
New Mesoporous Silica-Supported Organocatalysts Based on (2S)-(1,2,4-Triazol-3-yl)-Proline: Efficient, Reusable, and Heterogeneous Catalysts for the Asymmetric Aldol Reaction

 , ,
, ,
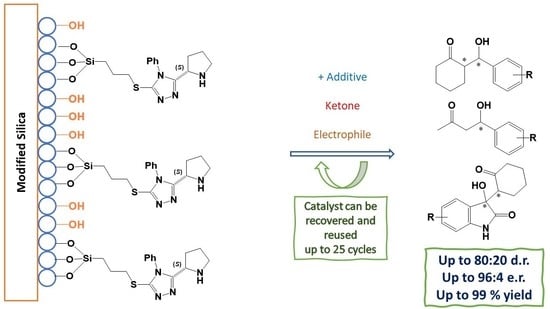
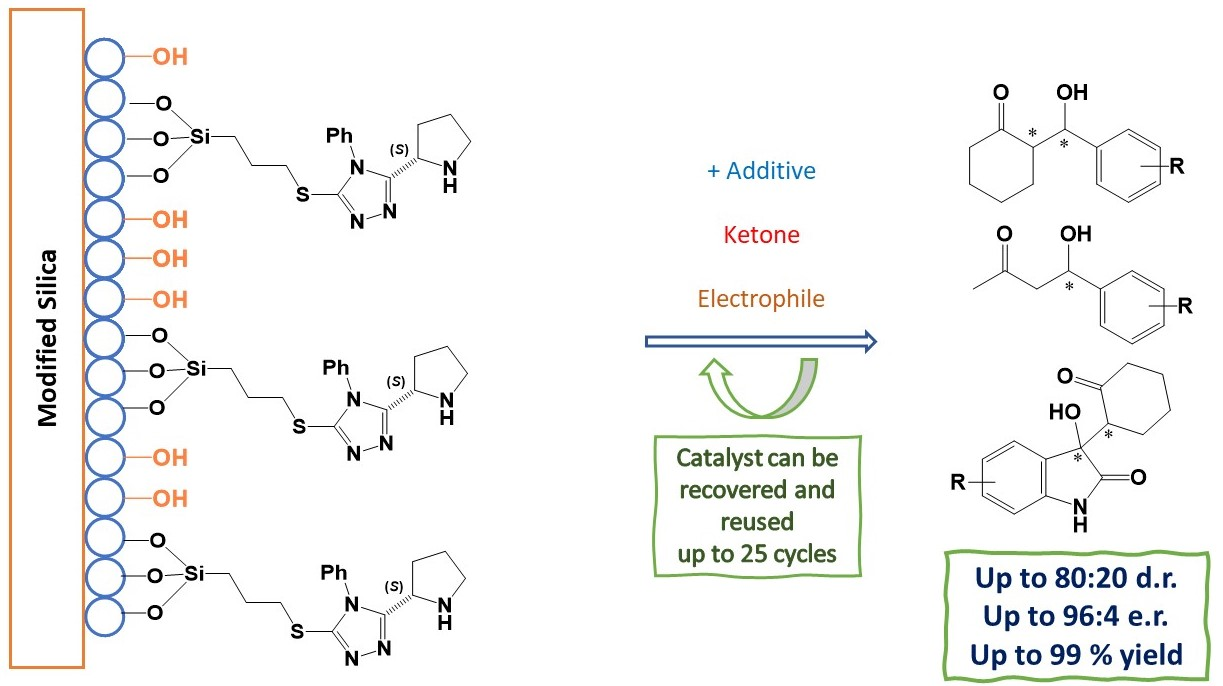
Abstract
:
1. Introduction
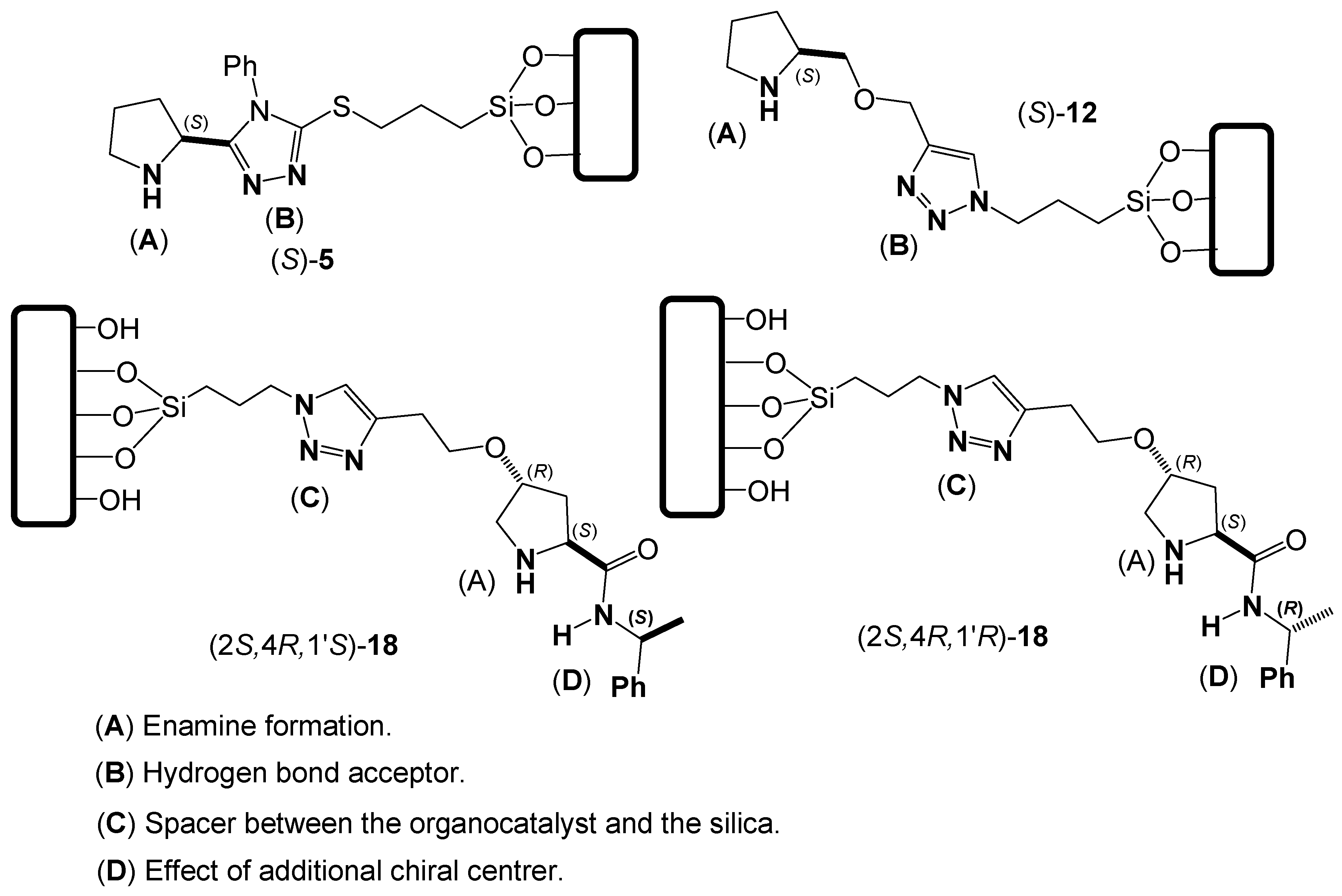
2. Results and Discussion
Evaluation of Organocatalysts (S)-5, (S)-12, (2S,4R,1′S)-18 and (2S,4R,1′R)-18
3. Materials and Methods
3.1. Starting Materials and Their Analysis
3.2. Procedures and General Methods
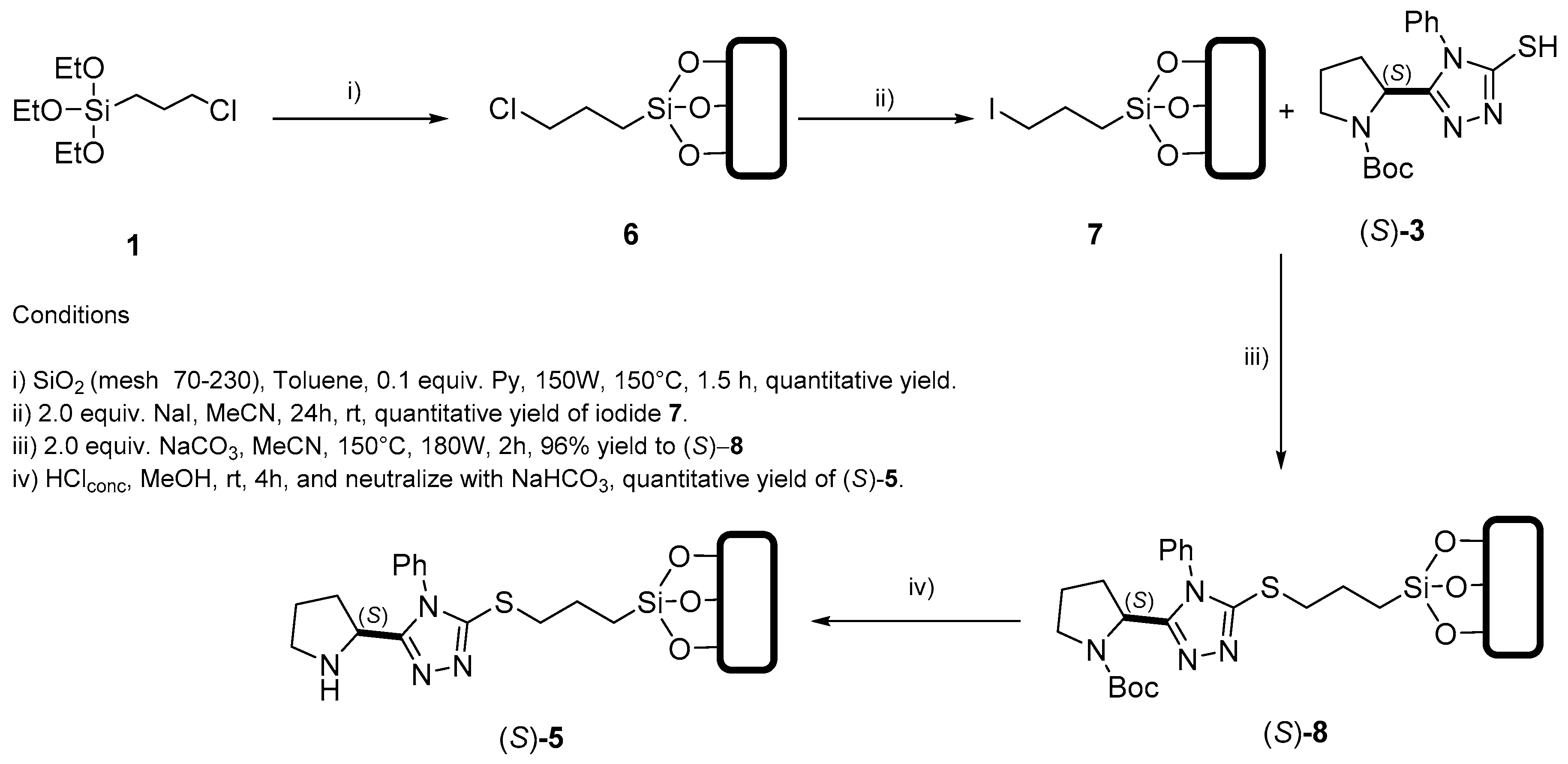
3.2.1. General Procedure 1: Finkelstein Reaction
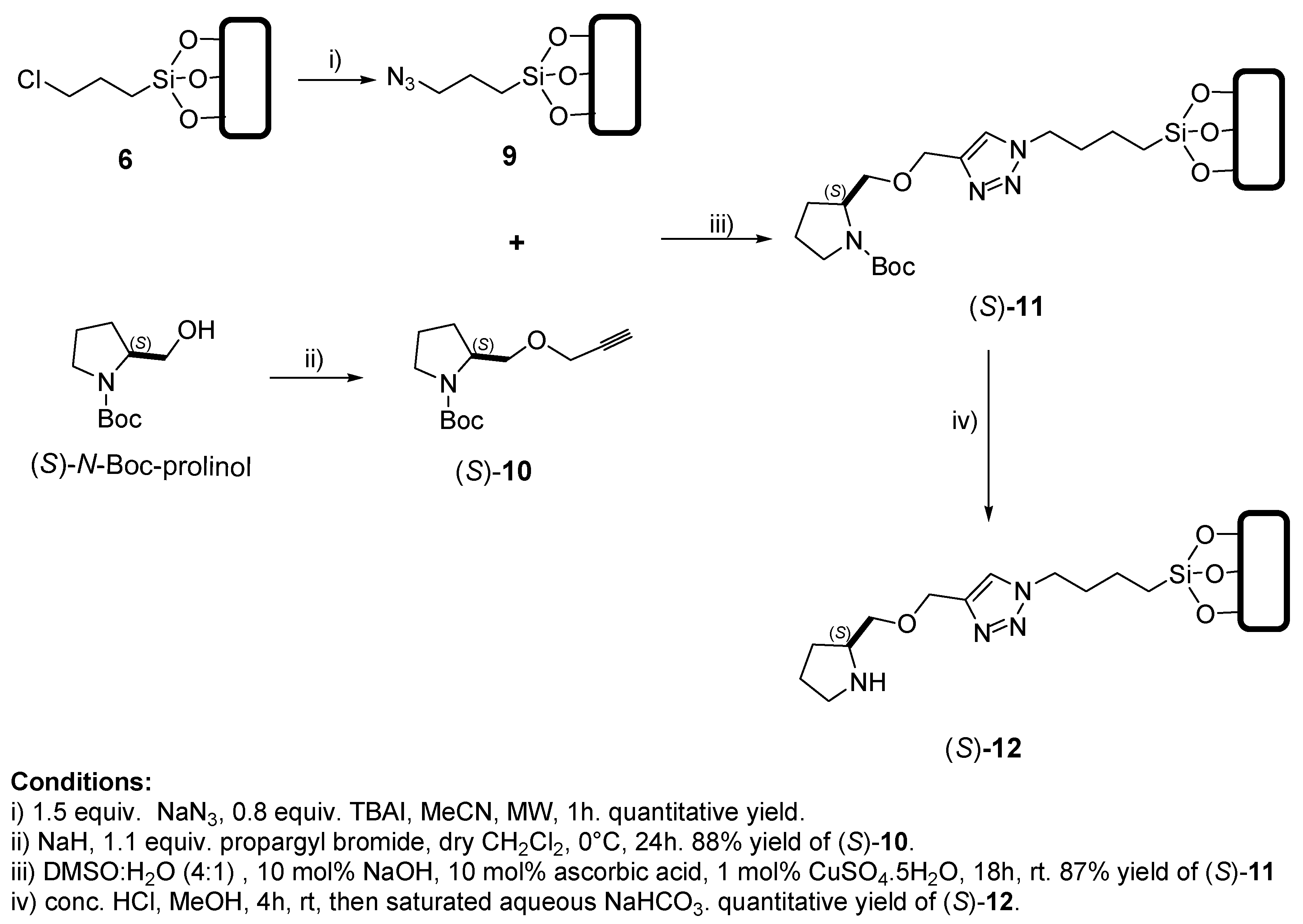
3.2.2. General Procedure 2: Propargylation Reaction, in the Synthesis of (S)-10, (2S,4R,1′-S)-16 and (2S,4R,1′-R)-16
3.2.3. General Procedure 3: Huisgen 1,3-dipolar Cycloaddition-Type Reaction (12h), in the Synthesis of (S)-11, (2S,4R,1′-S)-17 and (2S,4R,1′-R)-17
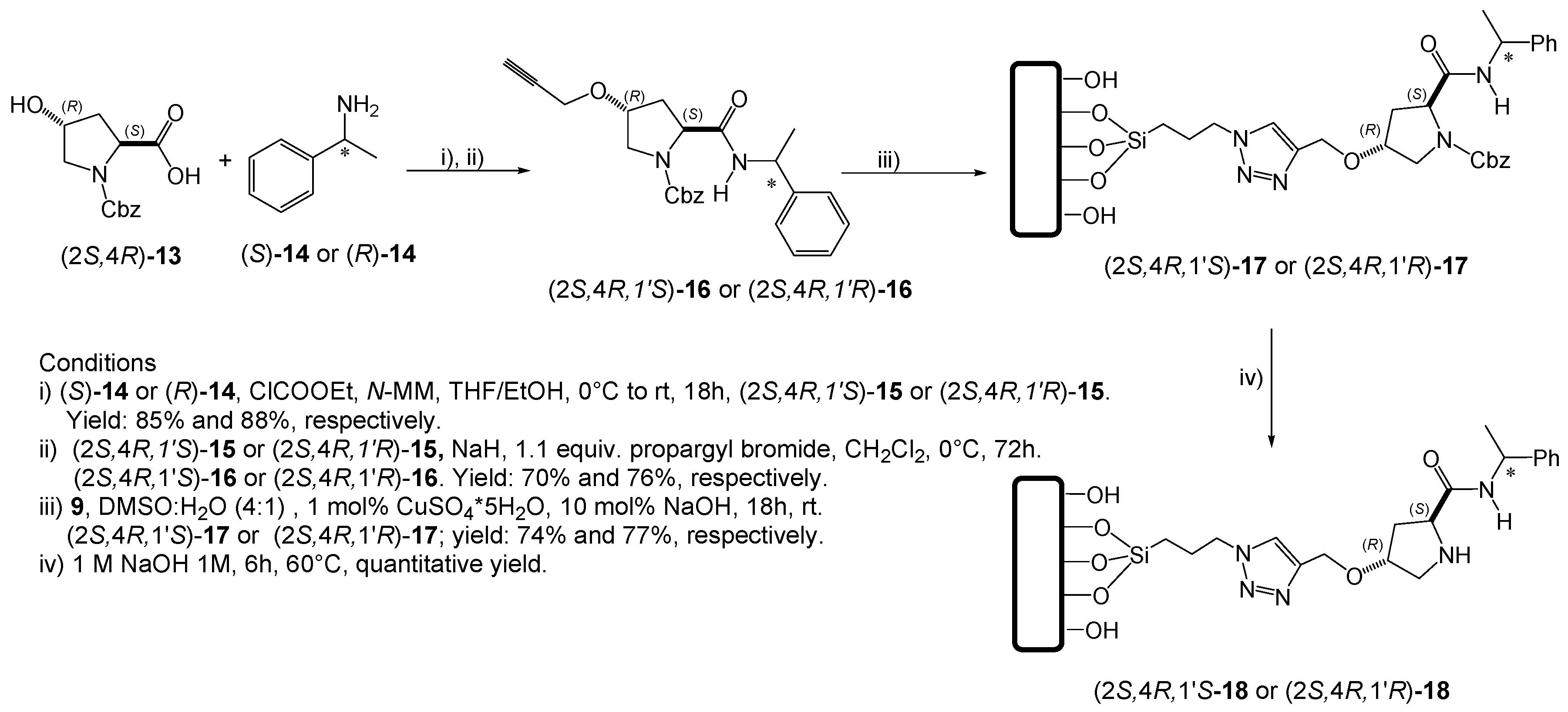
3.2.4. General Procedure 4: Coupling Reaction, in the Synthesis of (2S,4R,1′S)-15 or (2S,4R,1′R)-15
3.2.5. General Procedure 5: N-Boc Catalyst Deprotection Under Acidic Conditions
3.2.6. General Procedure 6: N-Cbz Catalyst Deprotection Under Basic Conditions
3.2.7. General Procedure 7: Activation of Catalysts (S)-5, (S)-12, (2S,4R,1′S)-18 and (2S,4R,1′R)-18.
General Procedure 7a: Reactivation of Used Silica-Supported Organocatalysts
3.2.8. General Procedure 8: Aldol Reaction
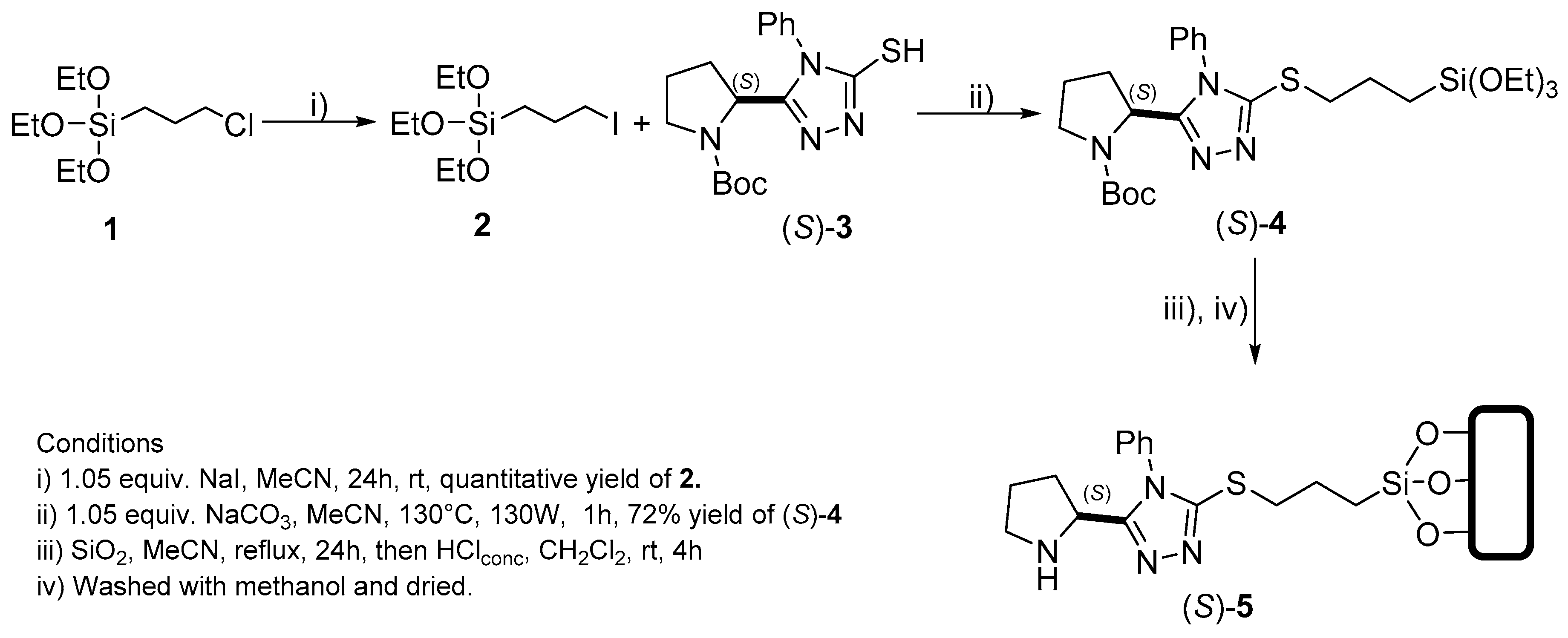
3.2.9. Procedure for Functionalization of Siloxy-OH Groups on Silica; Obtention of 6
3.3. Procedure for SN2 Type Reaction. Preparation of (S)-8
H and 13C NMR Spectra, Mass Spectra and Physical Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- List, B.; Lerner, L.A.; Barbas, C.F., III. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Dalko, P.I.; Moisan, L. The golden age of organocatalysis. Angew. Chem. Int. Ed. 2004, 43, 5138–5175. [Google Scholar] [CrossRef] [PubMed]
- Enders, D.; Grondal, C.; Huettl, M.R.M. Asymmetric Organocatalytic Domino Reactions. Angew. Chem. Int. Ed. 2007, 46, 1570–1581. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef]
- Trost, B.M.; Brindle, C.S. The direct catalytic asymmetric aldol reaction. Chem. Soc. Rev. 2010, 39, 1600–1632. [Google Scholar] [CrossRef] [Green Version]
- Benaglia, M.; Puglisi, A.; Cozzi, F. Polymer-Supported Organic Catalysts. Chem. Rev. 2003, 103, 3401–3429. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Giacalone, F.; Noto, R. Supported proline and proline-derivatives as recyclable organocatalysts. Chem. Soc. Rev. 2008, 37, 1666–1688. [Google Scholar] [CrossRef]
- Trindade, A.F.; Gois, P.M.P.; Afonso, C.A.M. Recyclable Stereoselective Catalysts. Chem. Rev. 2009, 109, 418–514. [Google Scholar] [CrossRef]
- Clapham, B.; Reger, T.S.; Janda, K.D. Polymer-supported catalysis in synthetic organic chemistry. Tetrahedron 2001, 57, 4637–4662. [Google Scholar] [CrossRef]
- Yan, J.; Wang, L. Merrifield Resin Supported Dipeptides: Efficient and Recyclable Organocatalysts for Asymmetric Aldol Reactions under Neat Reaction Conditions. Synthesis 2008, 2008, 2065–2072. [Google Scholar] [CrossRef]
- Hernández, J.G.; Juaristi, E. Recent efforts directed to the development of more sustainable asymmetric organocatalysis. Chem. Commun. 2012, 48, 5396–5409. [Google Scholar] [CrossRef] [PubMed]
- Thölke, S.; Zhu, H.; Jansen, D.; Octa-Smolin, F.; Thiele, M.; Kaupmees, K.; Leito, I.; Grimme, S.; Niemeyer, J. Cooperative Organocatalysis: A Systematic Investigation of Covalently Linked Organophosphoric Acids for the Stereoselective Transfer Hydrogenation of Quinolines. Eur. J. Org. Chem. 2019, 2019, 5190–5195. [Google Scholar] [CrossRef]
- Liu, N.; Xie, Y.-F.; Wang, C.; Li, S.-J.; Wei, D.; Li, M.; Dai, B. Cooperative Multifunctional Organocatalysts for Ambient Conversion of Carbon Dioxide into Cyclic Carbonates. ACS Catal. 2018, 8, 9945–9957. [Google Scholar] [CrossRef]
- Bukhryakov, K.V.; Desyatkin, V.G.; Rodionov, V.O. Cooperative organocatalysis of Mukaiyama-type aldol reactions by thioureas and nitro compounds. Chem. Commun. 2016, 52, 7576–7579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raja Mitra, R.; Niemeyer, J. Dual Brønsted-acid Organocatalysis: Cooperative Asymmetric Catalysis with Combined Phosphoric and Carboxylic Acids. ChemCatChem 2018, 10, 1221–1234. [Google Scholar] [CrossRef]
- Steiner, D.D.; Mase, N.; Barbas, C.F., III. Direct Asymmetric α-Fluorination of Aldehydes. Angew. Chem. Int. Ed. 2005, 44, 3706–3710. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.-T.; Harris, P.W.R.; Barker, D.; Brimble, M.A. Use of (S)-5-(2-Methylpyrrolidin-2-yl)-1H-tetrazole as a Novel and Enantioselective Organocatalyst for the Aldol Reaction. Eur. J. Org. Chem. 2008, 2008, 164–170. [Google Scholar] [CrossRef]
- Cruz-Hernández, C.; Landeros, J.M.; Juaristi, E. Multifunctional phosphoramide-(S)-prolinamide derivatives as efficient organocatalysts in asymmetric aldol and Michael reactions. New J. Chem. 2019, 43, 5455–5465. [Google Scholar] [CrossRef]
- Xu, L.W.; Lu, Y. Primary amino acids: Privileged catalysts in enantioselective organocatalysis. Org. Biomol. Chem. 2008, 6, 2047–2053. [Google Scholar] [CrossRef]
- Gerasimchuk, V.V.; Kucherenko, A.S.; Fakhrutdinov, A.N.; Medvedev, M.G.; Nelyubina, Y.V.; Zlotin, S.G. Towards Sustainable Amino Acid Derived Organocatalysts for Asymmetric syn-Aldol Reactions. Eur. J. Org. Chem. 2017, 2017, 2540–2544. [Google Scholar] [CrossRef]
- Kang, Q.K.; Selvakumar, S.; Maruoka, K. Asymmetric Synthesis of α-Amino Acids by Organocatalytic Biomimetic Transamination. Org. Lett. 2019, 21, 2294–2297. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, M.; Arrieta, A.; Arrastia, I.; Cossío, F.P. Organocatalysts Derived from Unnatural α-Amino Acids: Scope and Applications. Chem. Asian J. 2019, 14, 44–66. [Google Scholar] [CrossRef] [PubMed]
- Schnitzer, T.; Wennemers, H. Effect of β3-Amino Acids on the Performance of the Peptidic Catalyst H-d Pro-Pro-Glu-NH2. Helv. Chim. Acta 2019, 102, e1900070. [Google Scholar] [CrossRef]
- Schnitzer, T.; Budinská, A.; Wennemers, H. Organocatalysed conjugate addition reactions of aldehydes to nitroolefins with anti-selectivity. Nat. Catal. 2020, 3, 143–147. [Google Scholar] [CrossRef]
- Fingerhut, A.; Grau, D.; Tsogoeva, S.B. Peptides as Asymmetric Organocatalysts. In Sustainable Catalysis: Without Metals or Other Endangered Elements, Part 1; Royal Society of Chemistry: London, UK, 2015; Chapter 13; pp. 309–353. [Google Scholar] [CrossRef]
- Wiesner, M.; Revell, J.D.; Tonazzi, S.; Wennemers, H. Peptide catalyzed asymmetric conjugate addition reactions of aldehydes to nitroethylene—A convenient entry into γ2-amino acids. J. Am. Chem. Soc. 2008, 130, 5610–5611. [Google Scholar] [CrossRef] [PubMed]
- Davie, E.A.C.; Mennen, S.M.; Xu, Y.; Miller, S.J. Asymmetric catalysis mediated by synthetic peptides. Chem. Rev. 2007, 107, 5759–5812. [Google Scholar] [CrossRef]
- Machuca, E.; Juaristi, E. Organocatalytic activity of α,α-dipeptide derivatives of (S)-proline in the asymmetric aldol reaction in absence of solvent. Evidence for non-covalent π-π Interactions in the transition state. Tetrahedron Lett. 2015, 56, 1144–1148. [Google Scholar] [CrossRef]
- Machuca, E.; Rojas, Y.; Juaristi, E. Synthesis and Evaluation of (S)-Proline-Containing α,β-Dipeptides as Organocatalysts in Solvent-Free Asymmetric Aldol Reactions Under Ball-Milling Conditions. Asian J. Org. Chem. 2015, 4, 46–53. [Google Scholar] [CrossRef]
- Hernández, J.G.; Juaristi, E. Asymmetric aldol reaction organocatalyzed by (S)-proline-containing dipeptides: Improved stereoinduction under solvent-free conditions. J. Org. Chem. 2011, 76, 1464–1467. [Google Scholar] [CrossRef]
- Jiménez-González, E.; Gabriela Ávila-Ortiz, C.; González-Olvera, R.; Vargas-Caporali, J.; Dewynter, G.; Juaristi, E. Solution-phase synthesis of novel seven-membered cyclic dipeptides containing α- and β-amino acids. Tetrahedron 2012, 68, 9842–9852. [Google Scholar] [CrossRef]
- Koutoulogenis, G.; Kaplaneris, N.; Kokotos, C.G. (Thio)urea-mediated synthesis of functionalized six-membered rings with multiple chiral centers. Beilstein J. Org. Chem. 2016, 12, 462–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revelou, P.; Kokotos, C.G.; Moutevelis-Minakakis, P. Novel prolinamide-ureas as organocatalysts for the asymmetric aldol reaction. Tetrahedron 2012, 68, 8732–8738. [Google Scholar] [CrossRef]
- Bera, M.; Ghosh, T.K.; Akhuli, B.; Ghosh, P. Tris-ureas as versatile and highly efficient organocatalysts for Michael addition reactions of nitro-olefins: Mechanistic insight from in-situ diagnostics. J. Mol. Catal. A Chem. 2015, 408, 287–295. [Google Scholar] [CrossRef]
- Cruz-Hernández, C.; Martínez-Martínez, E.; Hernández-González, P.E.; Juaristi, E. Synthesis of a New N-Diaminophosphoryl-N’-[(2S)-2-pyrrolidinylmethyl]thiourea as a Chiral Organocatalyst for the Stereoselective Michael Addition of Cyclohexanone to Nitrostyrenes and Chalcones—Application in Cascade Processes for the Synthesis of Polycyclic Systems. Eur. J. Org. Chem. 2018, 2018, 6890–6900. [Google Scholar] [CrossRef]
- Wageling, N.B.; Decato, D.A.; Berryman, O.B. Steric effects of pH switchable, substituted (2-pyridinium)urea organocatalysts: A solution and solid phase study. Supramol. Chem. 2018, 30, 1004–1010. [Google Scholar] [CrossRef]
- Limnios, D.; Kokotos, C.G. Ureas and Thioureas as Asymmetric Organocatalysts. In Sustainable Catalysis: Without Metals or Other Endangered Elements, Part 2; Royal Society of Chemistry: London, UK, 2015; Chapter 19; pp. 196–255. [Google Scholar]
- Vakulya, B.; Varga, S.; Soós, T. Epi-cinchona based thiourea organocatalyst family as an efficient asymmetric Michael addition promoter: Enantioselective conjugate addition of nitroalkanes to chalcones and α,β-unsaturated N-acylpyrroles. J. Org. Chem. 2008, 73, 3475–3480. [Google Scholar] [CrossRef]
- Yoon, T.P.; Jacobsen, E.N. Highly enantioselective thiourea-catalyzed nitro-Mannich reactions. Angew. Chem. Int. Ed. 2005, 44, 466–468. [Google Scholar] [CrossRef]
- Gao, P.; Wang, C.; Wu, Y.; Zhou, Z.; Tang, C. Sugar-derived bifunctional thiourea organocatalyzed asymmetric Michael addition of acetylacetone to nitroolefins. Eur. J. Org. Chem. 2008, 2008, 4563–4566. [Google Scholar] [CrossRef]
- Lee, H.J.; Arumugam, N.; Almansour, A.I.; Kumar, R.S.; Maruoka, K. Design of New Amino Tf-Amide Organocatalysts: Environmentally Benign Approach to Asymmetric Aldol Synthesis. Synlett 2019, 30, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Lin, L.; Feng, X. Amide-based bifunctional organocatalysts in asymmetric reactions. Chem. Commun. 2009, 41, 6145–6158. [Google Scholar] [CrossRef]
- Cruz-Hernández, C.; Hernández-González, P.E.; Juaristi, E. Proline-Glycine Dipeptidic Derivatives of Chiral Phosphoramides as Organocatalysts for the Enantiodivergent Aldol Reaction of Aryl Aldehydes and Isatins with Cyclohexanone in the Presence of Water. Synthesis 2018, 50, 3445–3459. [Google Scholar] [CrossRef]
- Vega-Peñaloza, A.; Sánchez-Antonio, O.; Escudero-Casao, M.; Tasnádi, G.; Fülöp, F.; Juaristi, E. Stereoselective synthesis of chiral pyrrolidine derivatives of (+)-α-pinene containing a β-amino acid moiety. Synthesis 2013, 45, 2458–2468. [Google Scholar] [CrossRef]
- Gavendova, M.; Lennon, C.M.; Coffey, L.; Manesiotis, P.; Kinsella, M. Novel β-amino Amide Organocatalysts for the Synthesis of Pharmaceutically Relevant Oxindoles. ChemistrySelect 2019, 4, 8246–8254. [Google Scholar] [CrossRef]
- Groselj, U. Camphor-Derivatives in Asymmetric Organocatalysis—Synthesis and Application. Curr. Org. Chem. 2015, 19, 2048–2074. [Google Scholar] [CrossRef]
- Pan, L.; Ding, X.; Ding, J.; Li, D.; Chen, J.; Zuo, X.; An, R. Design and Synthesis of L-Proline Derivatives as Enantioselective Organocatalysts for Synthesis of the (S)-Wieland-Miescher Ketone. ChemistrySelect 2017, 2, 11999–12005. [Google Scholar] [CrossRef]
- Haldar, S.; Saha, S.; Mandal, S.; Jana, C.K. C-H functionalization enabled stereoselective Ugi-azide reaction to α-tetrazolyl alicyclic amines. Green Chem. 2018, 20, 3463–3467. [Google Scholar] [CrossRef]
- Rani, R.; Peddinti, R.K. Camphor-10-sulfonamide-based prolinamide organocatalyst for the direct intermolecular aldol reaction between ketones and aromatic aldehydes. Tetrahedron Asymmetry 2010, 21, 775–779. [Google Scholar] [CrossRef]
- Lin, J.-H.; Xiao, J.-C. Recent advances in asymmetric fluorination and fluoroalkylation reactions via organocatalysis. Tetrahedron Lett. 2014, 55, 6147–6155. [Google Scholar] [CrossRef]
- Obregón-Zúñiga, A.; Milán, M.; Juaristi, E. Improving the Catalytic Performance of (S)-Proline as Organocatalyst in Asymmetric Aldol Reactions in the Presence of Solvate Ionic Liquids: Involvement of a Supramolecular Aggregate. Org. Lett. 2017, 19, 1108–1111. [Google Scholar] [CrossRef]
- Reyes-Rangel, G.; Vargas-Caporali, J.; Juaristi, E. In search of diamine analogs of the α,α-diphenyl prolinol privileged chiral organocatalyst. Synthesis of diamine derivatives of α,α-diphenyl-(S)-prolinol and their application as organocatalysts in the asymmetric Michael and Mannich reactions. Tetrahedron 2016, 72, 379–391. [Google Scholar] [CrossRef]
- Reyes-Rangel, G.; Jiménez-González, E.; Olivares-Romero, J.L.; Juaristi, E. Enantioselective synthesis of β-amino acids using hexahydrobenzoxazolidinones as chiral auxiliaries. Tetrahedron Asymmetry 2008, 19, 2839–2849. [Google Scholar] [CrossRef]
- Luan, Y.; Zheng, N.; Qi, Y.; Tang, J.; Wang, G. Merging metal-organic framework catalysis with organocatalysis: A thiourea functionalized heterogeneous catalyst at the nanoscale. Catal. Sci. Technol. 2014, 4, 925–929. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, T.Y.; Telfer, S.G. Modulating the performance of an asymmetric organocatalyst by tuning its spatial environment in a metal-organic framework. J. Am. Chem. Soc. 2017, 139, 13936–13943. [Google Scholar] [CrossRef] [PubMed]
- Akagawa, K.; Takigawa, S.; Mano, E.; Kudo, K. Sequential oxidation/asymmetric aldol reaction of primary alcohols by resin-supported catalysts. Tetrahedron Lett. 2011, 52, 770–773. [Google Scholar] [CrossRef]
- Bartók, M. Advances in immobilized organocatalysts for the heterogeneous asymmetric direct aldol reactions. Catal. Rev. Sci. Eng. 2015, 57, 192–255. [Google Scholar] [CrossRef]
- Xie, Y.; Sharma, K.K.; Anan, A.; Wang, G.; Biradar, A.V.; Asefa, T. Efficient solid-base catalysts for aldol reaction by optimizing the density and type of organoamine groups on nanoporous silica. J. Catal. 2009, 265, 131–140. [Google Scholar] [CrossRef]
- Elmekawy, A.A.; Sweeney, J.B.; Brown, D.R. Efficient synthesis of supported proline catalysts for asymmetric aldol reactions. Catal. Sci. Technol. 2015, 5, 690–696. [Google Scholar] [CrossRef]
- Andreae, M.R.M.; Davis, A.P. Heterogeneous catalysis of the asymmetric aldol reaction by solid-supported proline-terminated peptides. Tetrahedron Asymmetry 2005, 16, 2487–2492. [Google Scholar] [CrossRef]
- Yang, Z.; Peng, H.; Wang, W.; Liu, T. Crystallization behavior of poly(ε-caprolactone)/layered double hydroxide nanocomposites. J. Appl. Polym. Sci. 2010, 116, 2658–2667. [Google Scholar] [CrossRef]
- Rostamnia, S.; Doustkhah, E. Nanoporous silica-supported organocatalyst: A heterogeneous and green hybrid catalyst for organic transformations. RSC Adv. 2014, 4, 28238–28248. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Giacalone, F.; Marculescu, A.M.; Noto, R. Novel prolinamide-supported polystyrene as highly stereoselective and recyclable organocatalyst for the aldol reaction. Adv. Synth. Catal. 2008, 350, 1397–1405. [Google Scholar] [CrossRef]
- Giacalone, F.; Gruttadauria, M.; Marculescu, A.M.; Noto, R. Polystyrene-supported proline and prolinamide. Versatile heterogeneous organocatalysts both for asymmetric aldol reaction in water and α-selenenylation of aldehydes. Tetrahedron Lett. 2007, 48, 255–259. [Google Scholar] [CrossRef]
- Price, G.A.; Hassan, A.; Chandrasoma, N.; Bogdan, A.R.; Djuric, S.W.; Organ, M.G. Pd-PEPPSI-IPent-SiO2: A Supported Catalyst for Challenging Negishi Coupling Reactions in Flow. Angew. Chem. Int. Ed. 2017, 56, 13347–13350. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, T.E.; Vestli, K.; Fredriksen, K.A.; Hansen, F.K.; Hansen, T. Synthesis of acrylic polymer beads for solid-supported proline-derived organocatalysts. Org. Lett. 2009, 11, 2968–2971. [Google Scholar] [CrossRef]
- Sasidharan, M.; Kiyozumi, Y.; Mal, N.K.; Mizukami, F. Synthesis, characterization, and application of mesoporous silica functionalized with alkyl-hydroperoxides. Adv. Funct. Mater. 2006, 16, 1853–1858. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Giacalone, F.; Marculescu, A.M.; Salvo, A.M.P.; Noto, R. Stereoselective aldol reaction catalyzed by a highly recyclable polystyrene supported substituted prolinamide catalyst. ARKIVOC 2009, 8, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Khalafi-Nezhad, A.; Shahidzadeh, E.S.; Sarikhani, S.; Panahi, F. A new silica-supported organocatalyst based on L-proline: An efficient heterogeneous catalyst for one-pot synthesis of spiroindolones in water. J. Mol. Catal. A Chem. 2013, 379, 1–8. [Google Scholar] [CrossRef]
- Ferré, M.; Cattoën, X.; Wong Chi Man, M.; Pleixats, R. Recyclable Silica-Supported Proline Sulphonamide Organocatalysts for Asymmetric Direct Aldol Reaction. ChemistrySelect 2016, 1, 6741–6748. [Google Scholar] [CrossRef]
- Rajput, A.P.; Nagarale, D.V. Modern Synthetic Tool L-Proline as an Organocatalyst. J. Chem. Pharm. Res. 2016, 8, 557–575. [Google Scholar]
- Corma, A.; Garcia, H. Silica-bound homogenous catalysts as recoverable and reusable catalysts in organic synthesis. Adv. Synth. Catal. 2006, 348, 1391–1412. [Google Scholar] [CrossRef]
- Rodríguez-Escrich, C.; Pericàs, M.A. Organocatalysis on Tap: Enantioselective Continuous Flow Processes Mediated by Solid-Supported Chiral Organocatalysts. Eur. J. Org. Chem. 2015, 2015, 1173–1188. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Antonio, O.; Juaristi, E. Synthesis of a new chiral organocatalyst derived from (S)-proline containing a 1,2,4-triazolyl moiety and its application in the asymmetric aldol reaction. Importance of one molecule of water generated in situ. Tetrahedron Lett. 2019, 60, 151128. [Google Scholar] [CrossRef]
- Han, Y.R.; Park, J.W.; Kim, H.; Ji, H.; Lim, S.H.; Jun, C.H. A one-step co-condensation method for the synthesis of well-defined functionalized mesoporous SBA-15 using trimethallylsilanes as organosilane sources. Chem. Commun. 2015, 51, 17084–17087. [Google Scholar] [CrossRef]
- Ferré, M.; Pleixats, R.; Wong Chi Man, M.; Cattoën, X. Recyclable organocatalysts based on hybrid silicas. In Proceedings of the Green Chemistry; Royal Society of Chemistry: London, UK, 2016; Volume 18, pp. 881–922. [Google Scholar] [CrossRef] [Green Version]
- Putz, A.M.; Almásy, L.; Len, A.; Ianăşi, C. Functionalized silica materials synthesized via co-condensation and post-grafting methods. Fuller. Nanotub. Carbon Nanostruct. 2019, 27, 323–332. [Google Scholar] [CrossRef]
- Scatena, G.S.; de la Torre, A.F.; Cass, Q.B.; Rivera, D.G.; Paixão, M.W. Multicomponent Approach to Silica-Grafted Peptide Catalysts: A 3 D Continuous-Flow Organocatalytic System with On-line Monitoring of Conversion and Stereoselectivity. ChemCatChem 2014, 6, 3208–3214. [Google Scholar] [CrossRef] [Green Version]
- Puglisi, A.; Benaglia, M.; Annunziata, R.; Chiroli, V.; Porta, R.; Gervasini, A. Chiral hybrid inorganic-organic materials: Synthesis, characterization, and application in stereoselective organocatalytic cycloadditions. J. Org. Chem. 2013, 78, 11326–11334. [Google Scholar] [CrossRef]
- Brühwiler, D. Postsynthetic functionalization of mesoporous silica. Nanoscale 2010, 2, 887–892. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.M.; Gao, J.S.; Yang, Q.H.; Li, C. Large-pore mesoporous ethane-silicas as efficient heterogeneous asymmetric catalysts. In Studies in Surface Science and Catalysis; Elsevier Inc.: Amsterdam, The Netherlands, 2007; Volume 170, pp. 1252–1259. [Google Scholar] [CrossRef]
- Sunada, K.; Takenaka, K.; Shiomi, T. Synthesis of polychloroprene-silica composites by sol-gel method in the presence of modified polychloroprene containing triethoxysilyl group. J. Appl. Polym. Sci. 2005, 97, 1545–1552. [Google Scholar] [CrossRef]
- Fischer, T.; Duong, Q.N.; García Mancheño, O. Triazole-Based Anion-Binding Catalysis for the Enantioselective Dearomatization of N-Heteroarenes with Phosphorus Nucleophiles. Chem. Eur. J. 2017, 23, 5983–5987. [Google Scholar] [CrossRef]
- Riente, P.; Yadav, J.; Pericàs, M.A. A click strategy for the immobilization of Macmillan organocatalysts onto polymers and magnetic nanoparticles. Org. Lett. 2012, 14, 3668–3671. [Google Scholar] [CrossRef]
- García Mancheño, O.; Asmus, S.; Zurro, M.; Fischer, T. Highly Enantioselective Nucleophilic Dearomatization of Pyridines by Anion-Binding Catalysis. Angew. Chem. Int. Ed. 2015, 54, 8823–8827. [Google Scholar] [CrossRef] [PubMed]
- Zurro, M.; Asmus, S.; Bamberger, J.; Beckendorf, S.; García Mancheño, O. Chiral Triazoles in Anion-Binding Catalysis: New Entry to Enantioselective Reissert-Type Reactions. Chem. Eur. J. 2016, 22, 3785–3793. [Google Scholar] [CrossRef] [PubMed]
- Goren, K.; Kehat, T.; Portnoy, M. Elucidation of Architectural Requirements from a Spacer in Supported Proline-Based Catalysts of Enantioselective Aldol Reaction. Adv. Synth. Catal. 2009, 351, 59–65. [Google Scholar] [CrossRef]
- Vega-Peñaloza, A.; Sánchez-Antonio, O.; Ávila-Ortiz, C.G.; Escudero-Casao, M.; Juaristi, E. An alternative synthesis of chiral (S)-proline derivatives that contain a thiohydantoin moiety and their application as organocatalysts in the asymmetric Michael addition reaction under solvent-free conditions. Asian J. Org. Chem. 2014, 3, 487–496. [Google Scholar] [CrossRef]
- Sousa, C.A.D.; Sampaio-Dias, I.E.; Rizzo-Aguiar, F.; Garcia-Mera, X.; Rodríguez-Borges, J.E. Enantiopure synthesis of 7-(1-pyrindanyl)propargyl ethers as rasagiline analogues via chemical or enzymatic resolution of 1-pyrindan-7-ol. RSC Adv. 2015, 5, 104509–104515. [Google Scholar] [CrossRef]
- Bew, S.P.; Hiatt-Gipson, G.D. Synthesis of C-propargylic esters of N-protected amino acids and peptides. J. Org. Chem. 2010, 75, 3897–3899. [Google Scholar] [CrossRef]
- Escudero-Casao, M.; Vega-Penaloza, A.; Juaristi, E. Enantiopure 1,2,3-Triazolyl-β-amino Acids via Click Cycloaddition Reaction on Racemic Alkynyl Precursors Followed by Separation of Stereoisomers. Curr. Top. Med. Chem. 2014, 14, 1257–1270. [Google Scholar] [CrossRef]
- Pellissier, H. Recent Developments in Enantioselective Organocatalytic Michael Reactions in Aqueous Media. Curr. Org. Chem. 2018, 21, 323–344. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Sun, J. How Understanding the Role of an Additive Can Lead to an Improved Synthetic Protocol without an Additive: Organocatalytic Synthesis of Chiral Diarylmethyl Alkynes. Angew. Chem. Int. Ed. 2017, 56, 11966–11970. [Google Scholar] [CrossRef]
- Hong, L.; Sun, W.; Yang, D.; Li, G.; Wang, R. Additive Effects on Asymmetric Catalysis. Chem. Rev. 2016, 116, 4006–4123. [Google Scholar] [CrossRef]
- Avila-Ortiz, C.G.; López-Ortiz, M.; Vega-Peñaloza, A.; Regla, I.; Juaristi, E. Use of (R)-Mandelic Acid as Chiral Co-Catalyst in the Michael Addition Reaction Organocatalyzed by (1S,4S)-2-Tosyl-2,5-diazabicyclo[2.2.1]heptane under Solvent-Free Conditions. Asymmetric Catal. 2015, 2, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Cobb, A.J.A.; Shaw, D.M.; Ley, S.V. 5-Pyrrolidin-2-yltetrazole: A New, Catalytic, More Soluble Alternative to Proline in an Organocatalytic Asymmetric Mannich-type Reaction. Synlett 2004, 15, 558–560. [Google Scholar] [CrossRef]
- Cobb, A.J.A.; Shaw, D.M.; Longbottom, D.A.; Gold, J.B.; Ley, S.V. Organocatalysis with proline derivatives: Improved catalysts for the asymmetric Mannich, nitro-Michael and aldol reactions. Org. Biomol. Chem. 2005, 3, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Torii, H.; Nakadai, M.; Ishihara, K.; Saito, S.; Yamamoto, H. Asymmetric direct aldol reaction assisted by water and a proline-derived tetrazole catalyst. Angew. Chem. Int. Ed. 2004, 43, 1983–1986. [Google Scholar] [CrossRef]
- Hartikka, A.; Arvidsson, P.I. Rational design of asymmetric organocatalysts—Increased reactivity and solvent scope with a tetrazolic acid. Tetrahedron Asymmetry 2004, 15, 1831–1834. [Google Scholar] [CrossRef]
- Nakashima, E.; Yamamoto, H. Process Catalyst Mass Efficiency by Using Proline Tetrazole Column-Flow System. Chem. Eur. J. 2018, 24, 1076–1079. [Google Scholar] [CrossRef]
- Nakashima, E.; Yamamoto, H. Asymmetric Aldol Synthesis: Choice of Organocatalyst and Conditions. Chem. Asian J. 2017, 12, 41–44. [Google Scholar] [CrossRef]
- Krištofíková, D.; Modrocká, V.; Mečiarová, M.; Šebesta, R. Green Asymmetric Organocatalysis. ChemSusChem 2020, 13, 2828–2858. [Google Scholar] [CrossRef]
- Tao, Y.; Dong, R.; Pavlidis, I.V.; Chen, B.; Tan, T. Using imidazolium-based ionic liquids as dual solvent-catalysts for sustainable synthesis of vitamin esters: Inspiration from bio- and organo-catalysis. Green Chem. 2016, 18, 1240–1248. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, P.; Singh Chimni, S. Mechanochemistry assisted asymmetric organo-catalysis: A sustainable approach. Beilstein J. Org. Chem. 2012, 8, 2132–2141. [Google Scholar] [CrossRef] [Green Version]
- Chowdari, N.S.; Ramachary, D.B.; Barbas, C.F., III. Organocatalysis in Ionic Liquids: Highly Efficient L-Proline-Catalyzed Direct Asymmetric Mannich Reactions Involving Ketone and Aldehyde Nucleophiles. Synlett 2003, 2003, 1906–1909. [Google Scholar] [CrossRef]
- Oliveira, M.R.; Deon, M.; Benvenutti, E.V.; Barros, V.A.; de Melo, D.C.; Franceschi, E.; Egues, S.M.; De Conto, J.F. Effect of microwave irradiation on the structural, chemical, and hydrophilicity characteristics of ordered mesoporous silica SBA-15. J. Sol Gel Sci. Technol. 2020, 94, 708–718. [Google Scholar] [CrossRef]
- Corresponding Specification Data about the Commercial Silica. Available online: https://www.mn-net.com/LCadsorbents/Irregularsilica/StandardsilicaforLC/tabid/6009/language/en-US/Default.aspx (accessed on 12 September 2020).
Sample Availability: Samples described in the manuscript are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst | Additive | Time (h) | dr[c] (anti/syn) | er[d] (anti/syn) | Yield % [e] |
|---|---|---|---|---|---|---|
| 1 | Commercial silica | None | 96 | - | - | - |
| 2 | (S)-5 [a] | None | 20 | 78:21 | 65:35/60:40 | 98 |
| 3 | (S)-5 [a] | H2O | 24 | 74:25 | 70:30/65:35 | 99 |
| 4 | (S)-5 [a] | p-nitro benzoic acid | 24 | 70:30 | 85:15/60:40 | 99 |
| 5 | (S)-5 [a] | salicylic acid | 24 | 70:30 | 65:35/60:40 | 99 |
| 6 | (S)-5 [a,f] | p-nitro-benzoic acid | 36 | 76:24 | 96:4/70:30 | 99 |
| 7 | (S)-5 [a,f] | salicylic acid | 36 | 75:25 | 95:5/60:40 | 98 |
| 8 | (S)-12 [b] | None | 36 | 75:25 | 90:10/80:20 | 99 |
| 9 | (S)-12 [b] | H2O | 36 | 80:20 | 90:10/80:20 | 96 |
| 10 | (S)-12 [b,f] | p-nitro- benzoic acid | 36 | 70:30 | 90:10/60:40 | 96 |
| 11 | (2S,4R,1′S)-18 [a,f] | None | 22 | 70:30 | 65:35/65:30 | 96 |
| 12 | (2S,4R,1′R)-18 [a,f] | None | 24 | 70:30 | 60:40/60:40 | 97 |
| Entry | Product | R | Time (h) | dr[a] (anti/syn) | er[b] (for anti isomer) | Yield % [c] |
|---|---|---|---|---|---|---|
| 1 | 20 | 2-NO2 | 36 | 70:30 | 70:30 | 95 |
| 2 | 21 | 3-NO2 | 36 | 75:25 | 70:30 | 95 |
| 3 | 22 | 2-CF3 | 72 | 85:15 | 60:40 | 95 |
| 4 | 23 | 4-Cl | 48 | 70:30 | 70:30 | 94 |
| 5 | 24 | 4-CF3 | 36 | 75:25 | 65:35 | 96 |
| 6 | 25 | 4-Br | 36 | 80:20 | 80:20 | 99 |
| 7 | 26 | -H | 48 | 70:30 | 60:40 | 94 |

| Entry | Product | R | Time (h) | er[a] | Yield % [b] |
|---|---|---|---|---|---|
| 1 | 27 | 4-NO2 | 48 | 70:30 | 98 |
| 2 | 28 | 2-NO2 | 48 | 65:35 | 98 |
| 3 | 29 | 2-Cl | 48 | 65:35 | 98 |
| 4 | 30c | 4-CF3 | 36 | 60:40 | 98 |
| 5 | 31 | 4-Br | 36 | 65:35 | 97 |
| 6 | 32c | 4-Cl | 72 | 70:30 | 97 |
| 7 | 33 | 4-F | 48 | 65:35 | 99 |
| 8 | 34 | -H | 96 | 60:40 | 98 |
| 9 | 35c | 4-CH3 | 96 | 60:40 | 98 |
| 10 | 36c | 3-Br | 48 | 60:40 | 99 |

| Product | -R | Time (h) | dr (anti) [a] | er (anti) [b] | Yield % [c] |
|---|---|---|---|---|---|
| 38 | -H | 10 | 70:30 | 70:30 | 88 |
| 39 | 5-NO2 | 9 | 75:25 | 60:40 | 85 |
| 40 | 7-Cl | 12 | 80:20 | 60:40 | 90 |

| Cycle | Er [a] | Yield % [b] |
|---|---|---|
| 1 | 70:30 | 98 |
| 2 | 65:35 | 98 |
| 3 | 65:35 | 99 |
| 4 | 65:35 | 98 |
| 5 | 66:34 | 98 |
| 6 | 65:35 | 97 |
| 7 | 65:35 | 97 |
| 8 | 65:35 | 99 |
| 9 | 60:40 | 98 |
| 10 | 60:40 | 98 |
| 11 | 60:40 | 98 |
| 12 | 60:40 | 99 |
| 13 | 60:40 | 96 |
| 14 | 60:40 | 97 |
| 15 | 60:40 | 96 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Antonio, O.; Romero-Sedglach, K.A.; Vázquez-Orta, E.C.; Juaristi, E. New Mesoporous Silica-Supported Organocatalysts Based on (2S)-(1,2,4-Triazol-3-yl)-Proline: Efficient, Reusable, and Heterogeneous Catalysts for the Asymmetric Aldol Reaction. Molecules 2020, 25, 4532. https://doi.org/10.3390/molecules25194532
Sánchez-Antonio O, Romero-Sedglach KA, Vázquez-Orta EC, Juaristi E. New Mesoporous Silica-Supported Organocatalysts Based on (2S)-(1,2,4-Triazol-3-yl)-Proline: Efficient, Reusable, and Heterogeneous Catalysts for the Asymmetric Aldol Reaction. Molecules. 2020; 25(19):4532. https://doi.org/10.3390/molecules25194532
Chicago/Turabian StyleSánchez-Antonio, Omar, Kevin A. Romero-Sedglach, Erika C. Vázquez-Orta, and Eusebio Juaristi. 2020. "New Mesoporous Silica-Supported Organocatalysts Based on (2S)-(1,2,4-Triazol-3-yl)-Proline: Efficient, Reusable, and Heterogeneous Catalysts for the Asymmetric Aldol Reaction" Molecules 25, no. 19: 4532. https://doi.org/10.3390/molecules25194532