.20) | C (20.58) H (7.24) | 1.47 | 13.31 |
| 2 | CH3 | Cl | 1.466 | C (−0.665) H (0.191, 0.187, 0.156) | −0.004 | 1.524 | 1.825 | C (1.70) H (1.20) | C (20.58) H (7.24) | 1.75 | 22.45 |
| 3 | F | CH3 | 0.398 | −0.108 | C (−0.309) H (0.190, 0.175, 0.169) | 1.421 | 1.510 | 1.47 | 13.31 | C (1.70) H (1.20) | C (20.58) H (7.24) |
| 5 | F | F | 0.387 | −0.076 | −0.074 | 1.375 | 1.360 | 1.47 | 13.31 | 1.47 | 13.31 |
| 6 | Cl | Cl | 0.982 | −0.254 | 0.237 | 1.811 | 1.790 | 1.75 | 22.45 | 1.75 | 22.45 |
| 7 | Br | Br | 0.971 | 0.004 | 0.074 | 1.984 | 1.959 | 1.85 | 26.52 | 1.85 | 26.52 |
| 8 | Cl | F | 0.347 | 0.119 | −0.019 | 1.803 | 1.367 | 1.75 | 22.45 | 1.47 | 13.31 |
| 9 | F | Cl | 0.468 | −0.026 | 0.222 | 1.384 | 1.784 | 1.47 | 13.31 | 1.75 | 22.45 |
| 10 | Cl | Br | 1.497 | −0.324 | 0.102 | 1.809 | 1.963 | 1.75 | 22.45 | 1.85 | 26.52 |
| 11 | Br | Cl | 1.406 | −0.007 | 0.033 | 1.986 | 1.787 | 1.85 | 26.52 | 1.75 | 22.45 |
| 12 | Br | F | 0.612 | 0.068 | −0.028 | 1.977 | 1.366 | 1.85 | 26.52 | 1.47 | 13.31 |
| 13 | F | Br | 0.621 | −0.033 | 0.048 | 1.384 | 1.956 | 1.47 | 13.31 | 1.85 | 26.52 |
| B3LYP/6-311+g(d,p) | ||||
|---|---|---|---|---|
| HCV GT | Significant Indices | Linear Pearson Coefficient | ||
| r | r2 | p | ||
| GT1a | Ionization Potential (I) | 0.67 | 0.44 | <0.05 |
| Electronegativity (χ) | 0.63 | 0.39 | <0.05 | |
| GT1b | Ionization Potential (I) | 0.72 | 0.52 | <0.05 |
| Electronegativity (χ) | 0.63 | 0.40 | <0.05 | |
| Significant Indices | Multiple Pearson Coefficient | |||
| GT1a | I and χ | 0.72 | 0.51 | <0.05 |
| GT1b | I and χ | 0.75 | 0.57 | <0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalil, A.; El-Khouly, A.S.; Elkaeed, E.B.; Eissa, I.H. The Inhibitory Potential of 2′-dihalo Ribonucleotides against HCV: Molecular Docking, Molecular Simulations, MM-BPSA, and DFT Studies. Molecules 2022, 27, 4530. https://doi.org/10.3390/molecules27144530
Khalil A, El-Khouly AS, Elkaeed EB, Eissa IH. The Inhibitory Potential of 2′-dihalo Ribonucleotides against HCV: Molecular Docking, Molecular Simulations, MM-BPSA, and DFT Studies. Molecules. 2022; 27(14):4530. https://doi.org/10.3390/molecules27144530
Chicago/Turabian StyleKhalil, Ahmed, Amany S. El-Khouly, Eslam B. Elkaeed, and Ibrahim H. Eissa. 2022. "The Inhibitory Potential of 2′-dihalo Ribonucleotides against HCV: Molecular Docking, Molecular Simulations, MM-BPSA, and DFT Studies" Molecules 27, no. 14: 4530. https://doi.org/10.3390/molecules27144530