
The Activating Effect of Strong Acid for Pd-Catalyzed Directed C–H Activation by Concerted Metalation-Deprotonation Mechanism


{kind=link}
{kind=link}
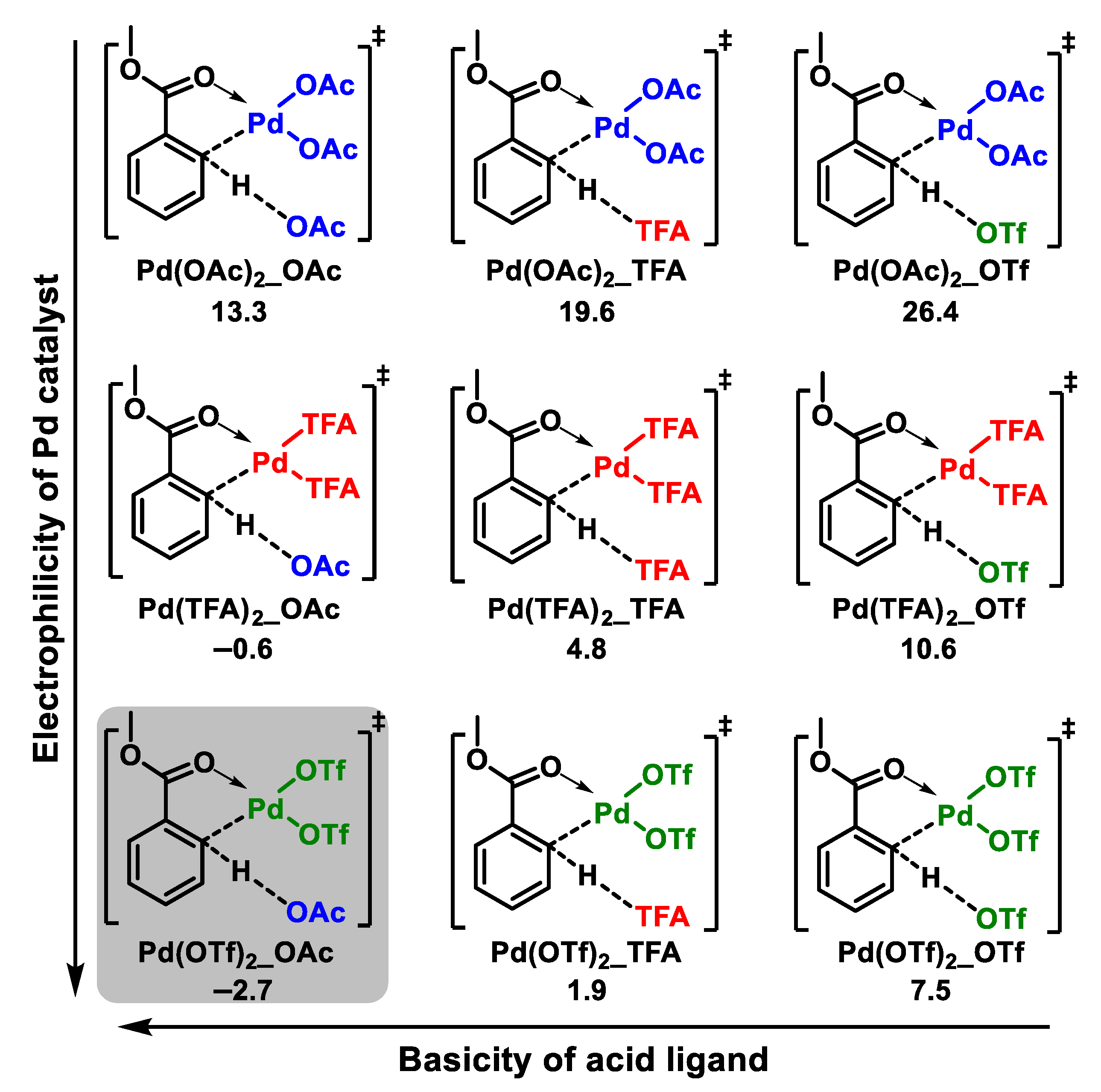{kind=link}
{kind=link}
{kind=link}
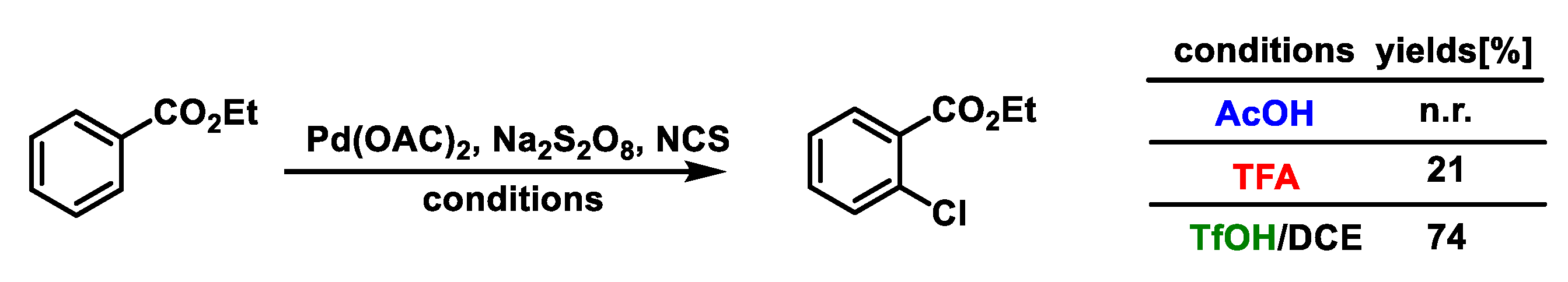{kind=link}
{kind=link}
{kind=link}
{kind=link}
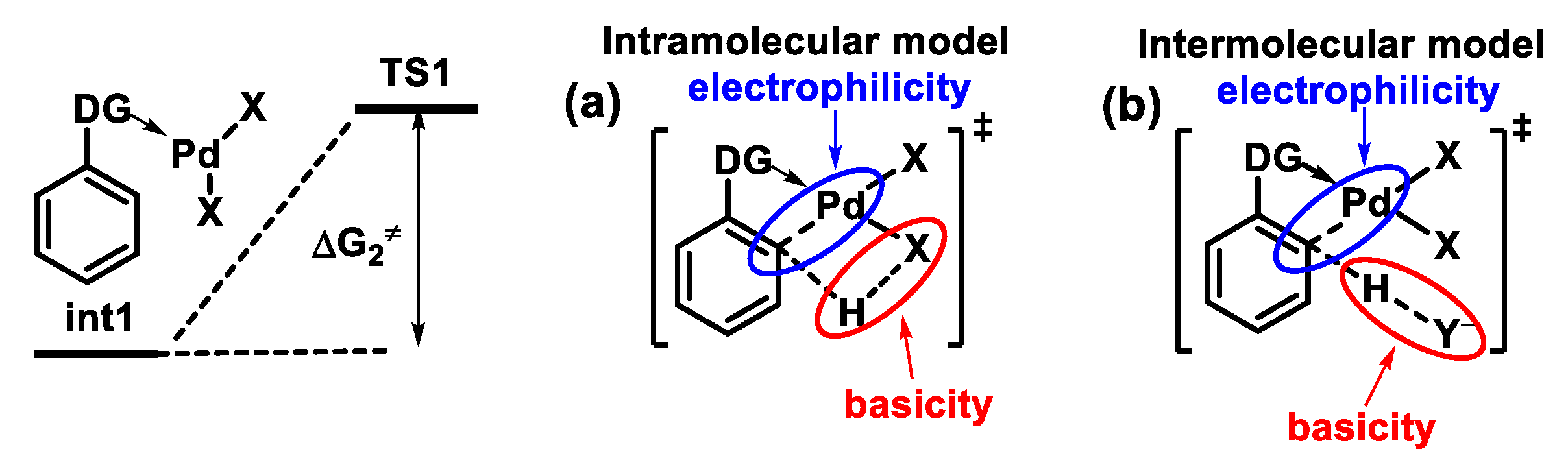{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Shilov, A.E.; Shul’pin, G.B. Activation of C–H Bonds by Metal Complexes. Chem. Rev. 1997, 97, 2879–2932. [Google Scholar] [CrossRef] [PubMed]
- Daugulis, O.; Do, H.-Q.; Shabashov, D. Palladium- and Copper-Catalyzed Arylation of Carbon−Hydrogen Bonds. Acc. Chem. Res. 2009, 42, 1074–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons, T.W.; Sanford, M.S. Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef] [Green Version]
- Neufeldt, S.R.; Sanford, M.S. Controlling Site Selectivity in Palladium-Catalyzed C–H Bond Functionalization. Acc. Chem. Res. 2012, 45, 936–946. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, L. Carboxylate-Assisted Ruthenium-Catalyzed Alkyne Annulations by C–H/Het–H Bond Functionalizations. Acc. Chem. Res. 2014, 47, 281–295. [Google Scholar] [CrossRef]
- Gao, K.; Yoshikai, N. Low-Valent Cobalt Catalysis: New Opportunities for C–H Functionalization. Acc. Chem. Res. 2014, 47, 1208–1219. [Google Scholar] [CrossRef]
- Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Transition Metals for C–H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef] [PubMed]
- Phipps, R.J.; Gaunt, M.J. A Meta-Selective Copper-Catalyzed C–H Bond Arylation. Science 2009, 323, 1593–1597. [Google Scholar] [CrossRef]
- Wang, D.-H.; Engle, K.M.; Shi, B.-F.; Yu, J.-Q. Ligand-Enabled Reactivity and Selectivity in a Synthetically Versatile Aryl C–H Olefination. Science 2010, 327, 315–319. [Google Scholar] [CrossRef] [Green Version]
- Brückl, T.; Baxter, R.D.; Ishihara, Y.; Baran, P.S. Innate and Guided C–H Functionalization Logic. Acc. Chem. Res. 2011, 45, 826–839. [Google Scholar] [CrossRef] [Green Version]
- Rousseau, G.; Breit, B. Removable Directing Groups in Organic Synthesis and Catalysis. Angew. Chem. Int. Ed. 2011, 50, 2450–2494. [Google Scholar] [CrossRef]
- Luo, J.; Preciado, S.; Larrosa, I. Overriding Ortho–Para Selectivity via a Traceless Directing Group Relay Strategy: The Meta-Selective Arylation of Phenols. J. Am. Chem. Soc. 2014, 136, 4109–4112. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.C.K.; Rovis, T. Complementary Strategies for Directed C(sp3)−H Functionalization: A Comparison of Transition-Metal-Catalyzed Activation, Hydrogen Atom Transfer, and Carbene/Nitrene Transfer. Angew. Chem. Int. Ed. 2018, 57, 62–101. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Sinha, S.K.; Achar, T.K.; Maiti, D. Accessing Remote meta-and para-C(sp2)–HBonds withCovalently Attached Directing Groups. Angew. Chem. Int. Ed. 2019, 58, 10820–10843. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.-Y.; Li, G.; Yu, J.-Q. Conformation-induced remote meta-C–H activation of amines. Nature 2014, 507, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Crimmin, M.R.; Toste, F.D.; Bergman, R.G. Ligand-Based Carbon–Nitrogen Bond Forming Reactions of Metal Dinitrosyl Complexes with Alkenes and Their Application to C–H Bond Functionalization. Acc. Chem. Res. 2014, 47, 517–529. [Google Scholar] [CrossRef]
- Rouquet, G.; Chatani, N. Catalytic Functionalization of C(sp2)-H and C(sp3)-H Bonds by Using Bidentate Directing Groups. Angew. Chem. Int. Ed. 2013, 52, 11726–11743. [Google Scholar] [CrossRef] [PubMed]
- Leow, D.; Li, G.; Mei, T.-S.; Yu, J.-Q. Activation of remote meta-C–H bonds assisted by an end-on template. Nature 2012, 486, 518–522. [Google Scholar] [CrossRef]
- Lee, S.; Lee, H.; Tan, K.L. Meta-Selective C–H Functionalization Using a Nitrile-Based Directing Group and Cleavable Si-Tether. J. Am. Chem. Soc. 2013, 135, 18778–18781. [Google Scholar] [CrossRef] [Green Version]
- Negretti, S.; Narayan, A.R.H.; Chiou, K.C.; Kells, P.M.; Stachowski, J.L.; Hansen, D.A.; Podust, L.M.; Montgomery, J.; Sherman, D.H. Directing Group-Controlled Regioselectivity in an Enzymatic C–H Bond Oxygenation. J. Am. Chem. Soc. 2014, 136, 4901–4904. [Google Scholar] [CrossRef]
- Sambiagio, C.; Schönbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnig, G.; Schaaf, P.; Wiesinger, T.; Zia, M.F.; Wencel-Delord, J.; Besset, T.; et al. A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry. Chem. Soc. Rev. 2018, 47, 6603–6743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Wang, B.; Zhang, J.; Yu, W.; Liu, Z.; Zhang, Y. Transition metal-catalyzed C–H bond functionalizations by the use of diverse directing groups. Org. Chem. Front. 2015, 2, 1107–1295. [Google Scholar] [CrossRef]
- Xu, Y.; Dong, G. sp3 C–H activation via exo-type directing groups. Chem. Sci. 2018, 9, 1424–1432. [Google Scholar] [CrossRef] [Green Version]
- Rej, S.; Ano, Y.; Chatani, N. Bidentate Directing Groups: An Efficient Tool in C–H Bond Functionalization Chemistry for the Expedient Construction of C–C Bonds. Chem. Rev. 2020, 120, 1788–1887. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-H.; Shi, B.-F.; Yu, J.-Q. Palladium(II)-Catalyzed ortho Alkylation of Benzoic Acids with Alkyl Halides. Angew. Chem. Int. Ed. 2009, 48, 6097–6100. [Google Scholar] [CrossRef] [Green Version]
- Dai, H.-X.; Stepan, A.F.; Plummer, M.S.; Zhang, Y.-H.; Yu, J.-Q. Divergent C–H Functionalizations Directed by Sulfonamide Pharmacophores: Late-Stage Diversification as a Tool for Drug Discovery. J. Am. Chem. Soc. 2011, 133, 7222–7228. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.M.; Mei, T.-S.; Wasa, M.; Yu, J.-Q. Weak Coordination as a Powerful Means for Developing Broadly Useful C–H Functionalization Reactions. Acc. Chem. Res. 2012, 45, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-C.; Hu, Y.; Bonacorsi, S.; Hong, Y.; Burrell, R.; Yu, J.-Q. Pd(II)-Catalyzed C–H Iodination Using Molecular I2 as the Sole Oxidant. J. Am. Chem. Soc. 2013, 135, 10326–10329. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Villa, G.; Thuy-Boun, P.S.; Homs, A.; Yu, J.-Q. Palladium-Catalyzed ortho-Selective C–H Deuteration of Arenes: Evidence for Superior Reactivity of Weakly Coordinated Palladacycles. Angew. Chem. Int. Ed. 2014, 53, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Gandeepan, P.; Ackermann, L. Transient Directing Groups for Transformative C–H Activation by Synergistic Metal Catalysis. Chem 2018, 4, 199–222. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.S.L.; Wasa, M.; Wang, X.; Yu, J.-Q. Palladium(II)-Catalyzed Selective Monofluorination of Benzoic Acids Using a Practical Auxiliary: A Weak-Coordination Approach. Angew. Chem. Int. Ed. 2011, 50, 9081–9084. [Google Scholar] [CrossRef] [PubMed]
- Tran, A.T.; Yu, J.-Q. Practical Alkoxythiocarbonyl Auxiliaries for Iridium(I)-Catalyzed C–H Alkylation of Azacycles. Angew. Chem. Int. Ed. 2017, 56, 10530–10534. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, D.-H.; Engle, K.M.; Yu, J.-Q. Pd(II)-Catalyzed Hydroxyl-Directed C–H Olefination Enabled by Monoprotected Amino Acid Ligands. J. Am. Chem. Soc. 2010, 132, 5916–5921. [Google Scholar] [CrossRef] [Green Version]
- Plata, R.E.; Hill, D.E.; Haines, B.E.; Musaev, D.G.; Chu, L.; Hickey, D.P.; Sigman, M.S.; Yu, J.-Q.; Blackmond, D.G. A Role for Pd(IV) in Catalytic Enantioselective C–H Functionalization with Monoprotected Amino Acid Ligands under Mild Conditions. J. Am. Chem. Soc. 2017, 139, 9238–9245. [Google Scholar] [CrossRef]
- Mei, T.-S.; Giri, R.; Maugel, N.; Yu, J.-Q. PdII-Catalyzed Monoselective ortho Halogenation of C–H Bonds Assisted by Counter Cations: A Complementary Method to Directed ortho-Lithiation. Angew. Chem. Int. Ed. 2008, 47, 5215–5219. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Lv, H.; Li, S. The C–H functionalization of organic cations: An interesting and fresh journey. Org. Biomol. Chem. 2020, 18, 8810–8826. [Google Scholar] [CrossRef]
- Pototschnig, G.; Maulide, N.; Schnürch, M. Direct Functionalization of C–H Bonds by Iron, Nickel, and Cobalt Catalysis. Chem. Eur. J. 2017, 23, 9206–9232. [Google Scholar] [CrossRef]
- Sun, X.; Shan, G.; Sun, Y.; Rao, Y. Regio- and Chemoselective C-H Chlorination/Bromination of Electron-Deficient Arenes by Weak Coordination and Study of Relative Directing-Group Abilities. Angew. Chem. Int. Ed. 2013, 52, 4440–4444. [Google Scholar] [CrossRef]
- Tischler, O.; Kovács, S.; Érsek, G.; Králl, P.; Daru, J.; Stirling, A.; Novák, Z. Study of Lewis acid accelerated palladium catalyzed Csingle bondH activation. J. Mol. Catal. A Chem. 2017, 426, 444–450. [Google Scholar] [CrossRef]
- Vana, J.; Bartacek, J.; Hanusek, J.; Roithová, J.; Sedlak, M. C–H Functionalizations by Palladium Carboxylates: The Acid Effect. J. Org. Chem. 2019, 84, 12746–12754. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Jia, C. New developments in transition metal-catalyzed synthetic reactions via C–H bond activation. Pure Appl. Chem. 2001, 73, 319–324. [Google Scholar] [CrossRef]
- Boele, M.D.K.; van Strijdonck, G.P.F.; de Vries, A.H.M.; Kamer, P.C.J.; de Vries, J.G.; van Leeuwen, P.W.N.M. Selective Pd-Catalyzed Oxidative Coupling of Anilides with Olefins through C–H Bond Activation at Room Temperature. J. Am. Chem. Soc. 2002, 124, 1586–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugulis, O.; Zaitsev, V.G. Anilide ortho-Arylation by Using C–H Activation Methodology. Angew. Chem. Int. Ed. 2005, 44, 4046–4048. [Google Scholar] [CrossRef] [PubMed]
- Brasche, G.; García-Fortanet, J.; Buchwald, S.L. Twofold C–H Functionalization: Palladium-Catalyzed Ortho Arylation of Anilides. Org. Lett. 2008, 10, 2207–2210. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Mei, T.-S.; Yu, J.-Q. Versatile Pd(OTf)2·2H2O-Catalyzed ortho-Fluorination Using NMP as a Promoter. J. Am. Chem. Soc. 2009, 131, 7520–7521. [Google Scholar] [CrossRef] [PubMed]
- Houlden, C.E.; Hutchby, M.; Bailey, C.D.; Ford, J.G.; Tyler, S.N.G.; Gagné, M.R.; Lloyd-Jones, G.C.; Booker-Milburn, K.I. Room-Temperature Palladium-Catalyzed C–H Activation: Ortho-Carbonylation of Aniline Derivatives. Angew. Chem. Int. Ed. 2009, 48, 1830–1833. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Truesdale, L.; Yu, J.-Q. Pd(II)-Catalyzed ortho-Trifluoromethylation of Arenes Using TFA as a Promoter. J. Am. Chem. Soc. 2010, 132, 3648–3649. [Google Scholar] [CrossRef]
- Xiao, B.; Gong, T.-J.; Xu, J.; Liu, Z.-J.; Liu, L. Palladium-Catalyzed Intermolecular Directed C–H Amidation of Aromatic Ketones. J. Am. Chem. Soc. 2011, 133, 1466–1474. [Google Scholar] [CrossRef]
- Shan, G.; Yang, X.; Ma, L.; Rao, Y. Pd-Catalyzed C-H Oxygenation with TFA/TFAA: Expedient Access to Oxygen-Containing Heterocycles and Late-Stage Drug Modification. Angew. Chem. Int. Ed. 2012, 51, 13070–13074. [Google Scholar] [CrossRef]
- Nishikata, T.; Abela, A.R.; Huang, S.; Lipshutz, B.H. Cationic Palladium(II) Catalysis: C–H Activation/Suzuki-Miyaura Couplings at Room Temperature. J. Am. Chem. Soc. 2010, 132, 4978–4979. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yang, X.; Zhang, Y.; Xue, Y. Computational Mechanism Study on Allylic Oxidation of cis-Internal Alkenes: Insight into the Lewis Acid-Assisted Brønsted Acid (LBA) Catalysis in Heteroene Reactions. J. Org. Chem. 2018, 83, 13344–13355. [Google Scholar] [CrossRef]
- Ghaderi, A.; Iwasaki, T.; Kambe, N. Pivalic Acid-Assisted Rh(III)-Catalyzed C–H Functionalization of 2-Arylpyridine Derivatives Using Arylsilanes. Asian J. Org. Chem. 2019, 8, 1344–1347. [Google Scholar] [CrossRef]
- Jia, C.; Kitamura, T.; Fujiwara, Y. Catalytic Functionalization of Arenes and Alkanes via C–H Bond Activation. Acc. Chem. Res. 2001, 34, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.M.; Wang, D.-H.; Yu, J.-Q. Ligand-Accelerated C–H Activation Reactions: Evidence for a Switch of Mechanism. J. Am. Chem. Soc. 2010, 132, 14137–14151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labinger, J.A.; Bercaw, J.E. Understanding and exploiting C–H bond activation. Nature 2002, 417, 507–514. [Google Scholar] [CrossRef]
- Ackermann, L. Carboxylate-Assisted Transition-Metal-Catalyzed C–H Bond Functionalizations: Mechanism and Scope. Chem. Rev. 2011, 111, 1315–1345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Shi, L.; Ding, Y. Theoretical Analysis of the Mechanism of Palladium(II) Acetate-Catalyzed Oxidative Heck Coupling of Electron-Deficient Arenes with Alkenes: Effects of the Pyridine-Type Ancillary Ligand and Origins of the meta-Regioselectivity. J. Am. Chem. Soc. 2011, 133, 20218–20229. [Google Scholar] [CrossRef]
- Yang, Y.-F.; Cheng, G.-J.; Liu, P.; Leow, D.; Sun, T.-Y.; Chen, P.; Zhang, X.; Yu, J.-Q.; Wu, Y.-D.; Houk, K.N. Palladium-Catalyzed Meta-Selective C–H Bond Activation with a Nitrile-Containing Template: Computational Study on Mechanism and Origins of Selectivity. J. Am. Chem. Soc. 2014, 136, 344–355. [Google Scholar] [CrossRef]
- Cheng, G.-J.; Yang, Y.-F.; Liu, P.; Chen, P.; Sun, T.-Y.; Li, G.; Zhang, X.; Houk, K.N.; Yu, J.-Q.; Wu, Y.-D. Role of N-Acyl Amino Acid Ligands in Pd(II)-Catalyzed Remote C–H Activation of Tethered Arenes. J. Am. Chem. Soc. 2014, 136, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Gorelsky, S.I.; Lapointe, D.; Fagnou, K. Analysis of the Concerted Metalation-Deprotonation Mechanism in Palladium-Catalyzed Direct Arylation Across a Broad Range of Aromatic Substrates. J. Am. Chem. Soc. 2008, 130, 10848–10849. [Google Scholar] [CrossRef] [PubMed]
- Potavathri, S.; Pereira, K.C.; Gorelsky, S.I.; Pike, A.; LeBris, A.P.; DeBoef, B. Regioselective Oxidative Arylation of Indoles Bearing N-Alkyl Protecting Groups: Dual C–H Functionalization via a Concerted Metalation-Deprotonation Mechanism. J. Am. Chem. Soc. 2010, 132, 14676–14681. [Google Scholar] [CrossRef] [Green Version]
- Anand, M.; Sunoj, R.B. Palladium(II)-Catalyzed Direct Alkoxylation of Arenes: Evidence for Solvent-Assisted Concerted Metalation Deprotonation. Org. Lett. 2011, 13, 4802–4805. [Google Scholar] [CrossRef]
- Gorelsky, S.I.; Lapointe, D.; Fagnou, K. Analysis of the Palladium-Catalyzed (Aromatic)C–H Bond Metalation–Deprotonation Mechanism Spanning the Entire Spectrum of Arenes. J. Org. Chem. 2012, 77, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.; Macgregor, S.A.; McMullin, C.L. Computational Studies of Carboxylate-Assisted C–H Activation and Functionalization at Group 8–10 Transition Metal Centers. Chem. Rev. 2017, 117, 8649–8709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, C.; Zhu, L.; Qu, L.-B.; Bai, R.; Lan, Y. Mechanistic view of Ru-catalyzed C–H bond activation and functionalization: Computational advances. Chem. Soc. Rev. 2018, 47, 7552–7576. [Google Scholar] [CrossRef]
- Geometries were optimized with M06/(LANL2DZ+f: Pd; 6-31G(d): Other atoms). Single-point energies were calculated with M06/(SDD: Pd; 6-311++G(d, p): Other atoms) with the SMD solvation model (dichloroethane was chosen as the solvent). All calculations were performed with Gaussian 09. (Frisch, M.J.; et al.) See SI for more computational details.
- Skapski, A.C.; Smart, M.L. The crystal structure of trimeric palladium(II) acetate. J. Chem. Soc. D 1970, 11, b658–b659. [Google Scholar] [CrossRef]
- Kurzeev, S.A.; Kazankov, G.M.; Ryabov, A.D. Second- and inverse order pathways in the mechanism of orthopalladation of primary amines. Inorg. Chim. Acta 2002, 340, 192–196. [Google Scholar] [CrossRef]
- Bakhmutov, V.I.; Berry, J.F.; Cotton, F.A.; Ibragimov, S.; Murillo, C.A. Non-trivial behavior of palladium(ii) acetate. Dalton Trans. 2005, 1989–1992. [Google Scholar] [CrossRef]
- Gandeepan, P.; Parthasarathy, K.; Cheng, C.-H. Synthesis of Phenanthrone Derivatives from sec-Alkyl Aryl Ketones and Aryl Halides via a Palladium-Catalyzed Dual C–H Bond Activation and Enolate Cyclization. J. Am. Chem. Soc. 2010, 132, 8569–8571. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.S.; Zhao, X.; Borduas, N.; Dong, V.M. Pd-catalyzed ortho-arylation of phenylacetamides, benzamides, and anilides with simple arenes using sodium persulfate. Chem. Sci. 2010, 1, 331–336. [Google Scholar] [CrossRef]
- Zhao, X.; Yeung, C.S.; Dong, V.M. Palladium-Catalyzed Ortho-Arylation of O-Phenylcarbamates with Simple Arenes and Sodium Persulfate. J. Am. Chem. Soc. 2010, 132, 5837–5844. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.-B.; Wang, G.-W. Synthesis of [60]Fullerene-Fused Tetralones via Palladium- Catalyzed Ketone-Directed sp2 C-H Activation and sp3 C-H Functionalization. Adv. Synth. Catal. 2016, 358, 1548–1554. [Google Scholar] [CrossRef]
- Tóth, B.L.; Kovács, S.; Sályi, G.; Novák, Z. Mild and Efficient Palladium-Catalyzed Direct Trifluoroethylation of Aromatic Systems by C–H Activation. Angew. Chem. Int. Ed. 2016, 55, 1988–1992. [Google Scholar] [CrossRef]
- Guner, V.A.; Houk, K.N.; Davies, I.W. Computational Studies on the Electrocyclizations of 1-Amino-1,3,5-hexatrienes. J. Org. Chem. 2004, 69, 8024–8028. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.-Q.; Fu, Y.; Liu, L.; Guo, Q.-X. How to Promote Sluggish Electrocyclization of 1,3,5-Hexatrienes by Captodative Substitution. J. Org. Chem. 2006, 71, 6157–6164. [Google Scholar] [CrossRef]
- Xue, L.; Su, W.; Lin, Z. Mechanism of silver- and copper-catalyzed decarboxylation reactions of aryl carboxylic acids. Dalton Trans. 2011, 40, 11926–11936. [Google Scholar] [CrossRef]
- Ryabov, A.D. Cyclopalladated Complexes in Organic Synthesis. Synthesis 1985, 233–252. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, H.; Sun, T.-Y. The Activating Effect of Strong Acid for Pd-Catalyzed Directed C–H Activation by Concerted Metalation-Deprotonation Mechanism. Molecules 2021, 26, 4083. https://doi.org/10.3390/molecules26134083
Jiang H, Sun T-Y. The Activating Effect of Strong Acid for Pd-Catalyzed Directed C–H Activation by Concerted Metalation-Deprotonation Mechanism. Molecules. 2021; 26(13):4083. https://doi.org/10.3390/molecules26134083
Chicago/Turabian StyleJiang, Heming, and Tian-Yu Sun. 2021. "The Activating Effect of Strong Acid for Pd-Catalyzed Directed C–H Activation by Concerted Metalation-Deprotonation Mechanism" Molecules 26, no. 13: 4083. https://doi.org/10.3390/molecules26134083