
Recent Advances in Asymmetric Synthesis of Pyrrolidine-Based Organocatalysts and Their Application: A 15-Year Update

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
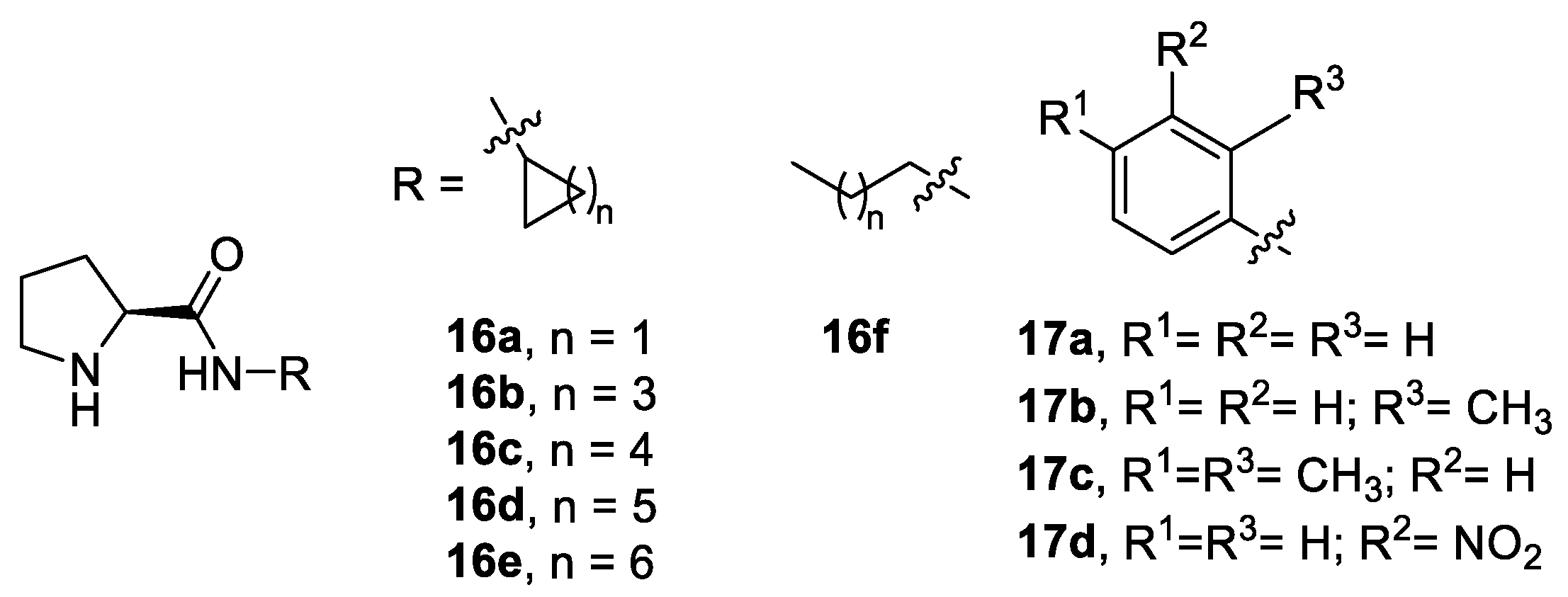{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
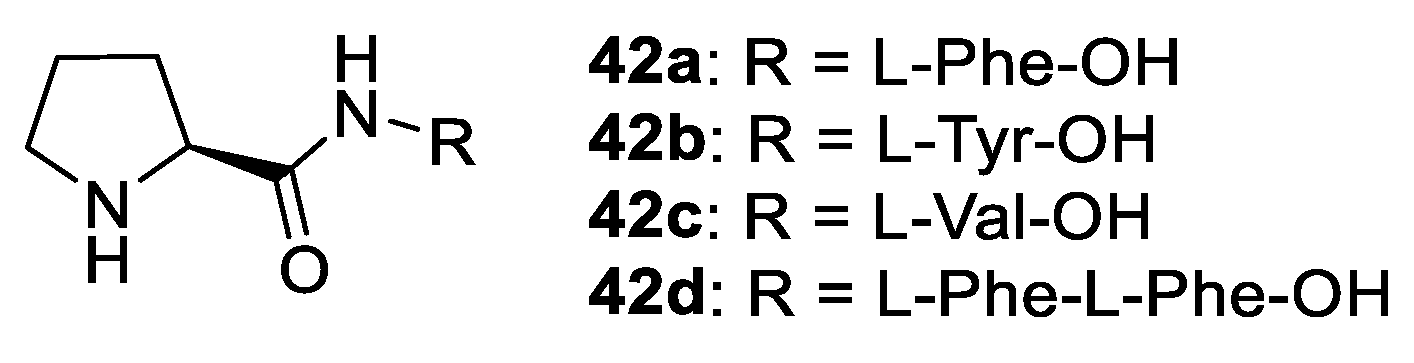{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
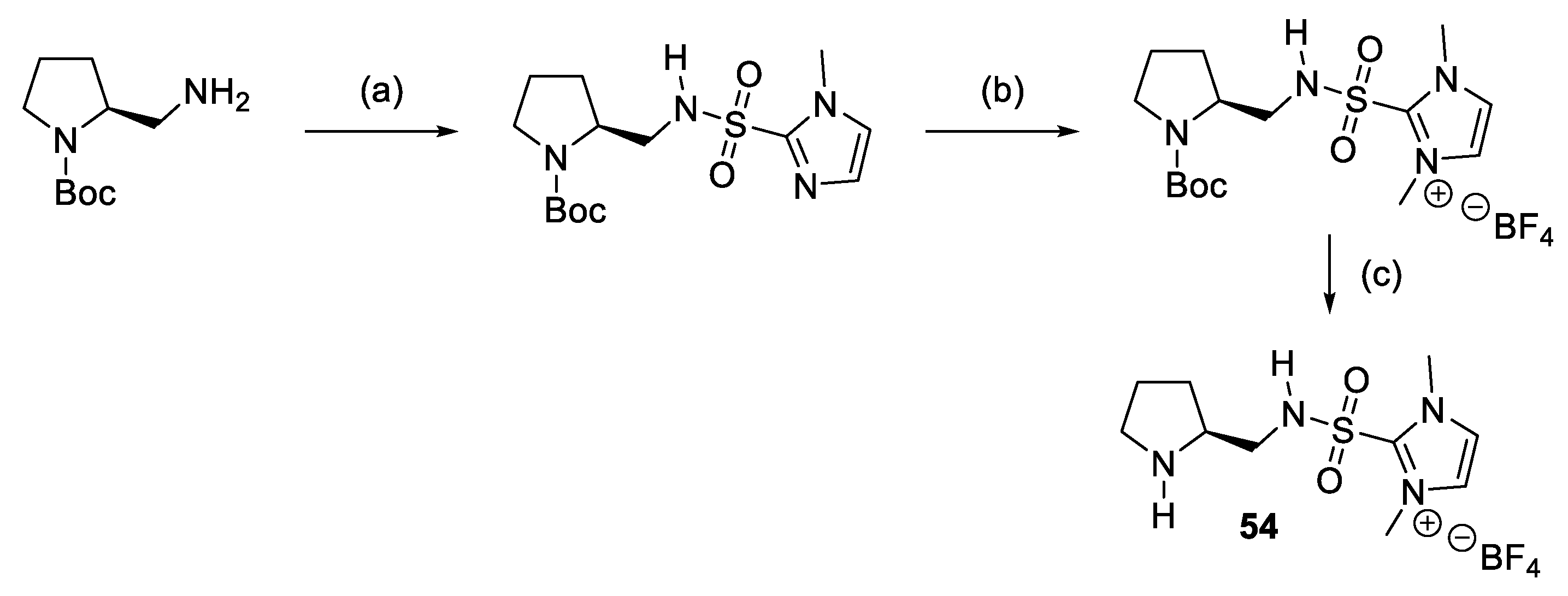{kind=link}
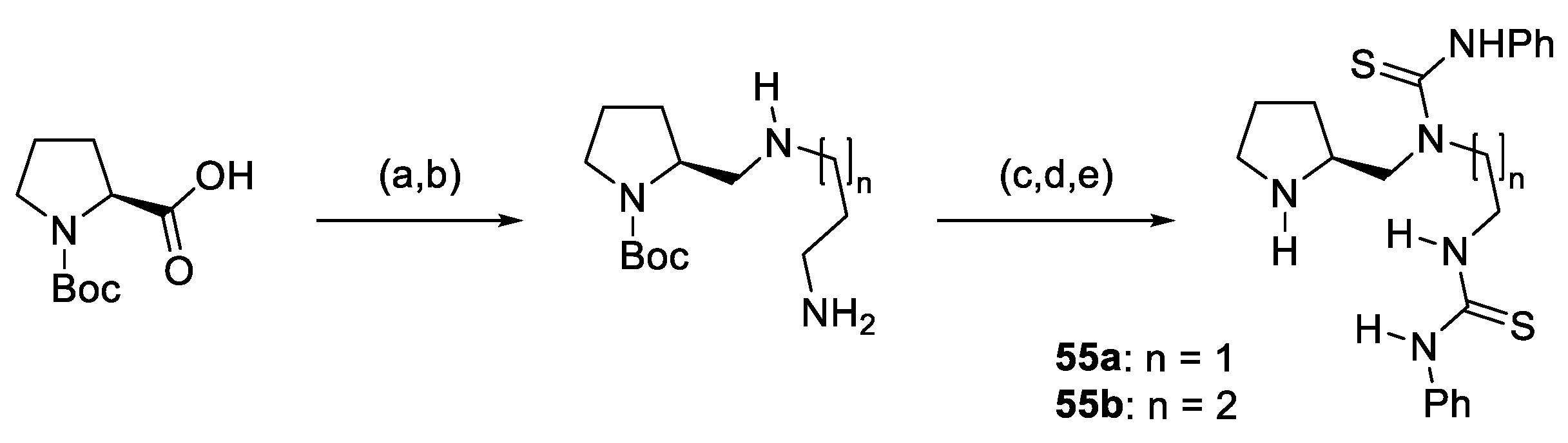{kind=link}
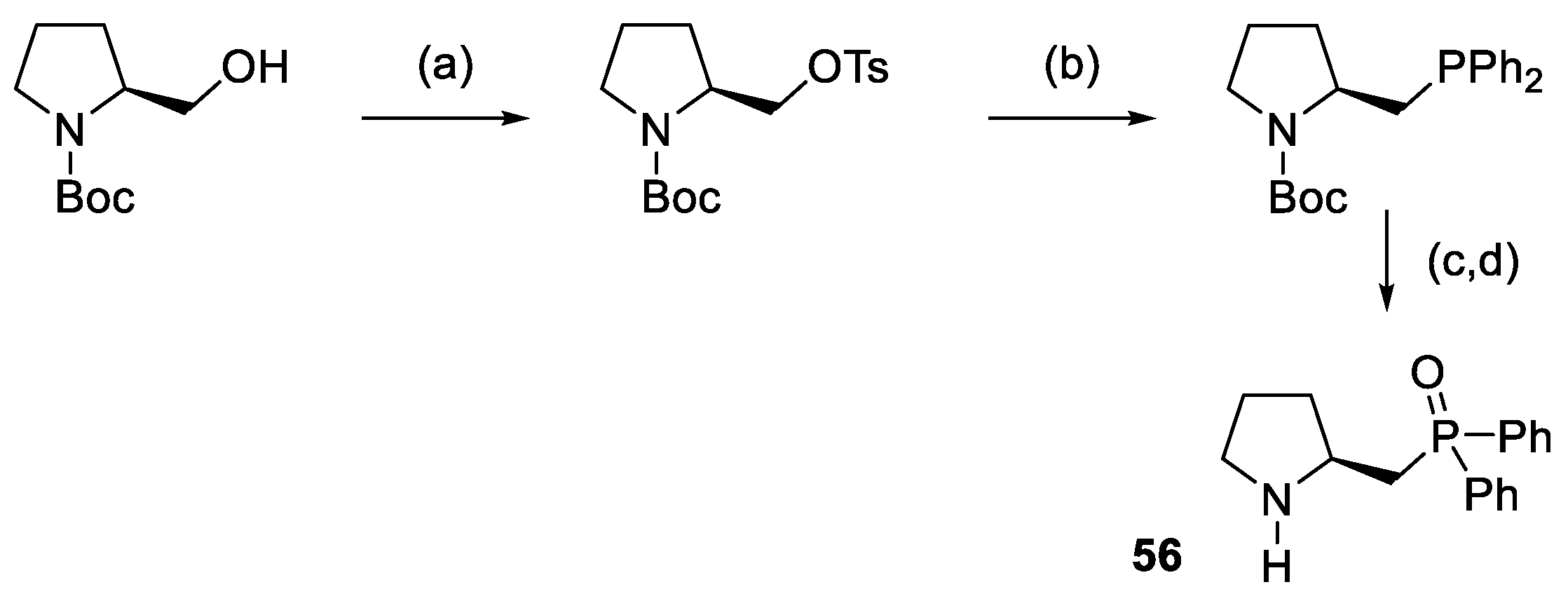{kind=link}
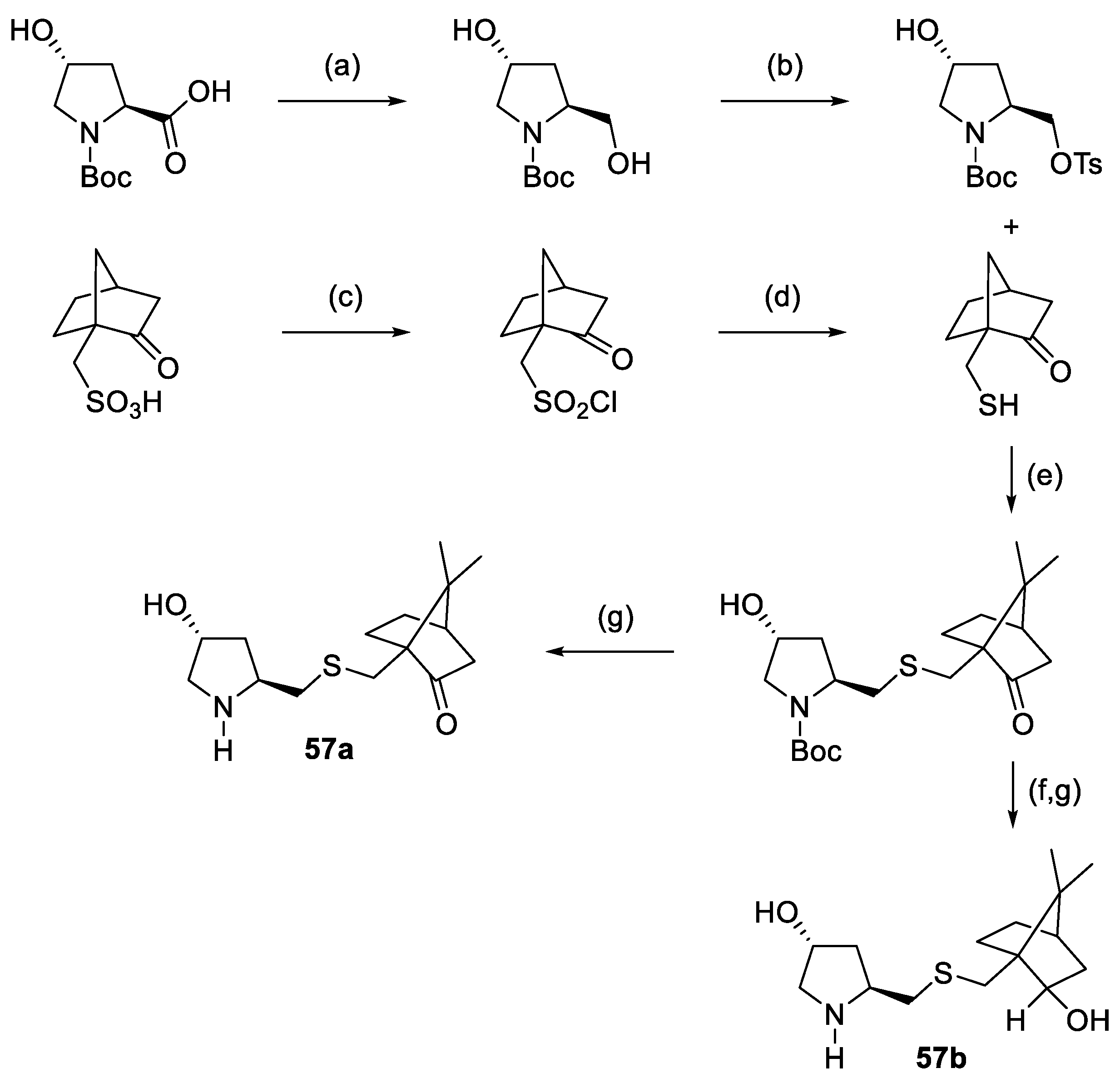{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
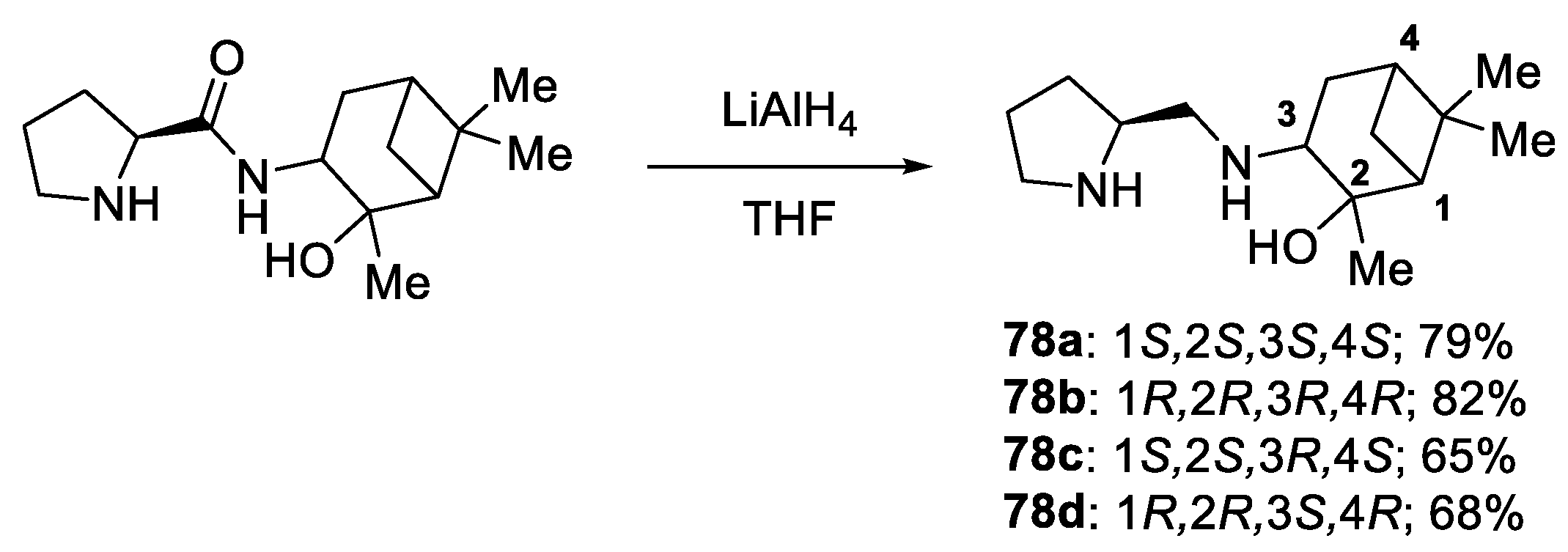{kind=link}
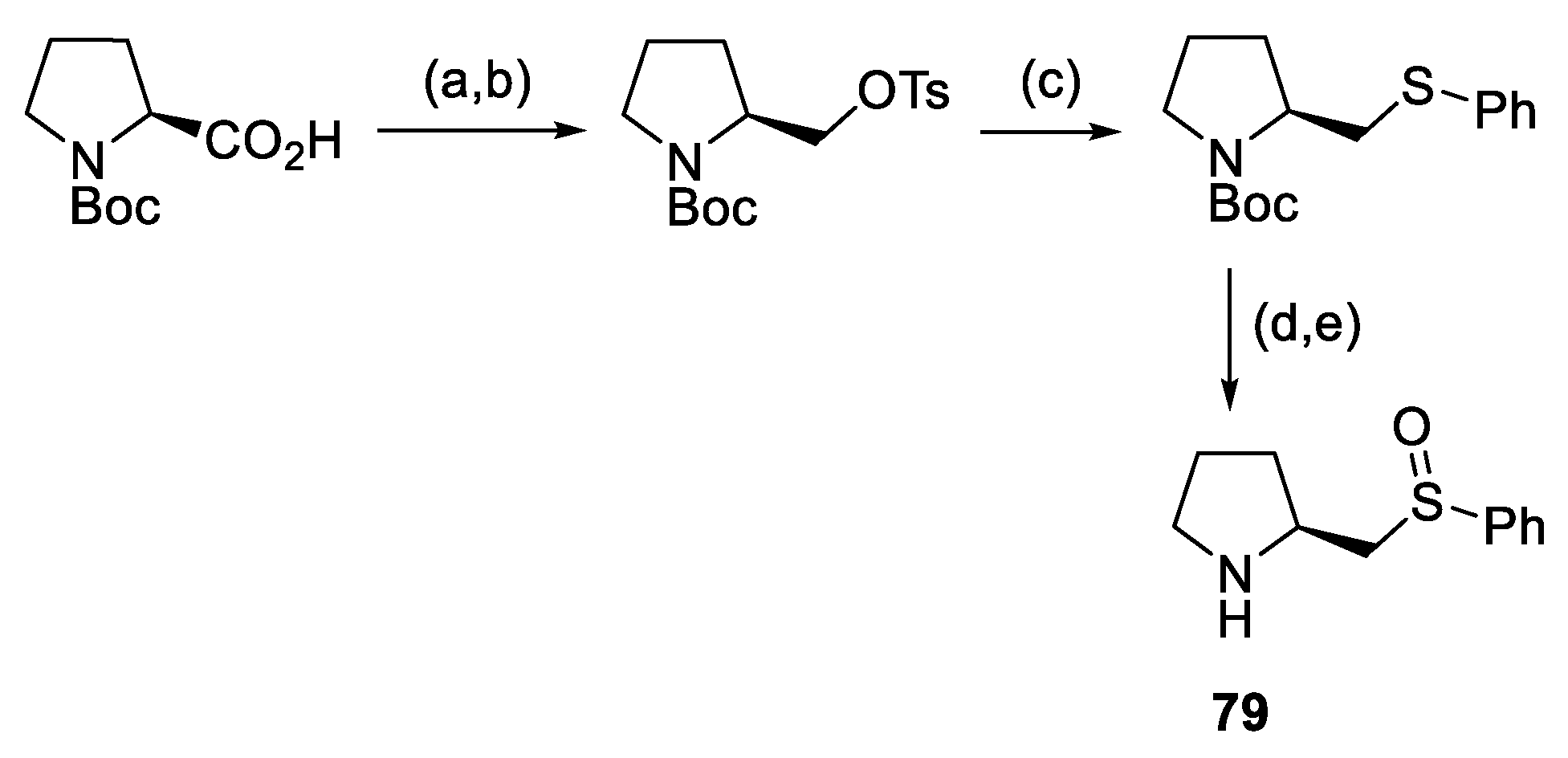{kind=link}
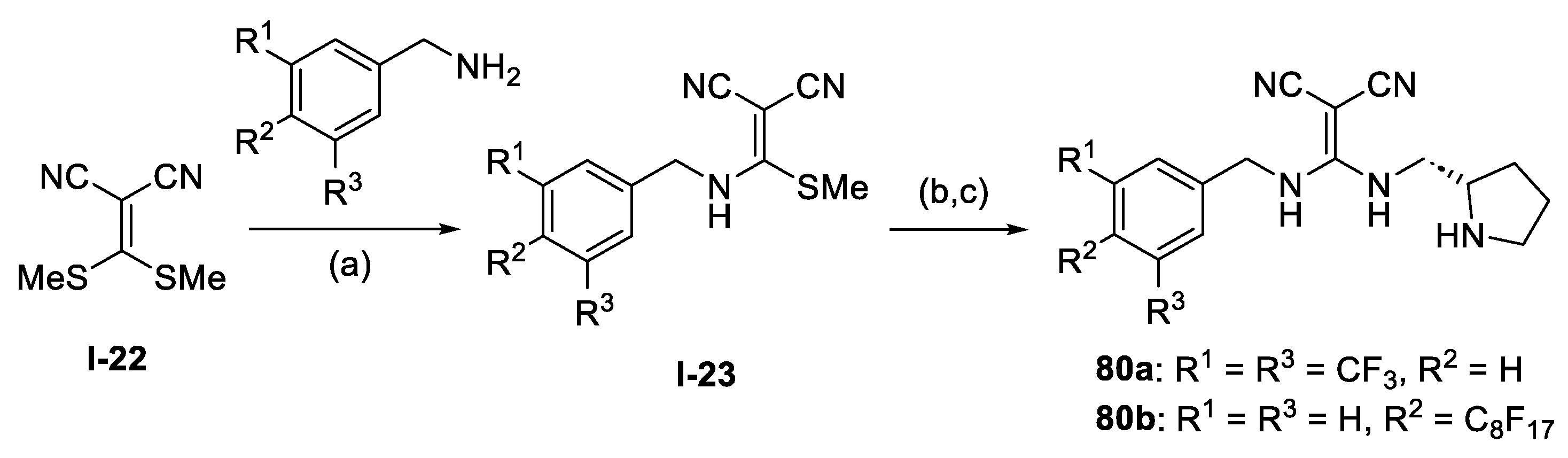{kind=link}
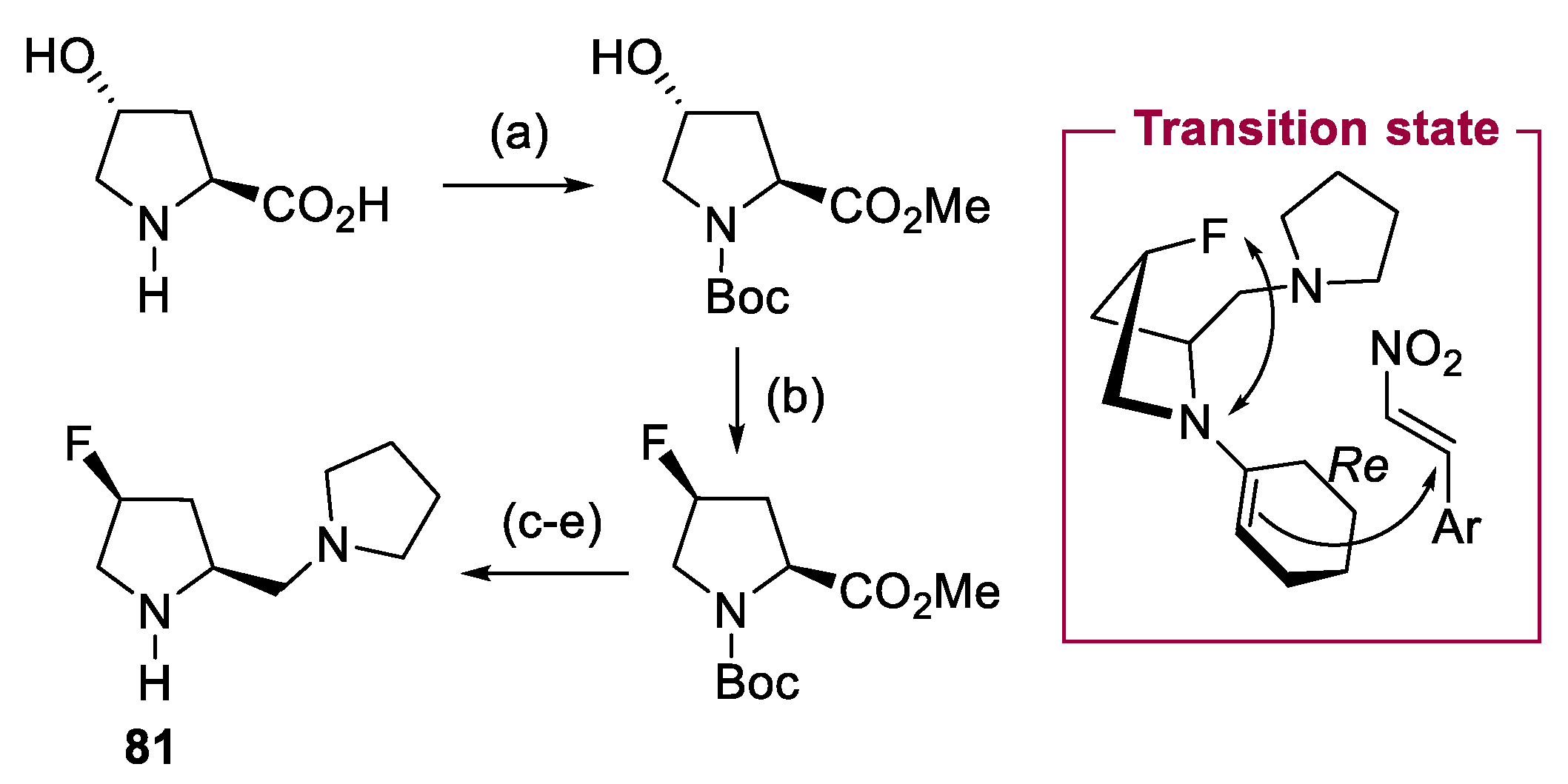{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
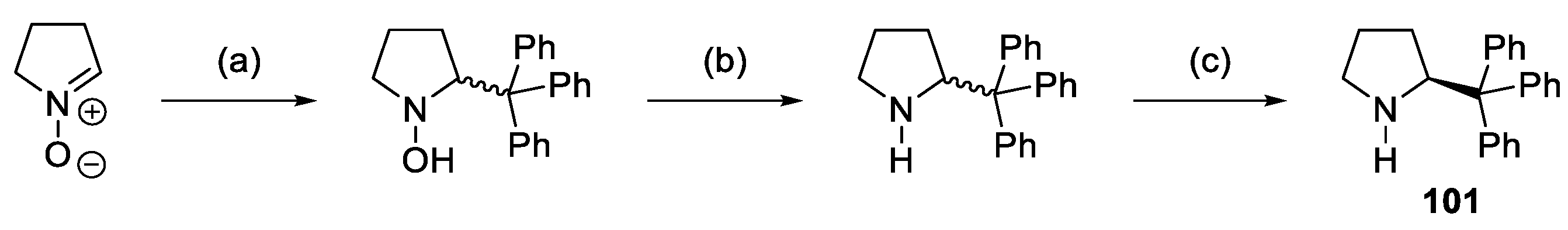{kind=link}
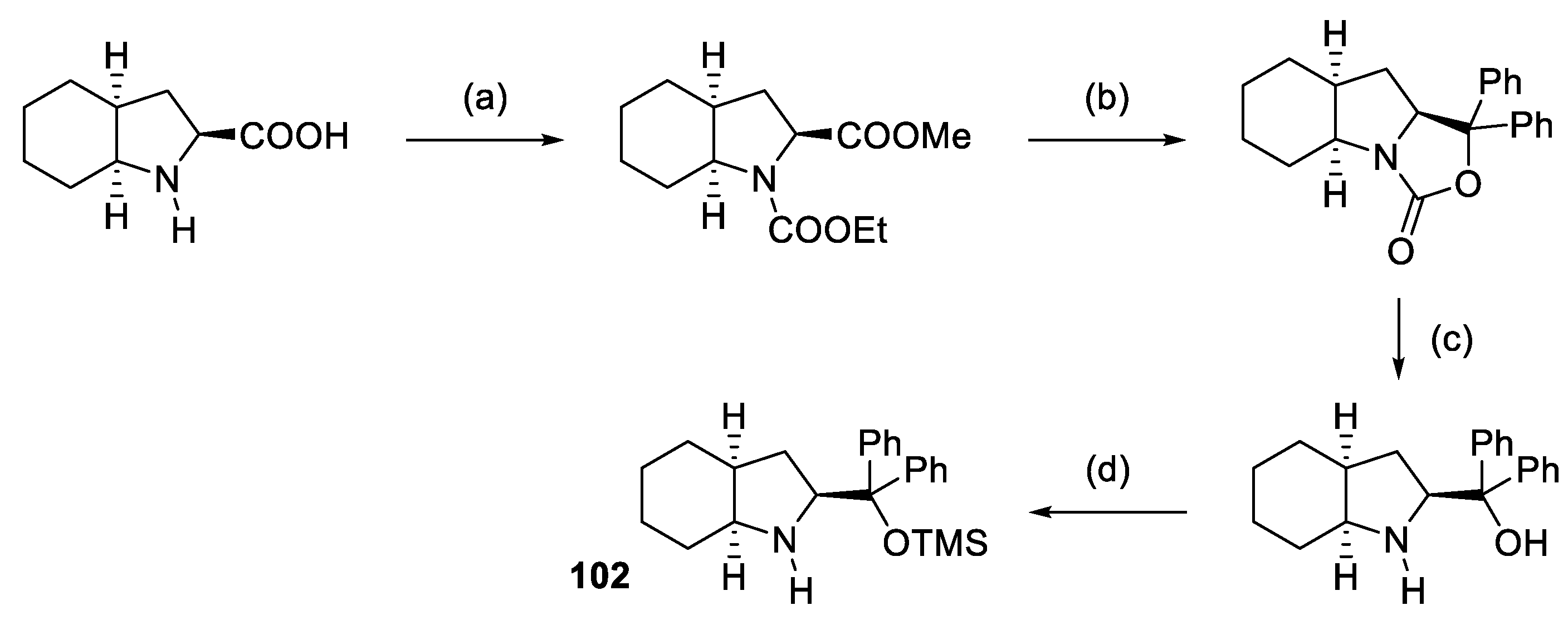{kind=link}
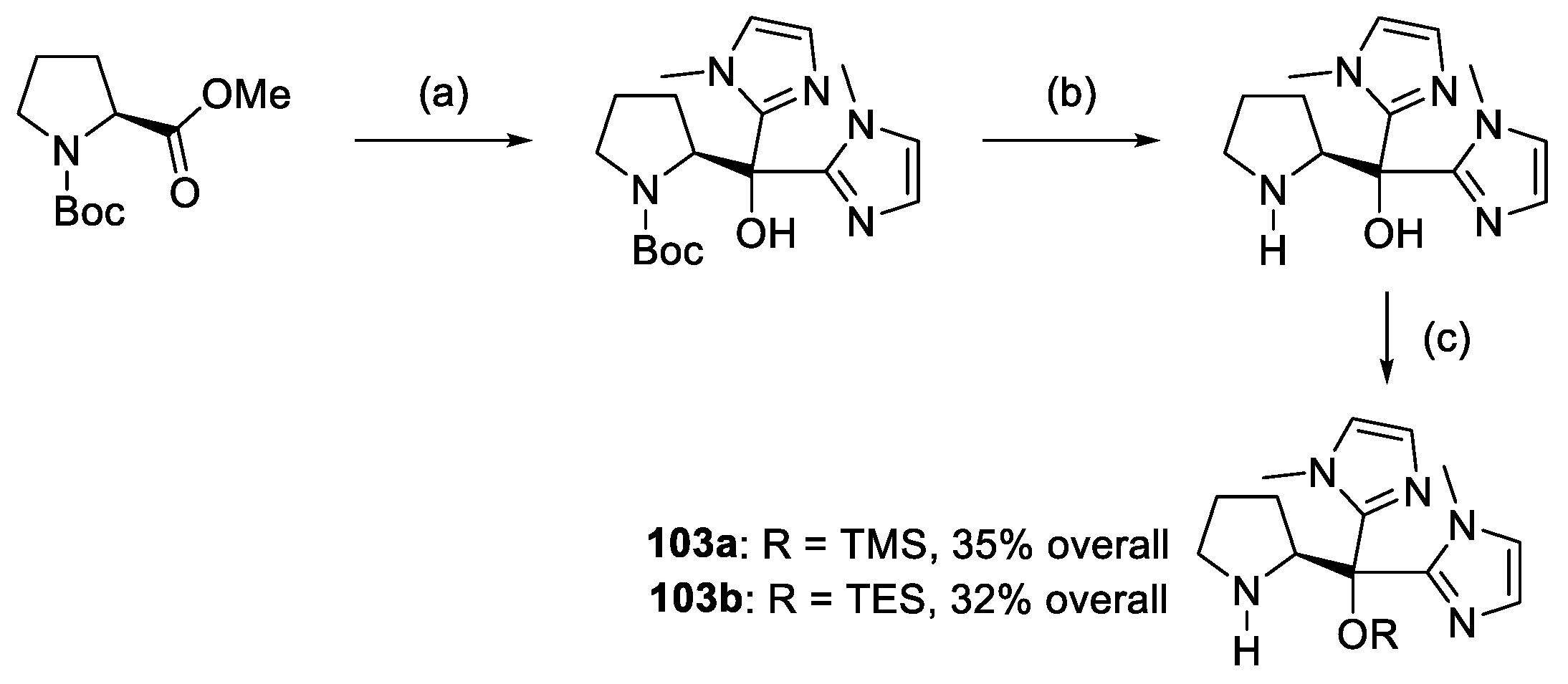{kind=link}
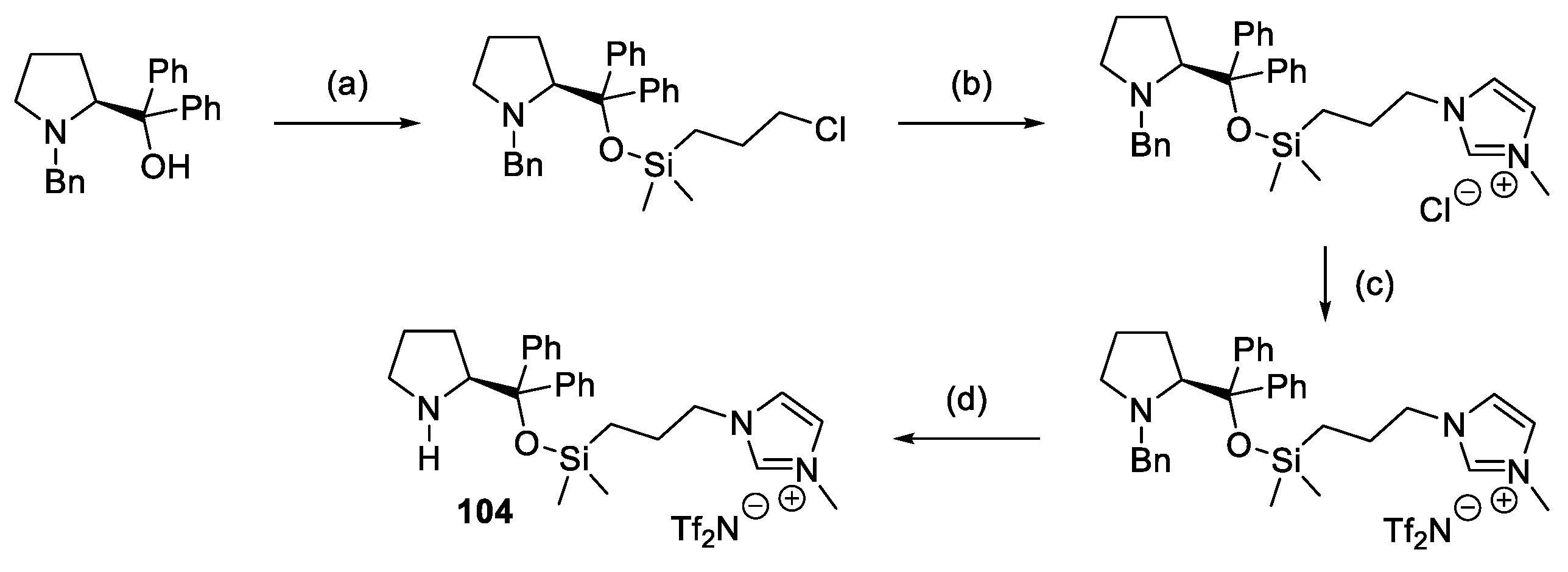{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
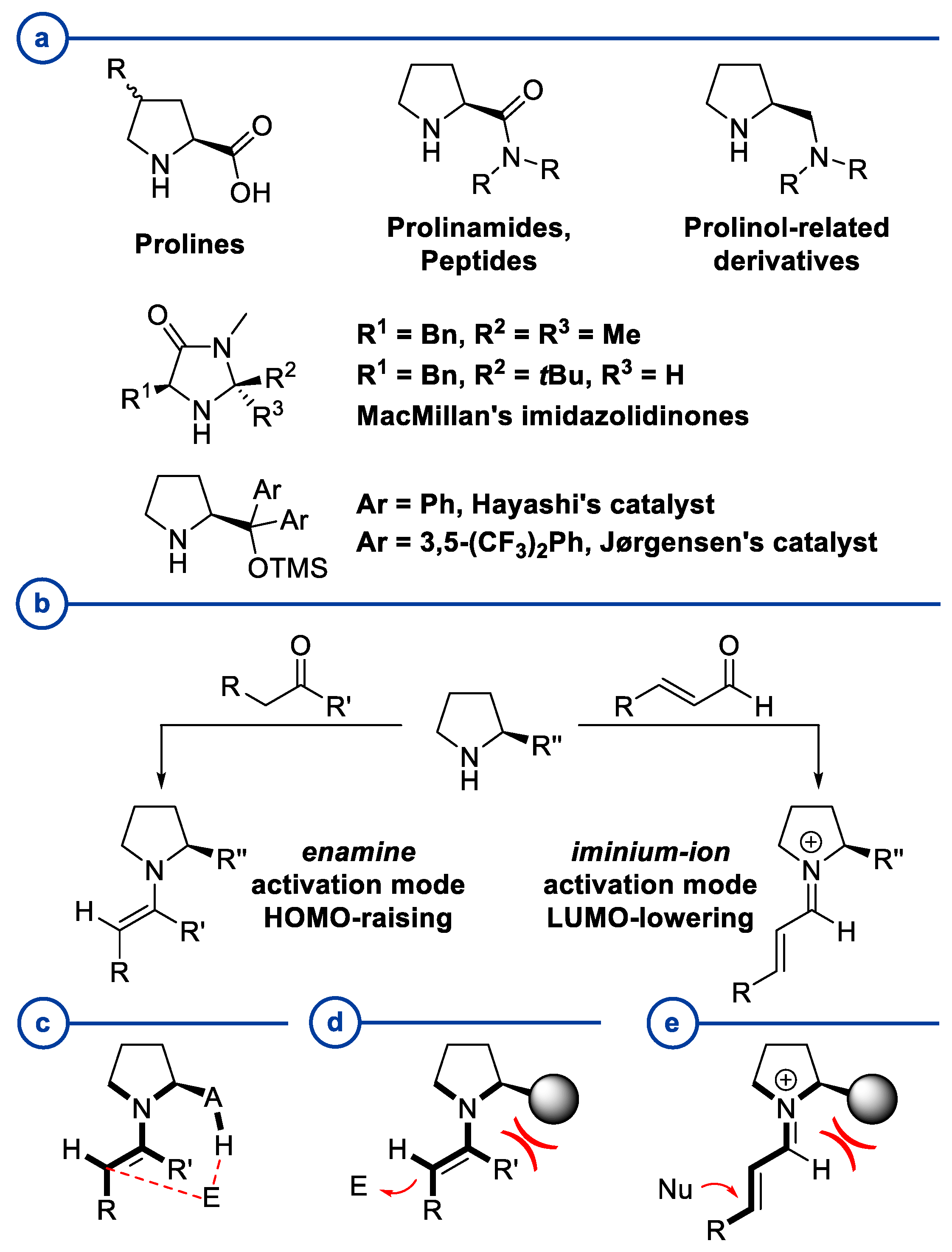
:1. Introduction
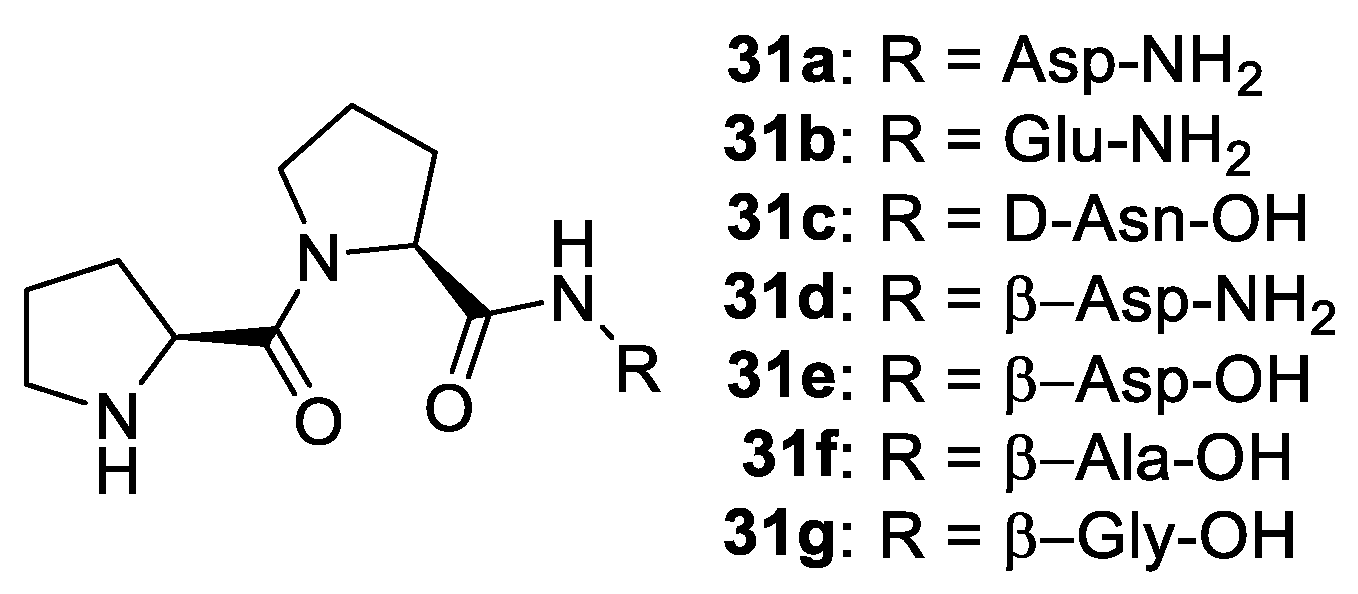
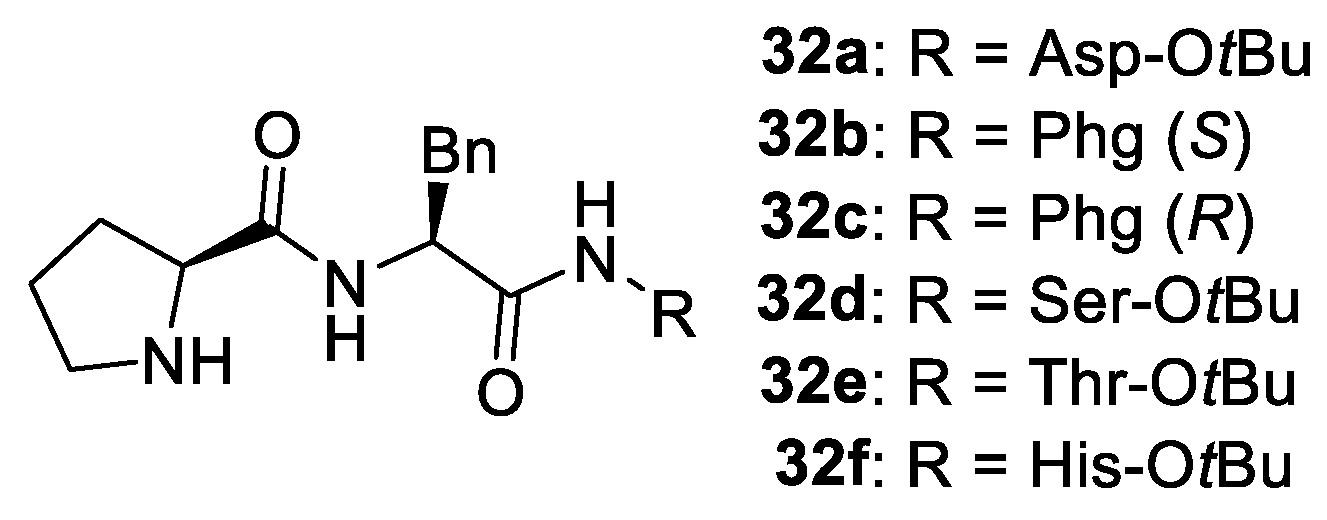
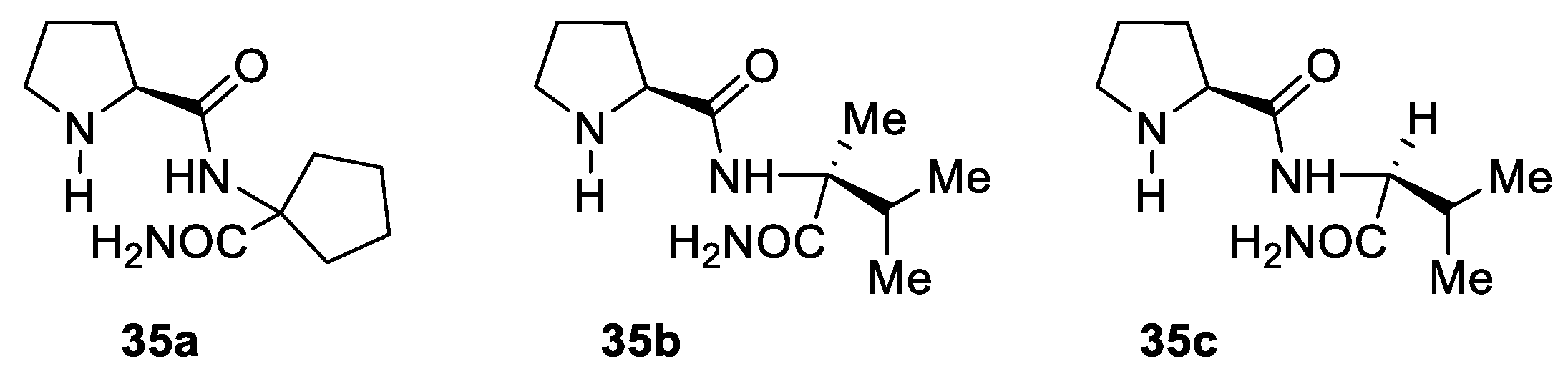
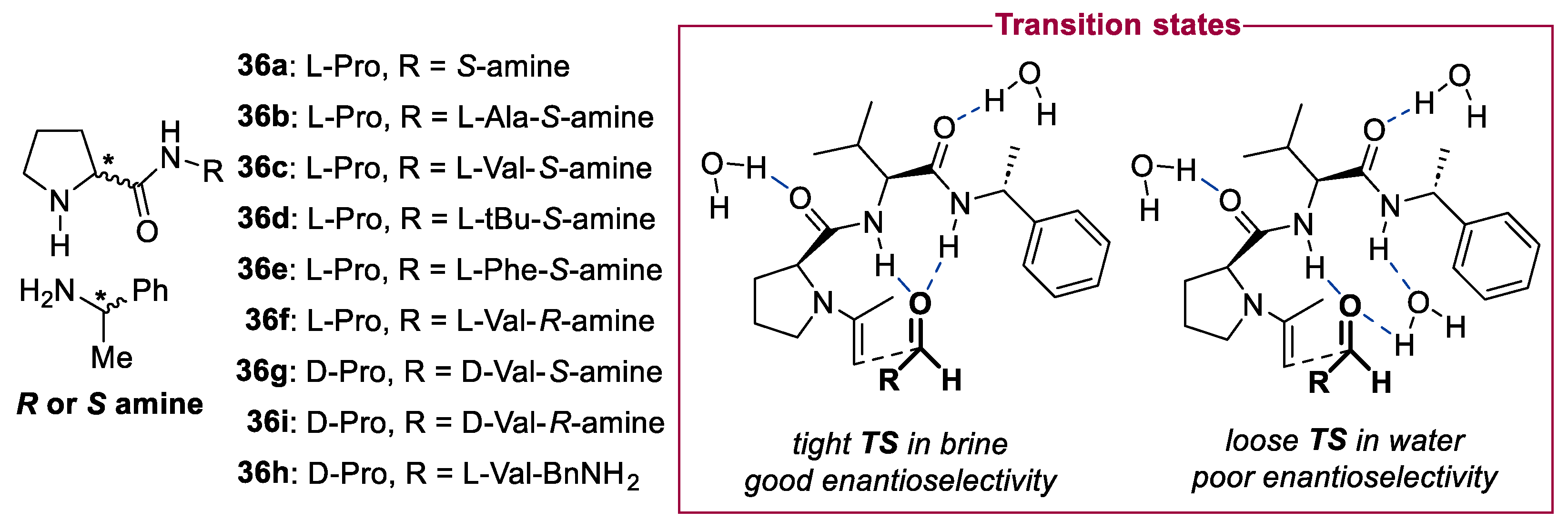
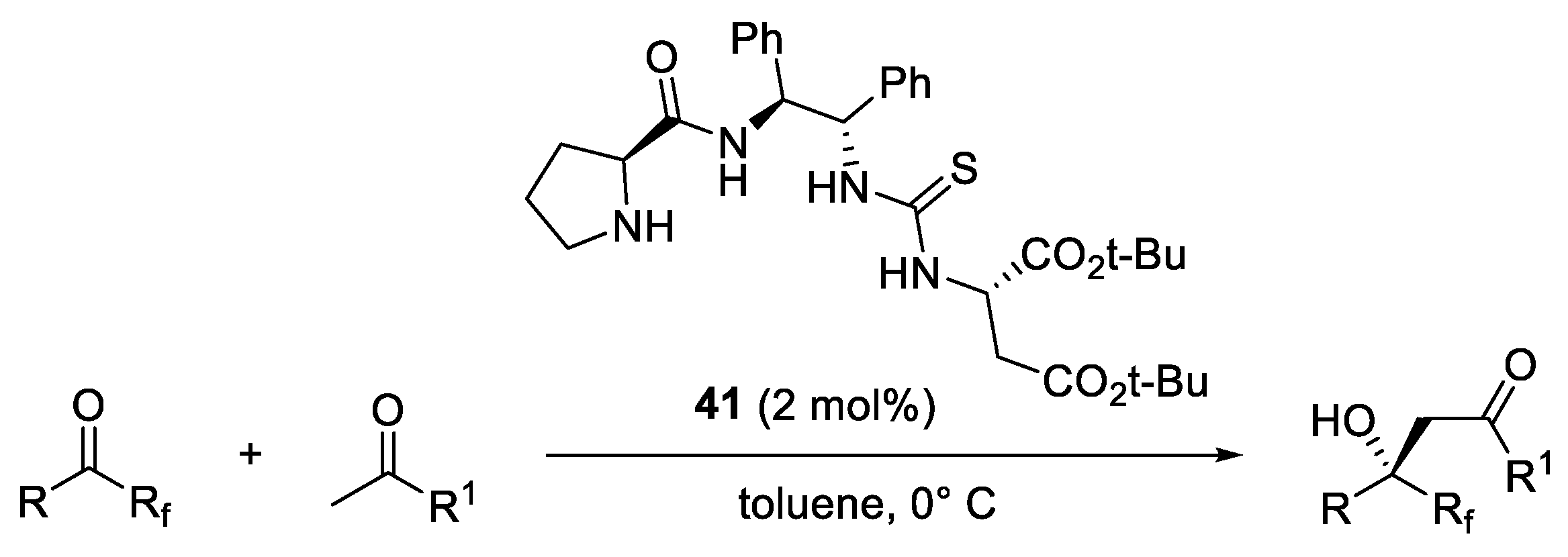
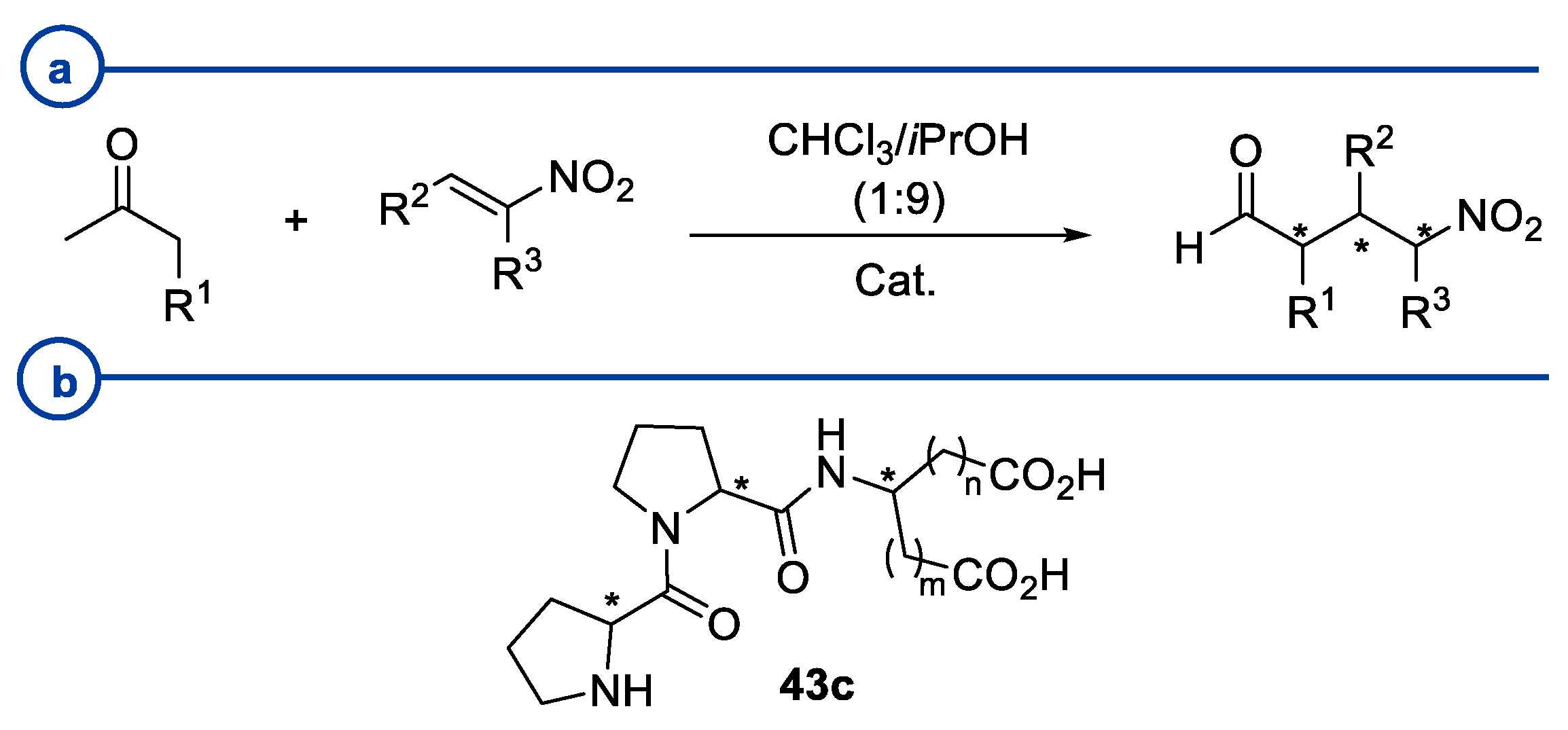
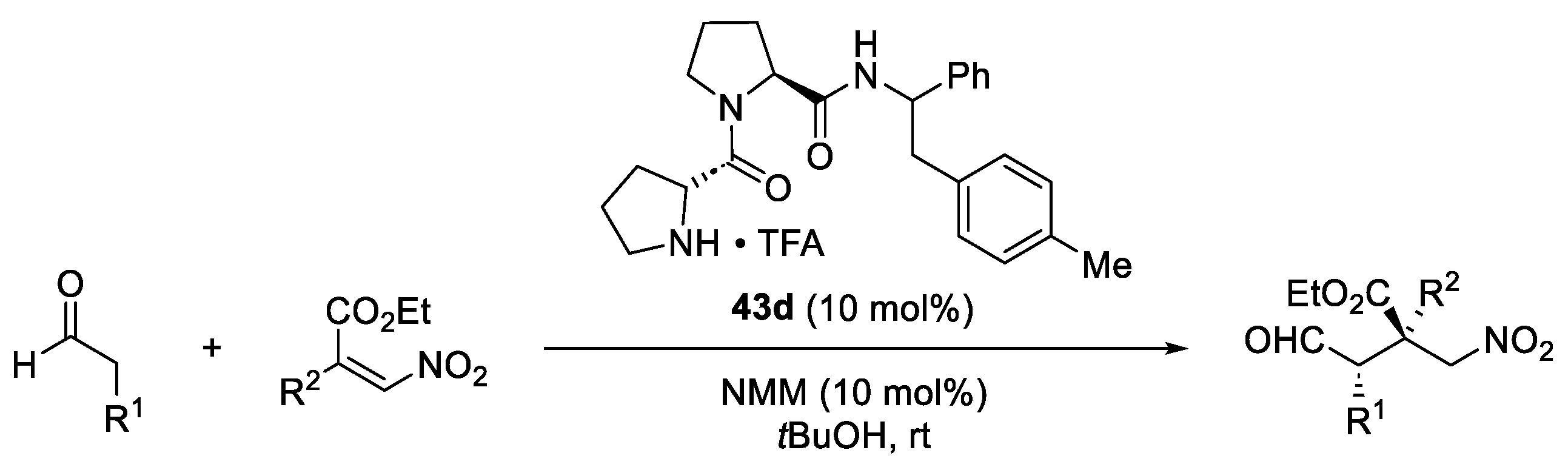
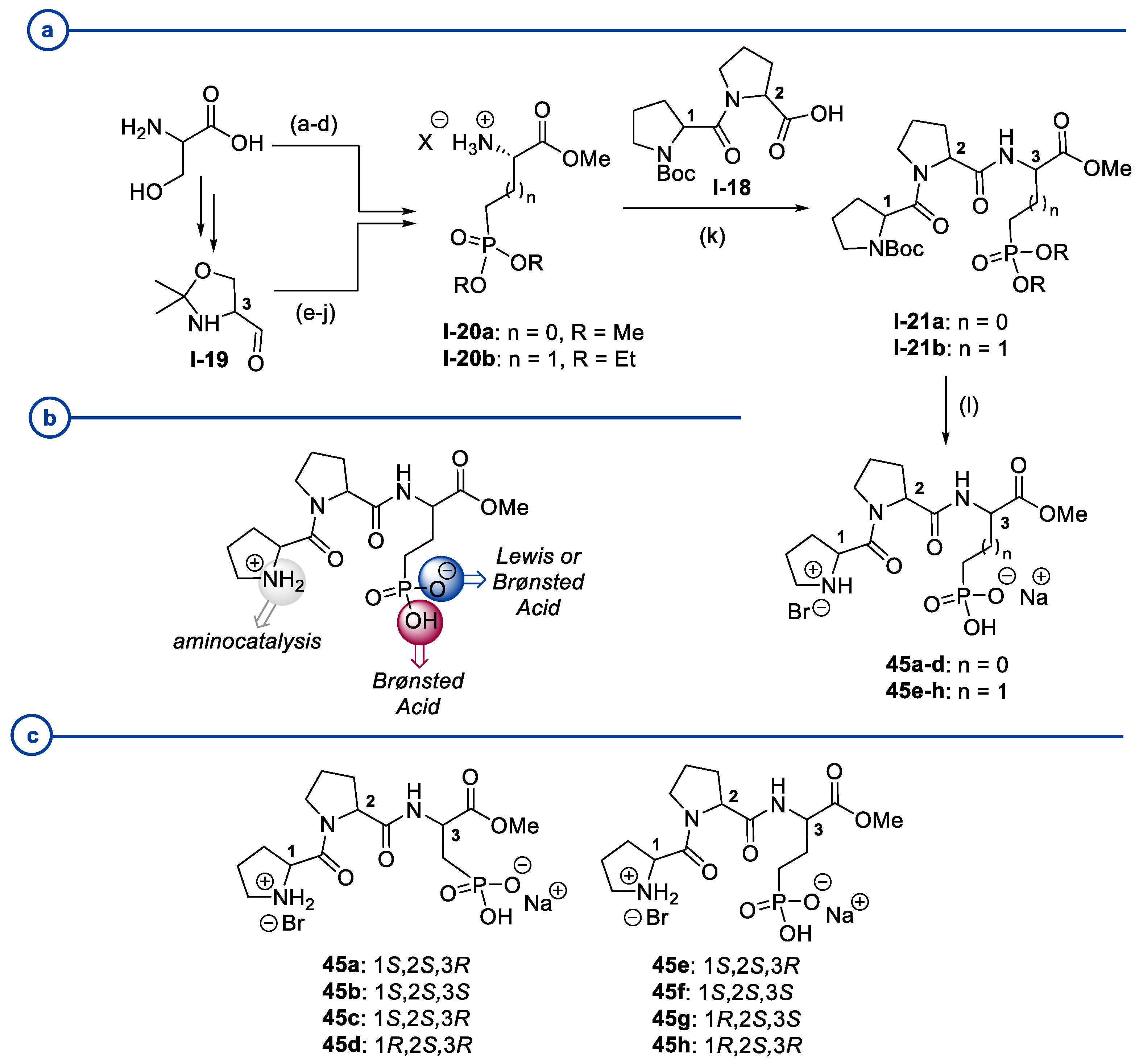
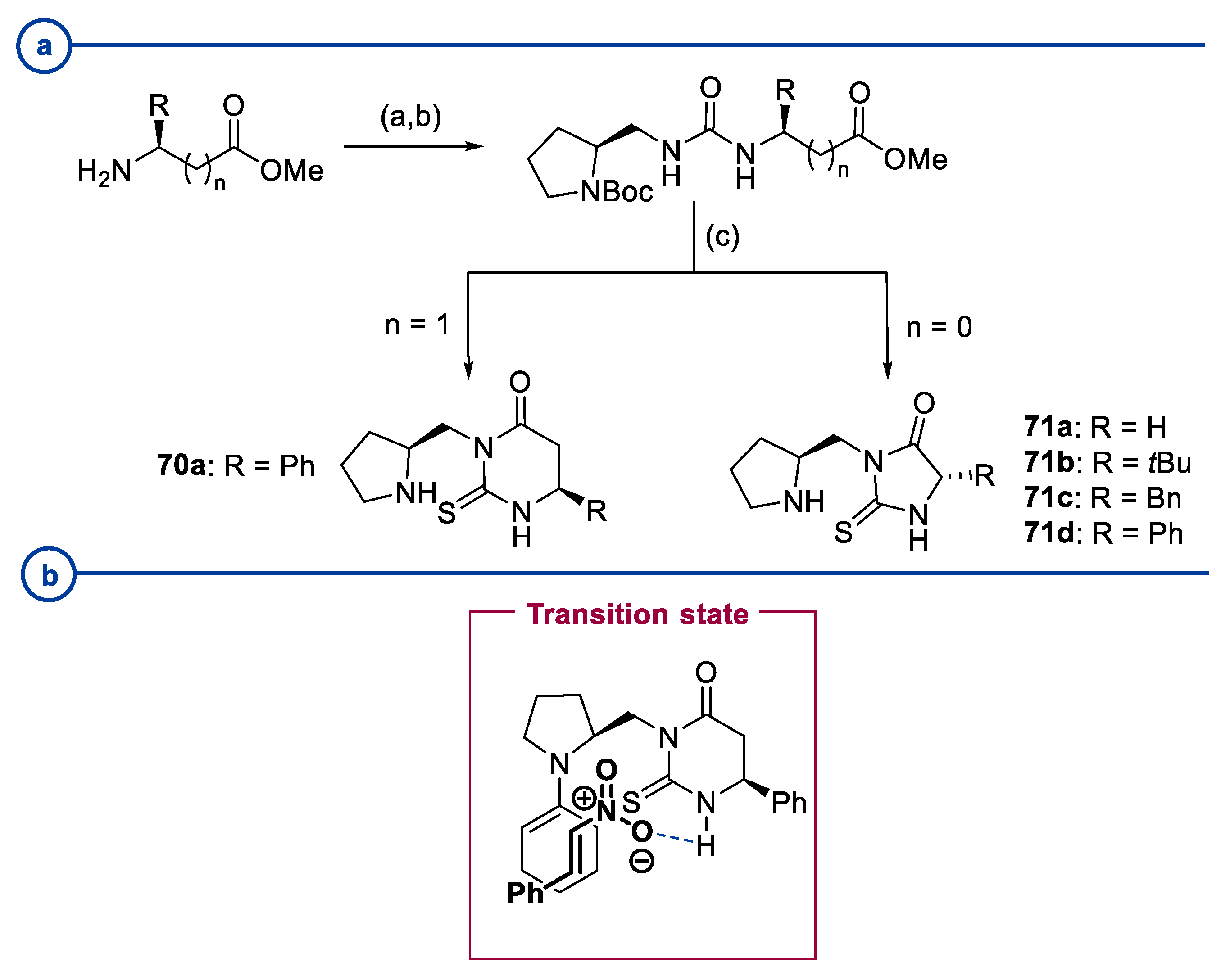
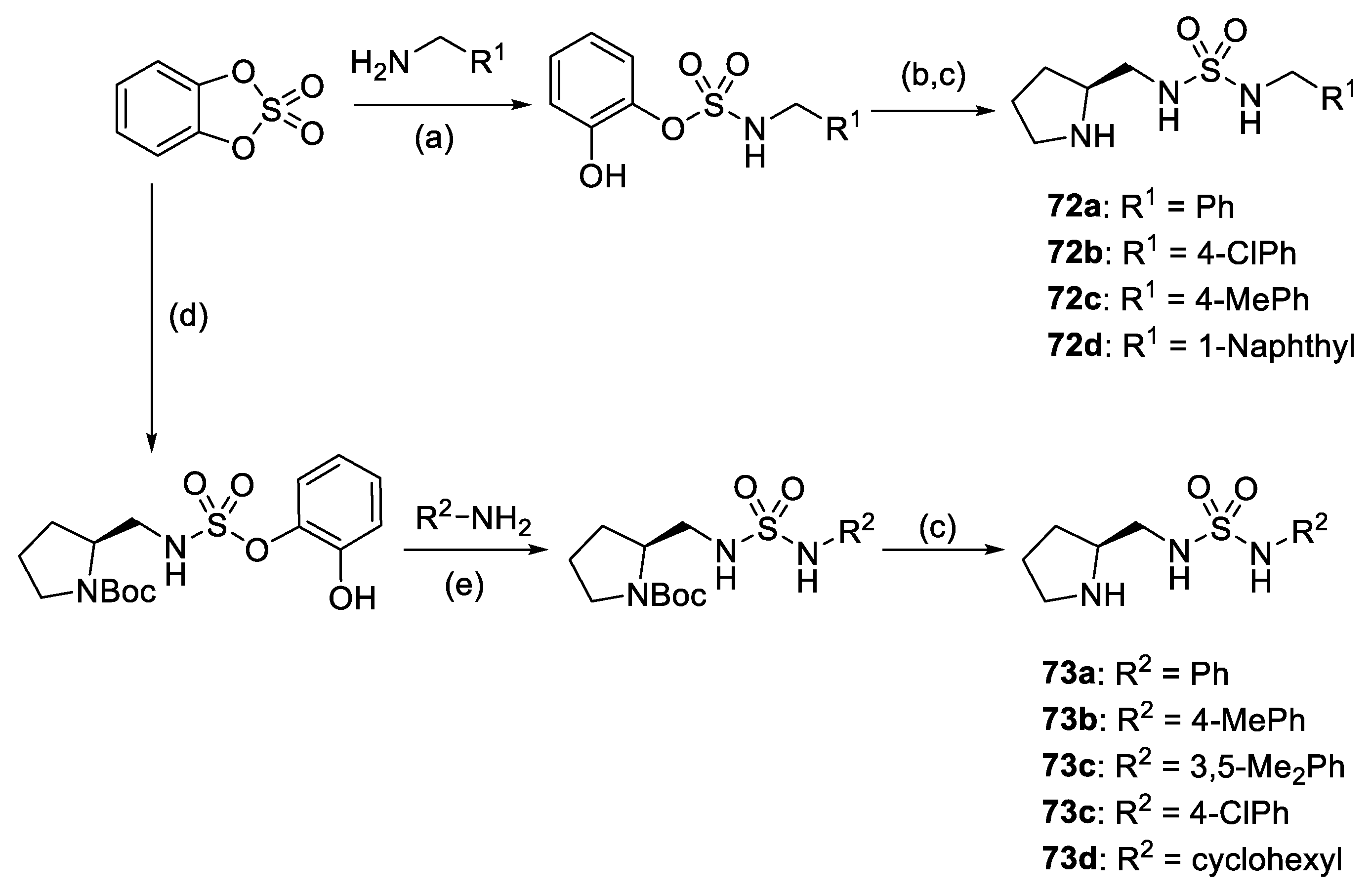
2. Proline-Related Organocatalysts
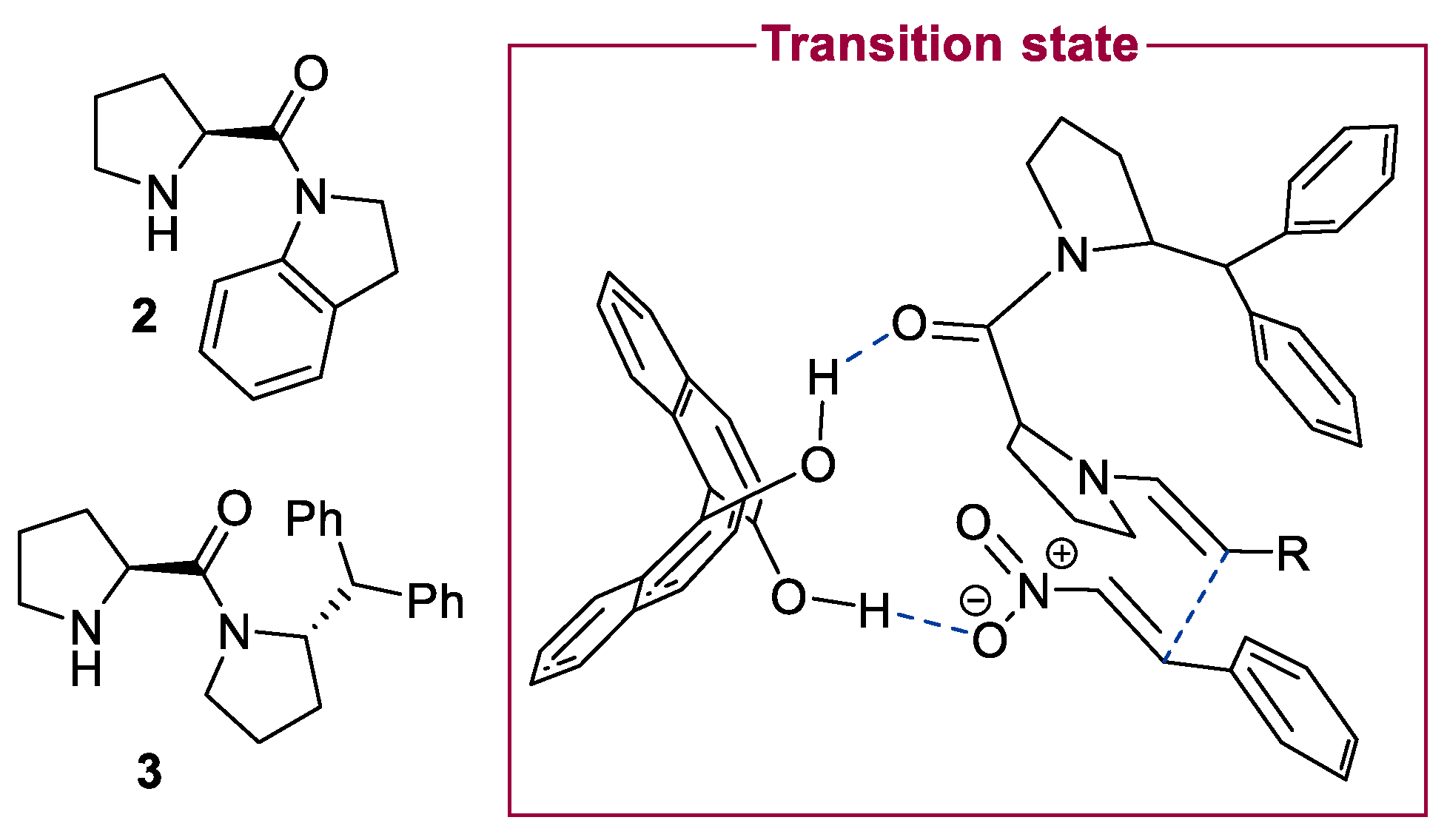
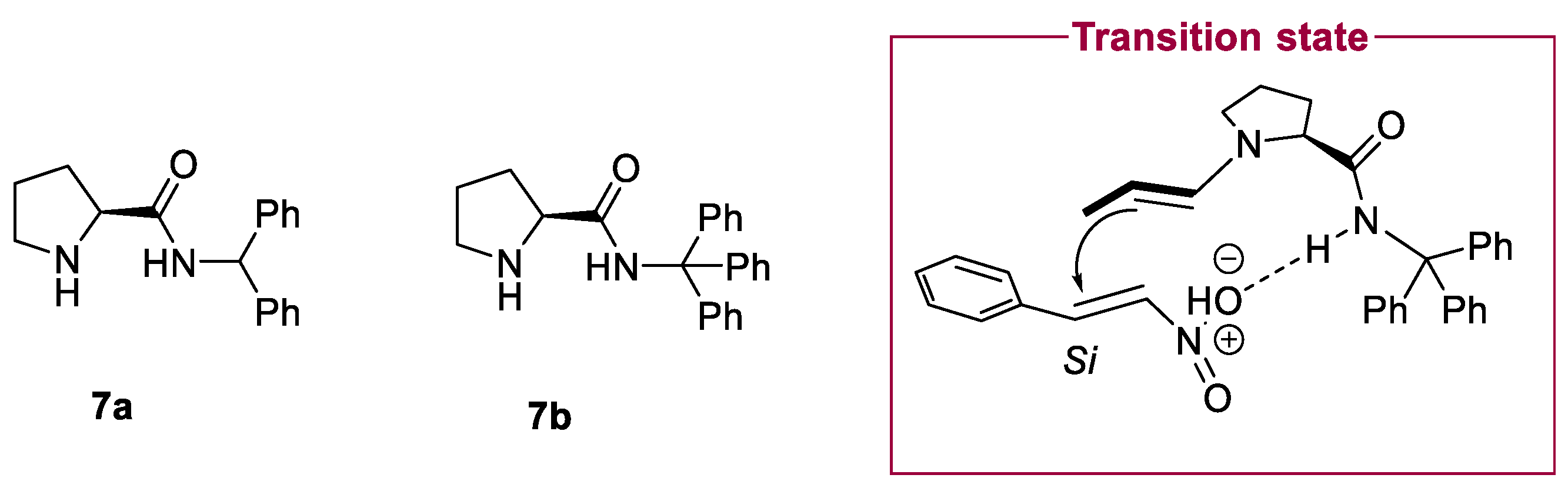
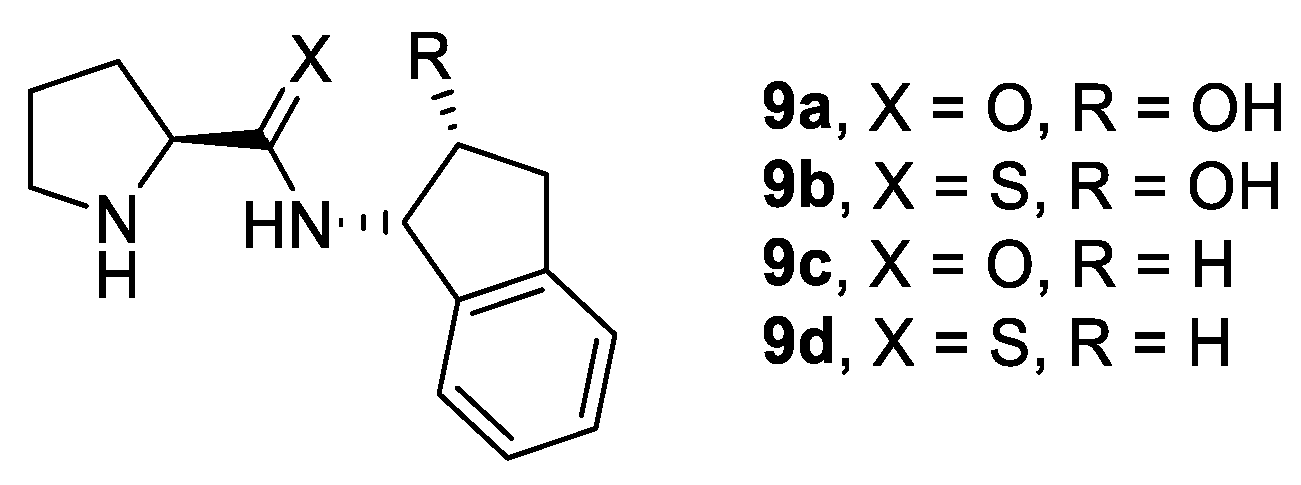
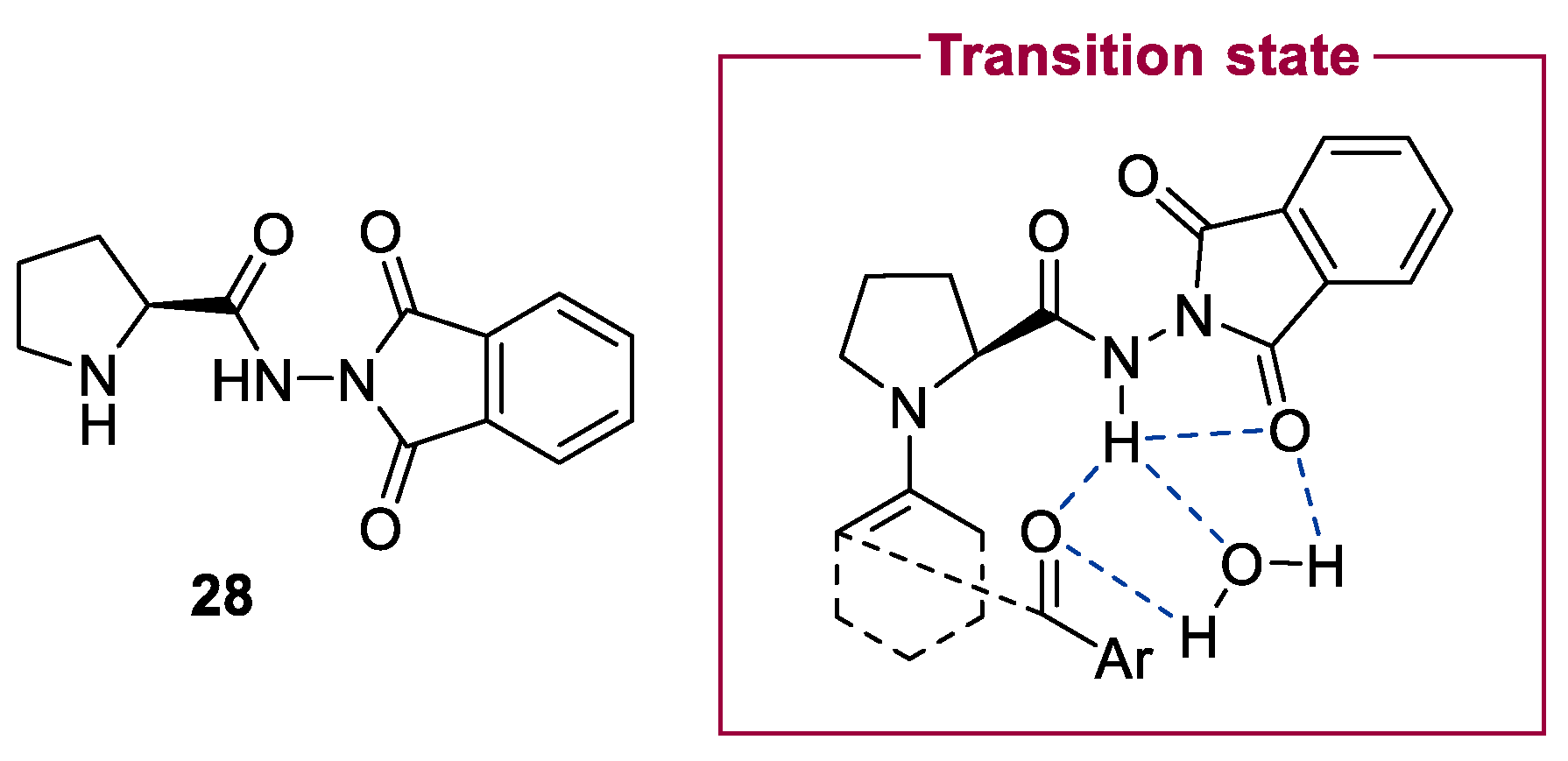
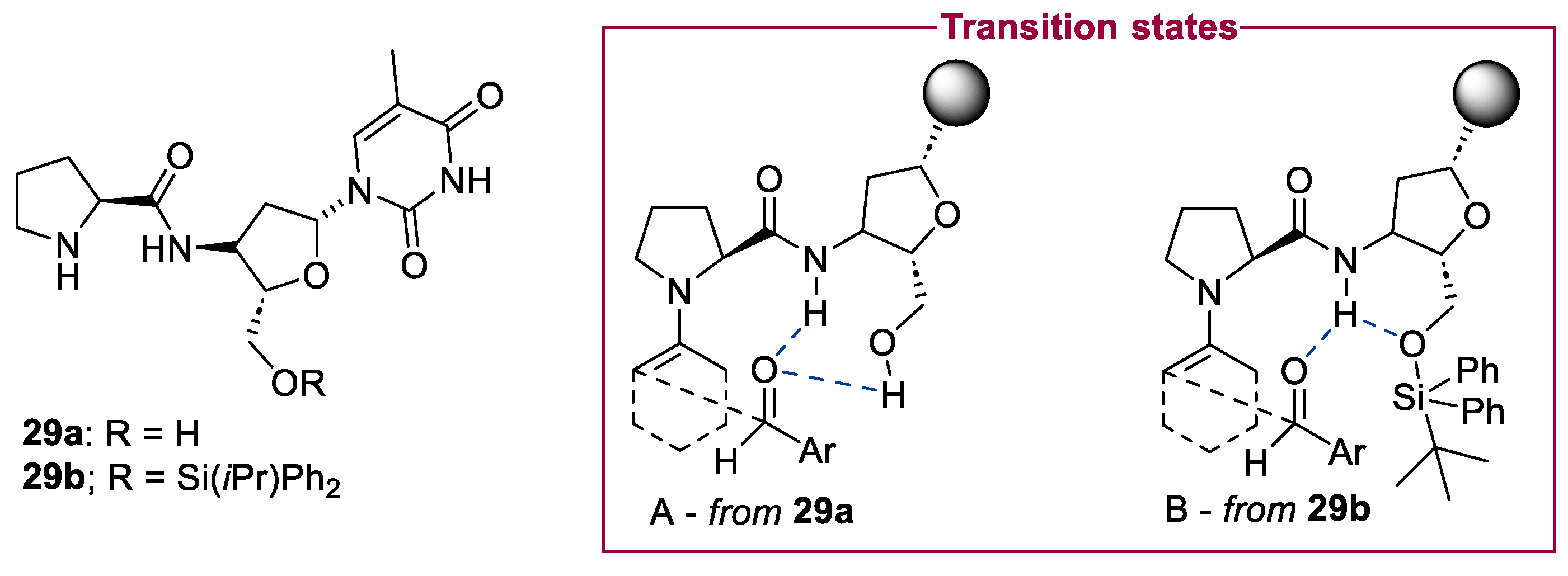
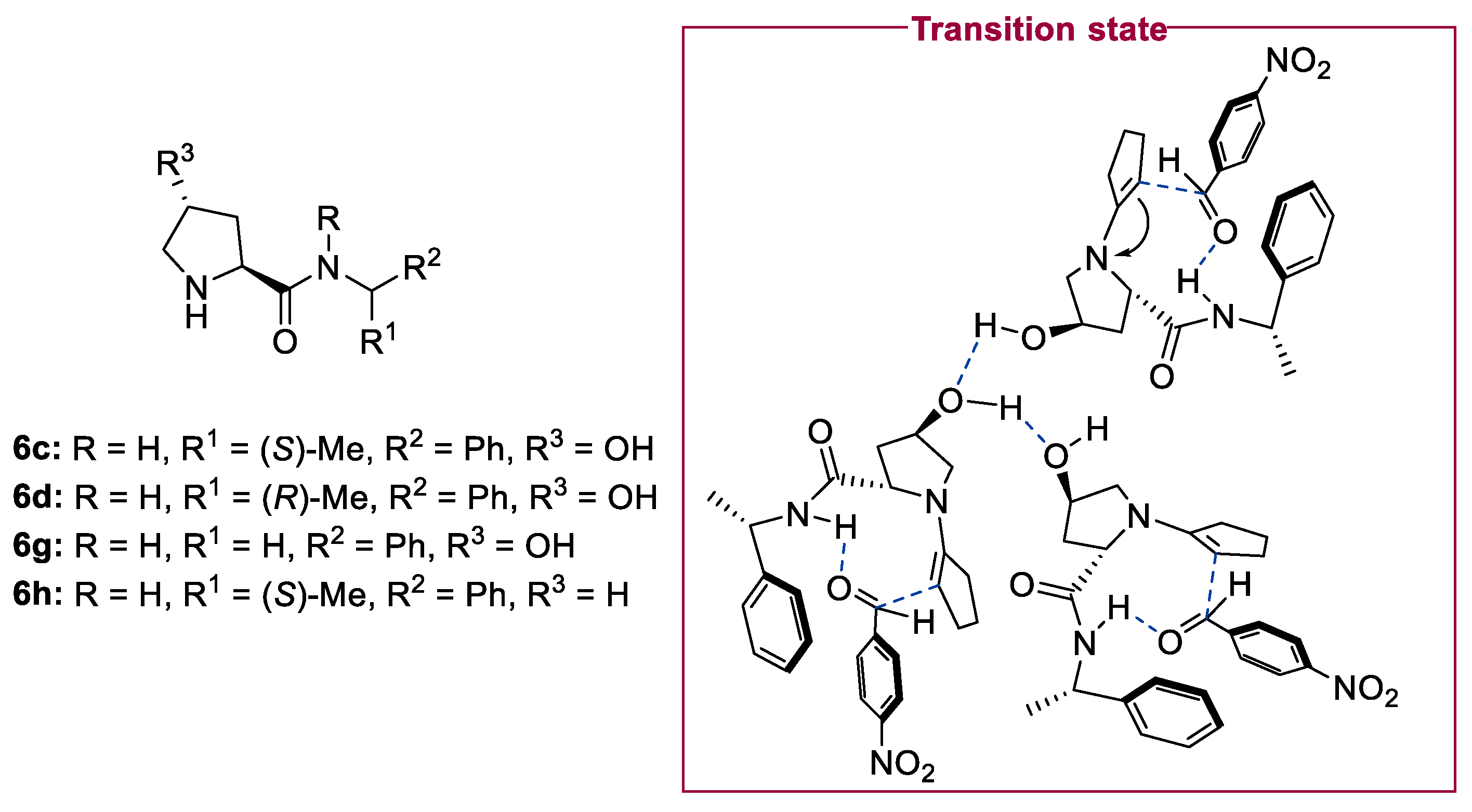
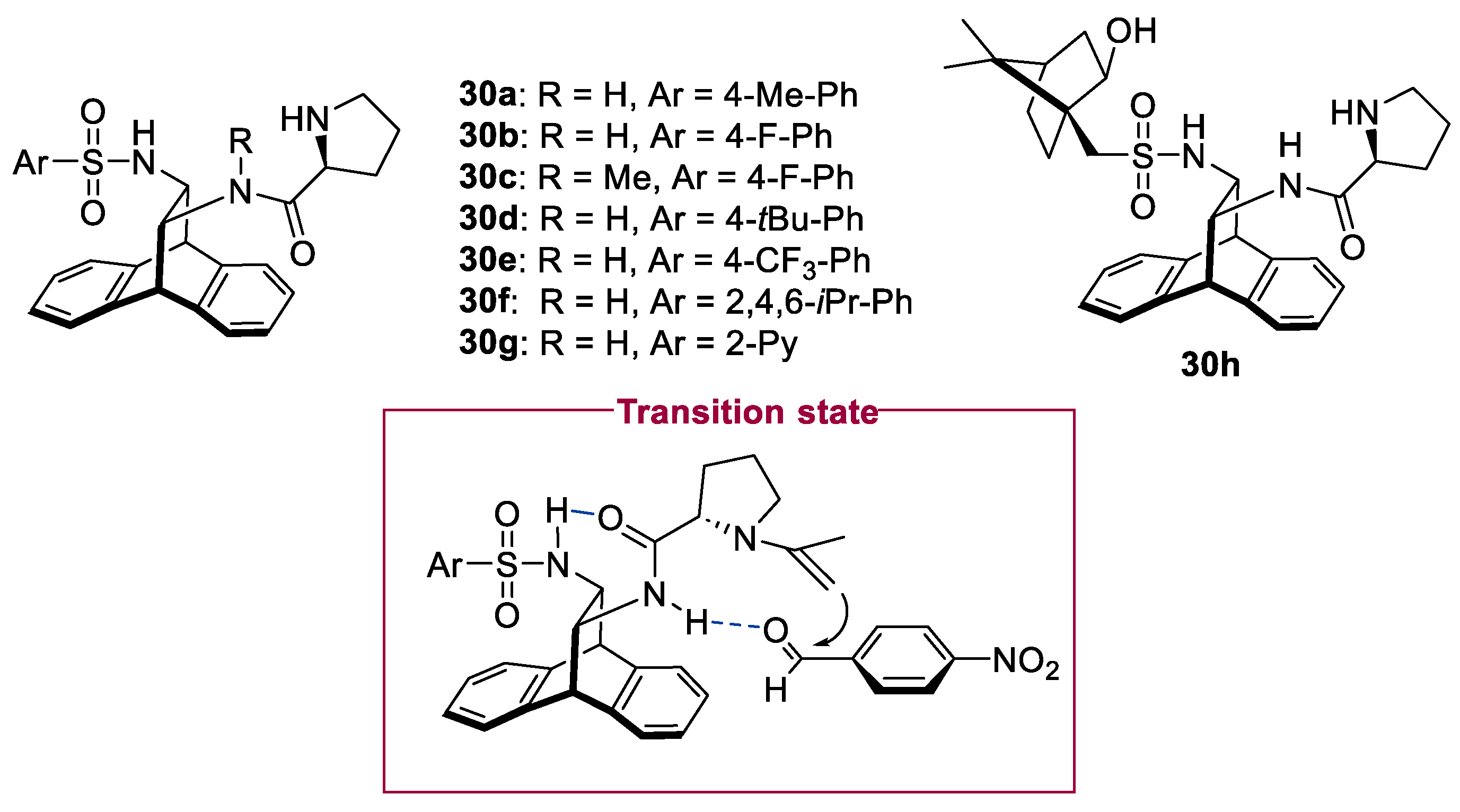
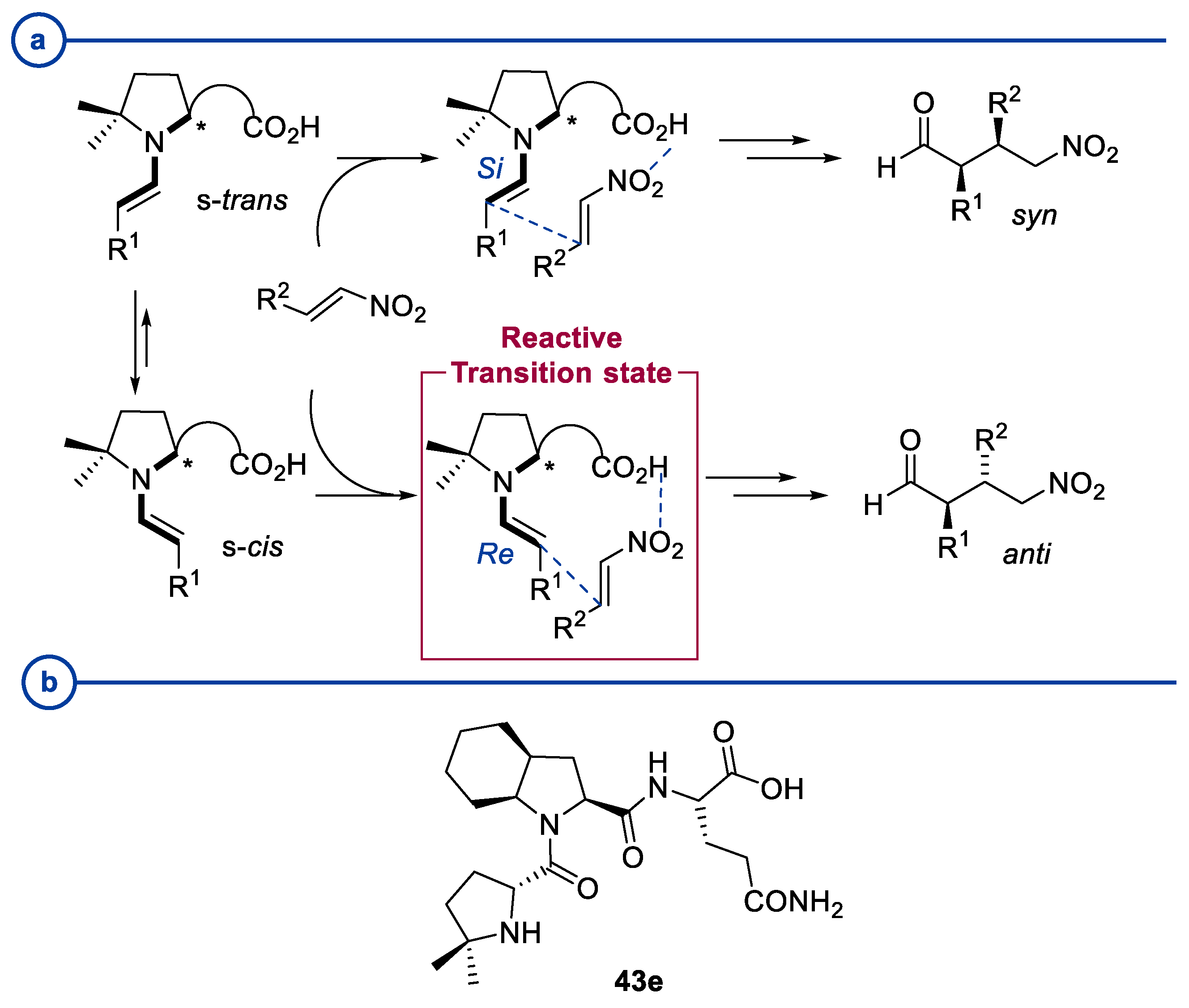
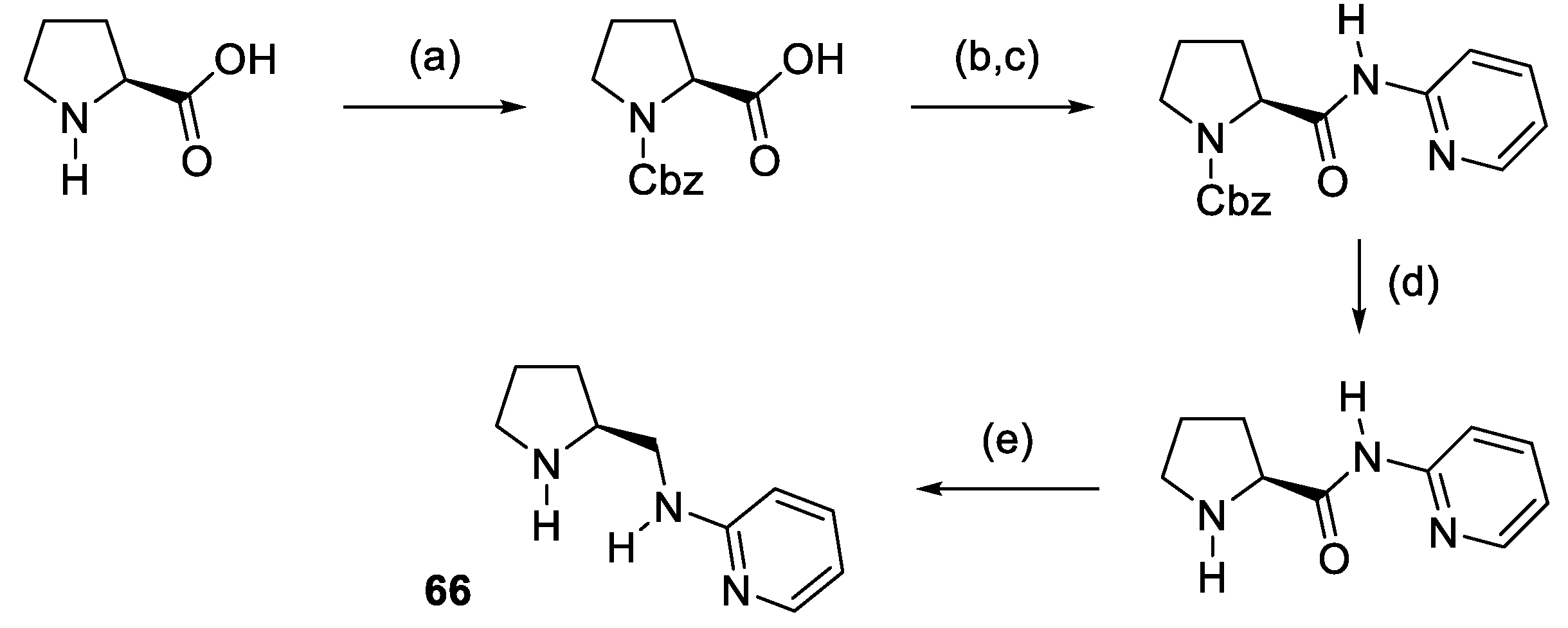
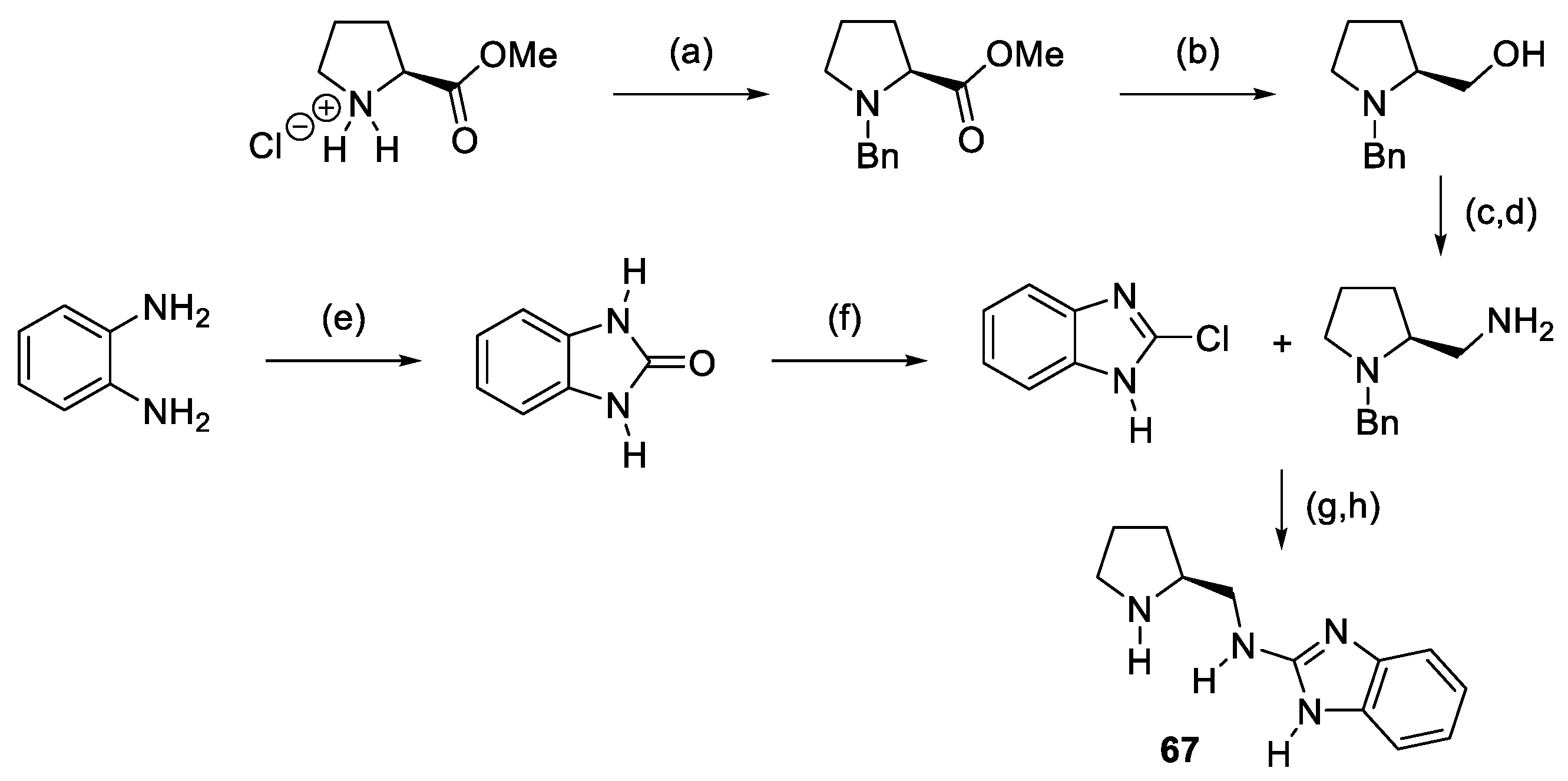
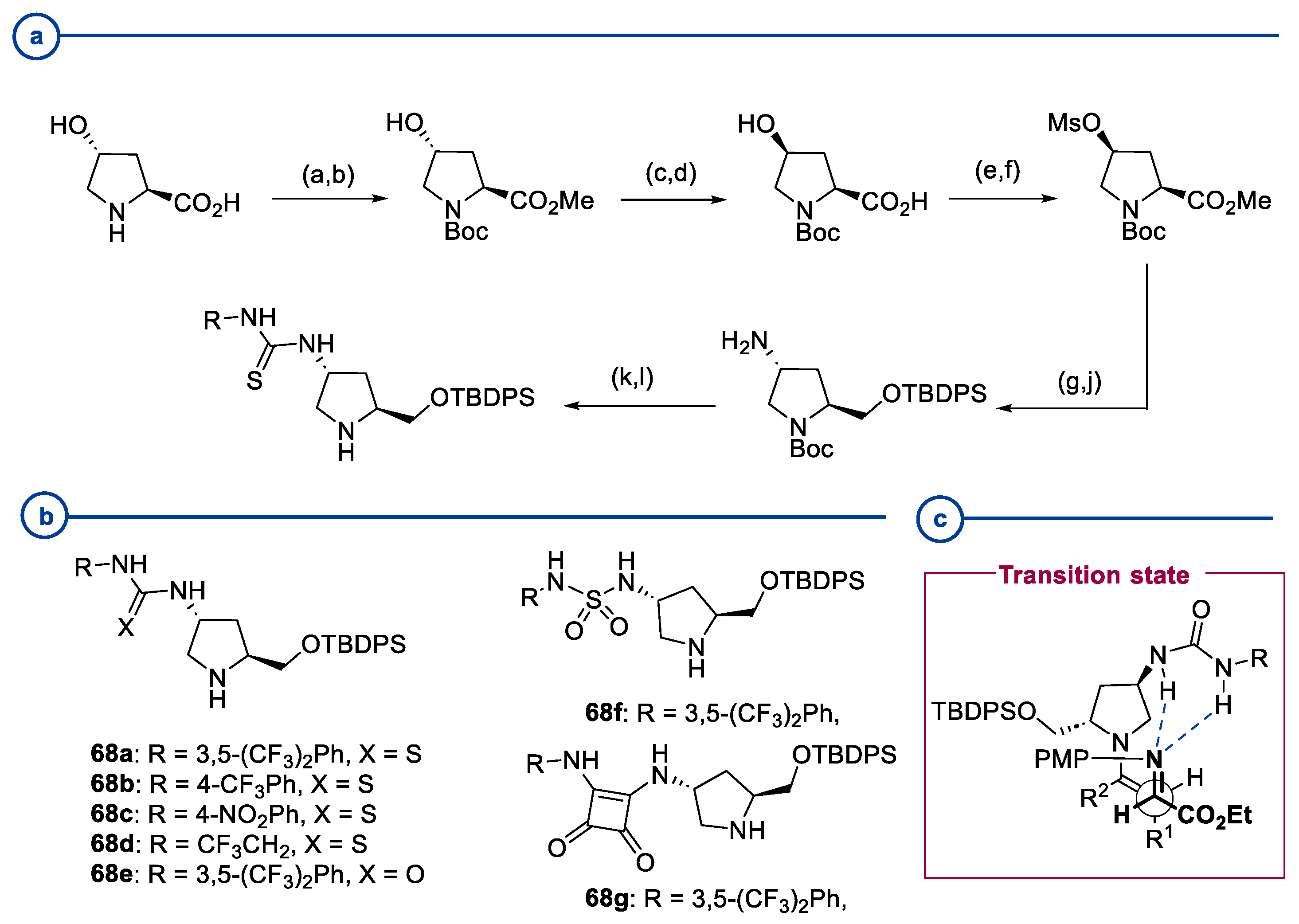
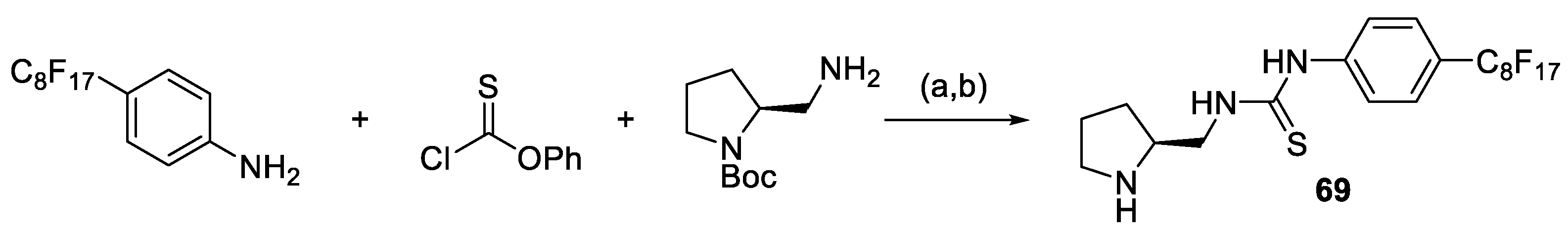
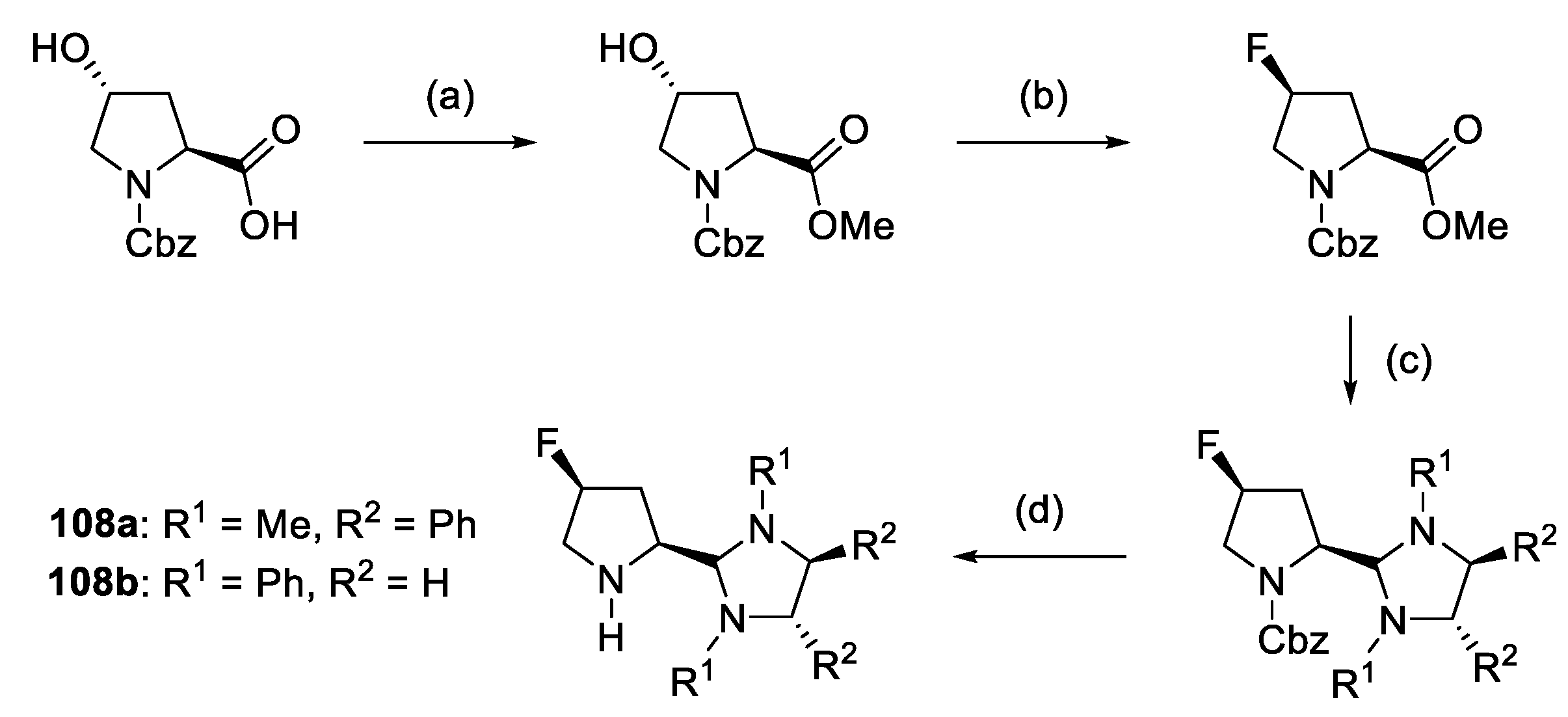
2.1. Prolinamides
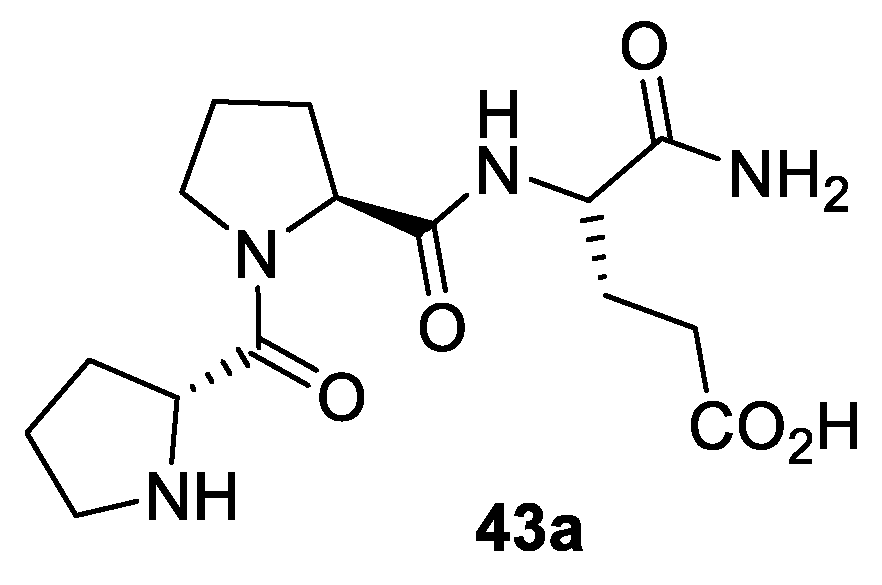
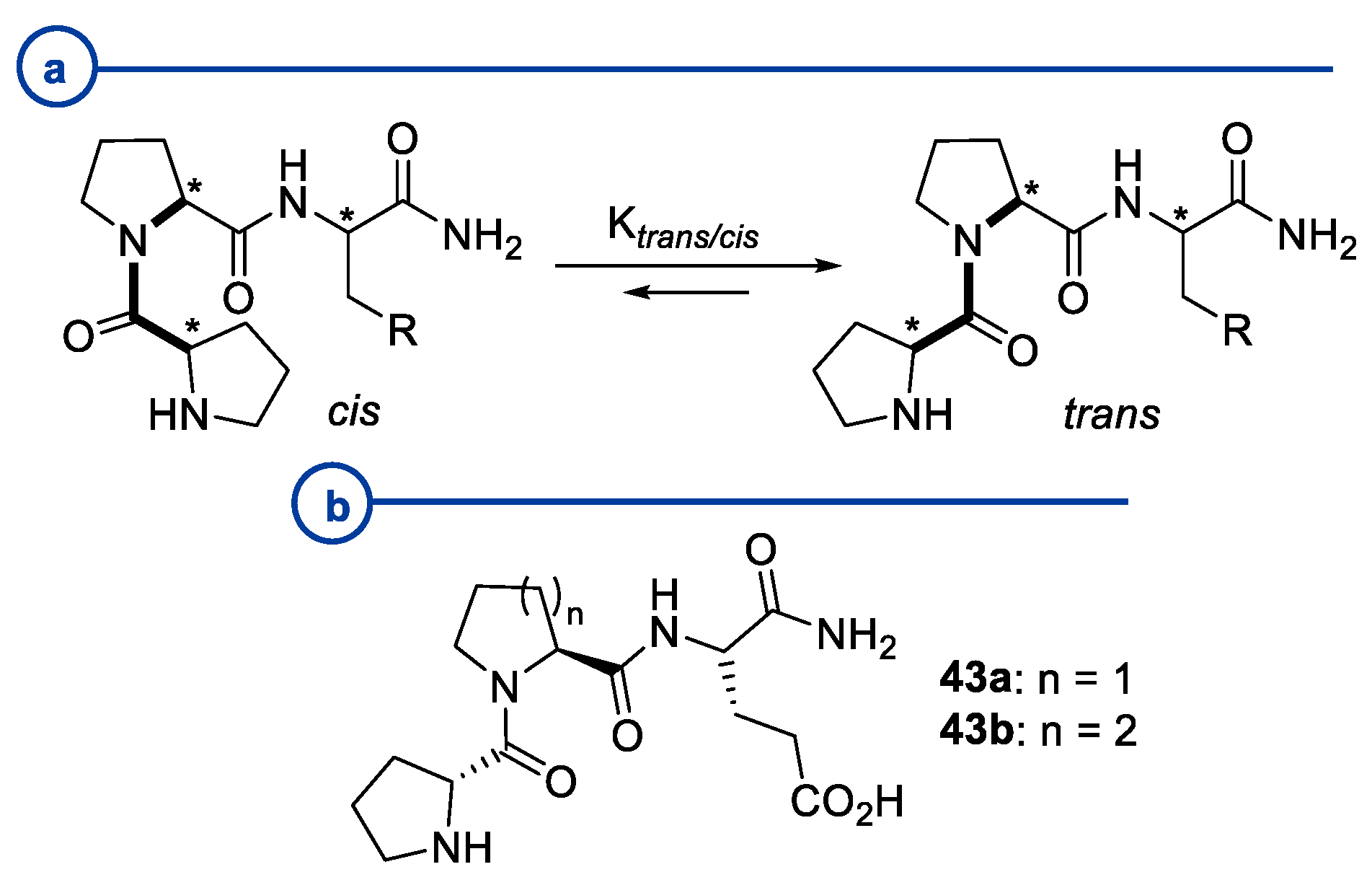
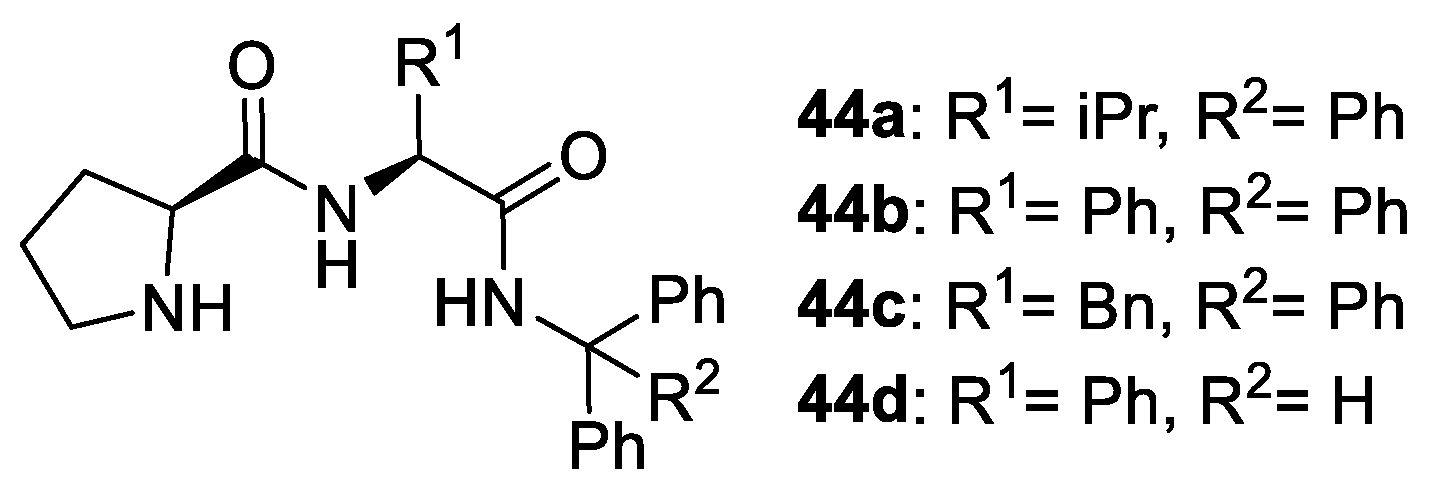
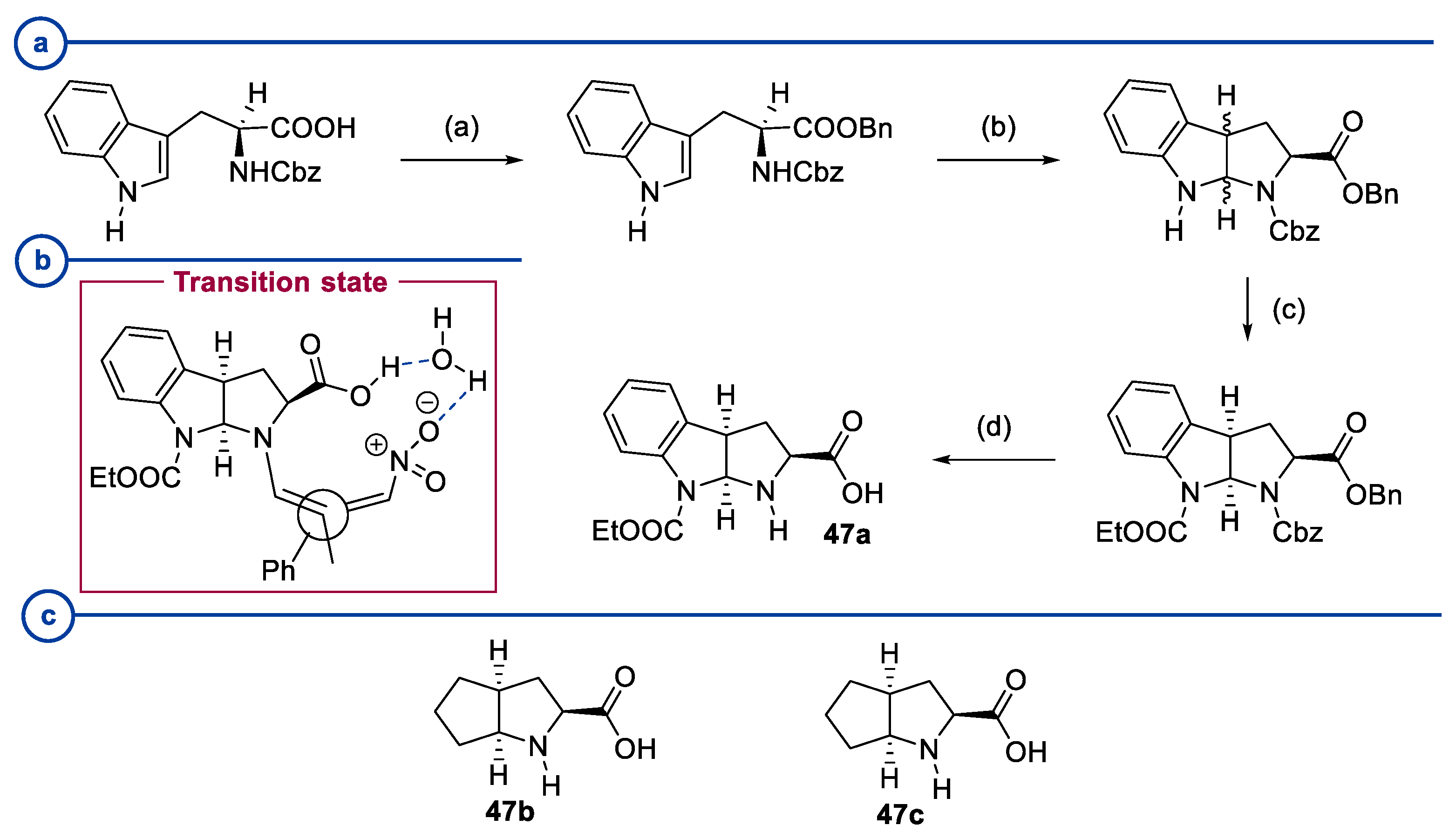
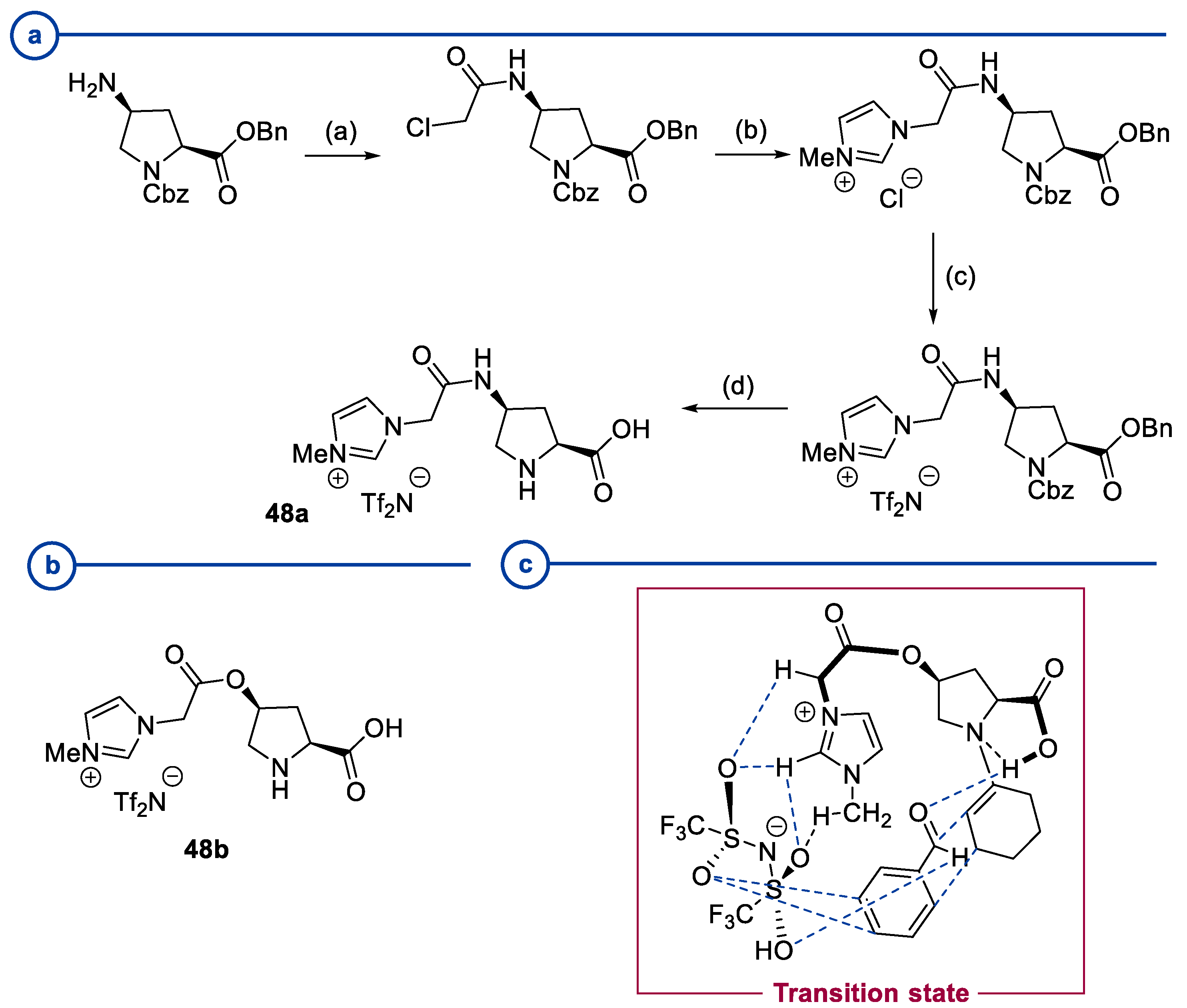
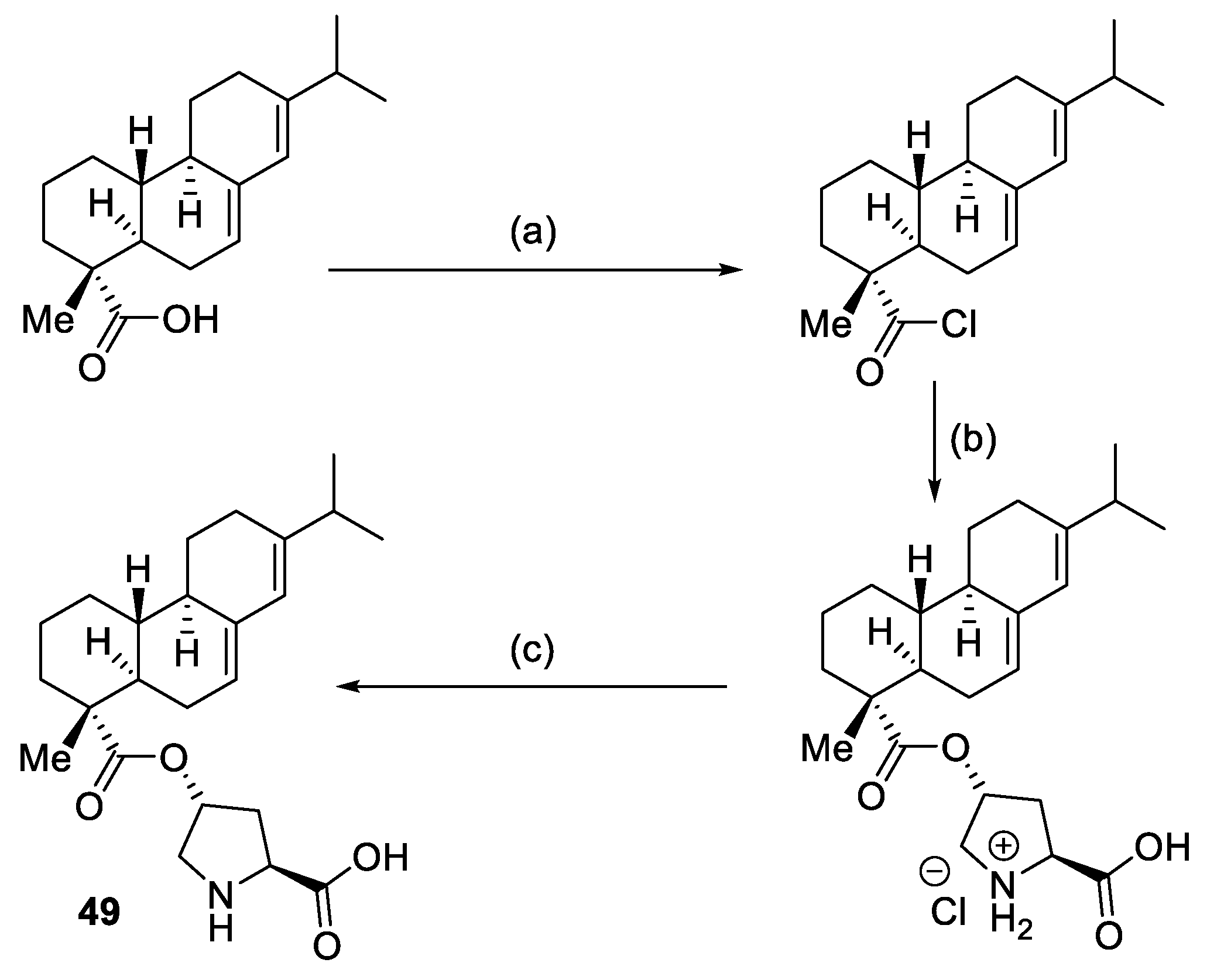
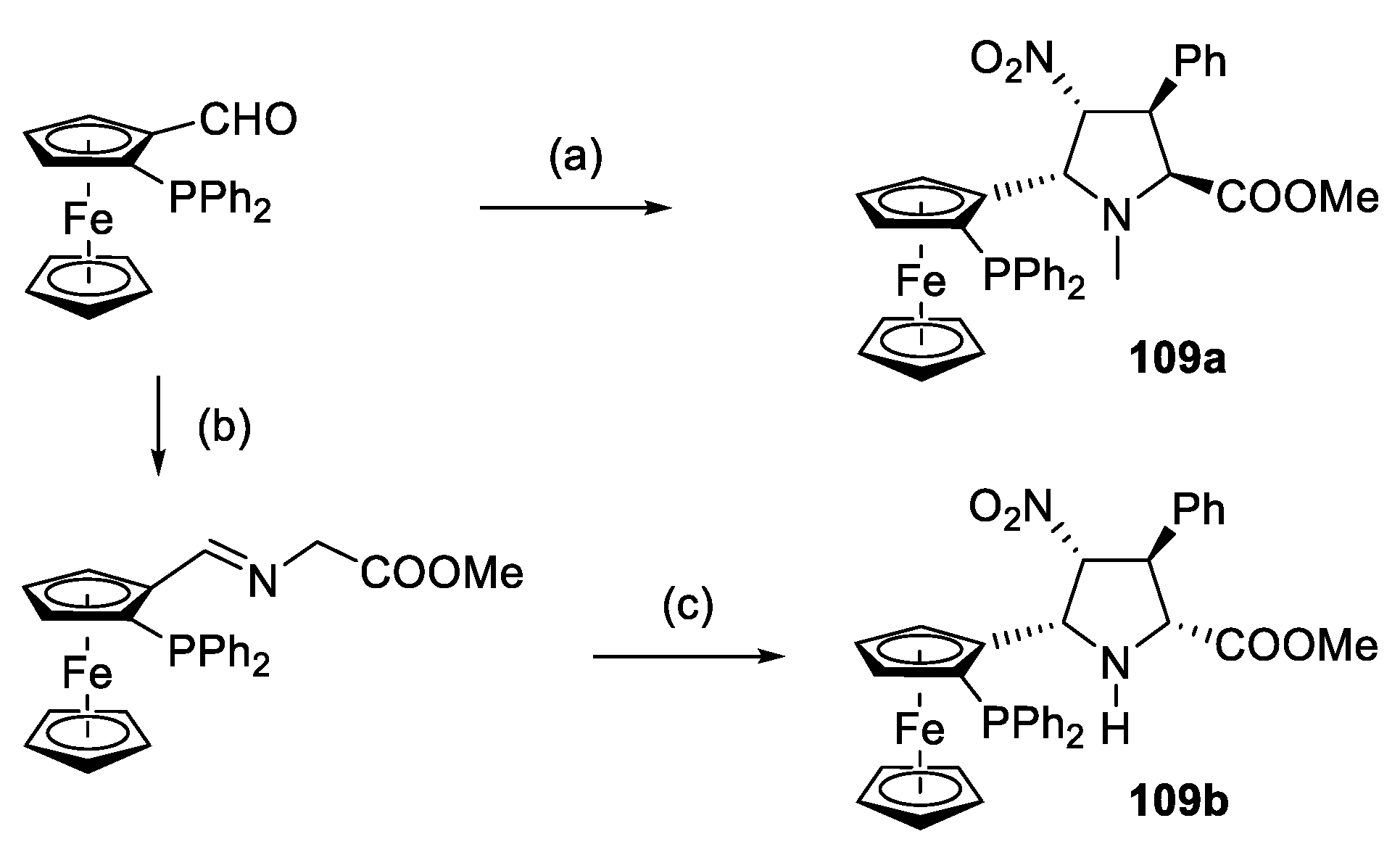
2.2. Peptides
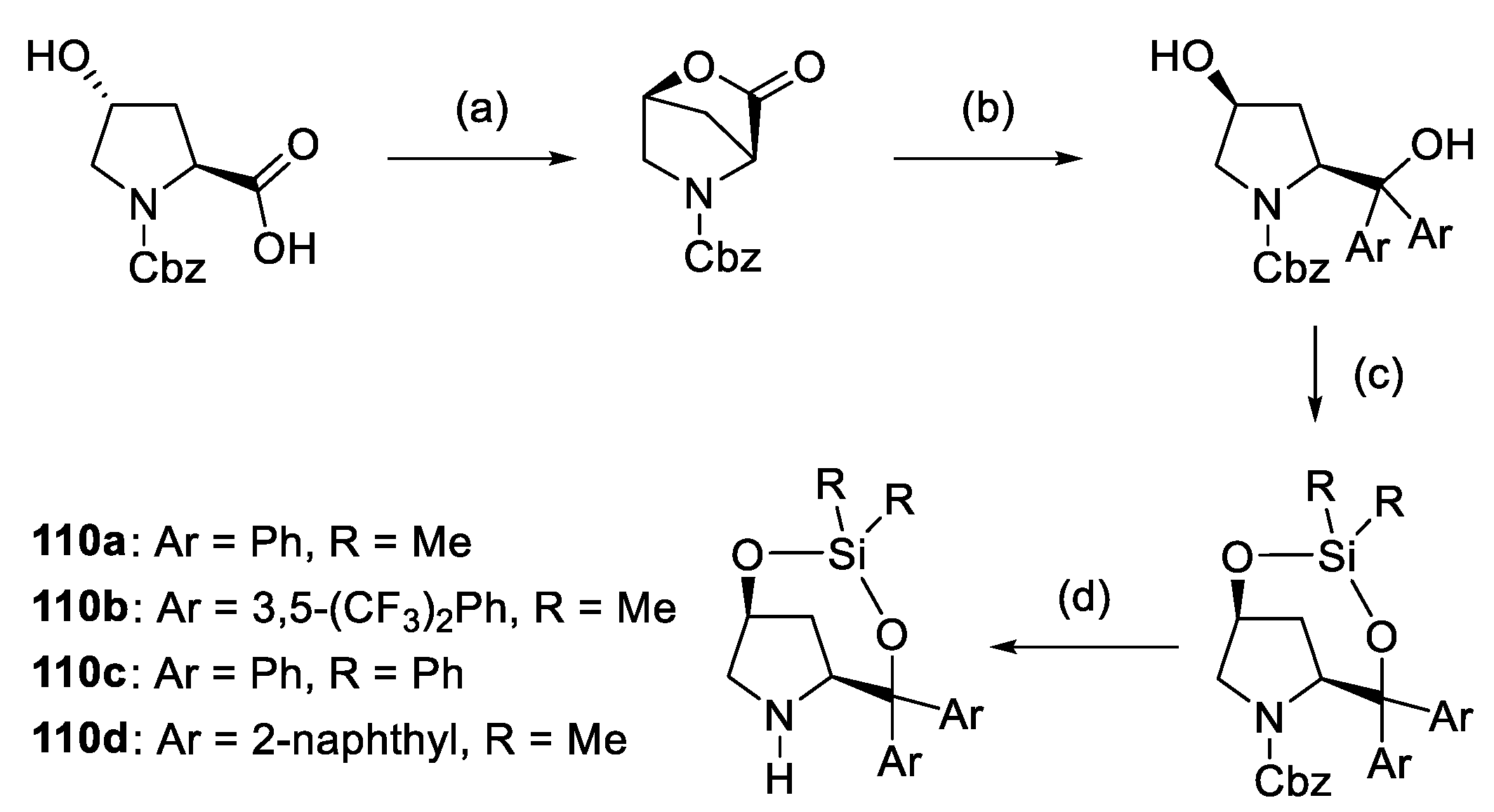
2.3. Substituted Prolines
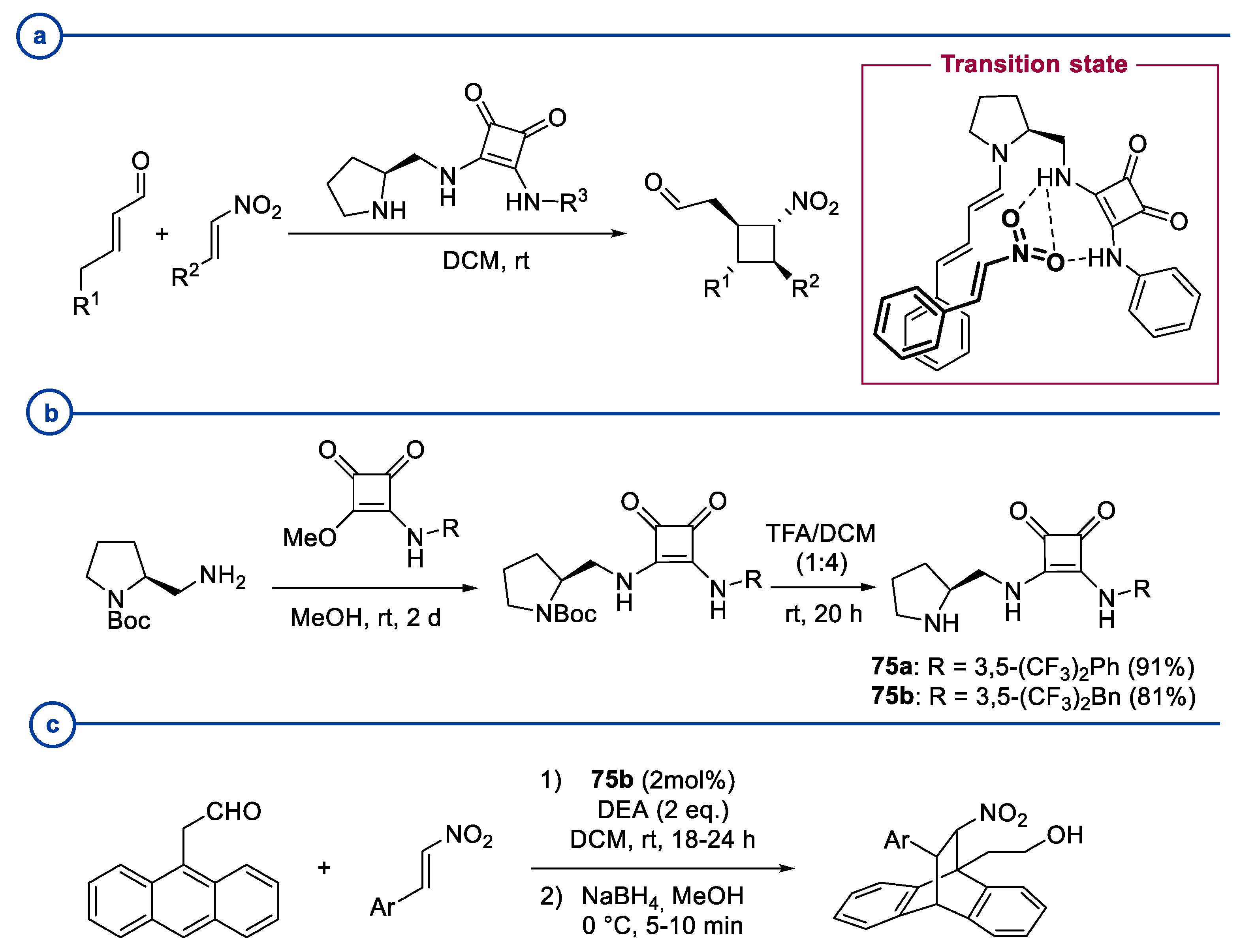
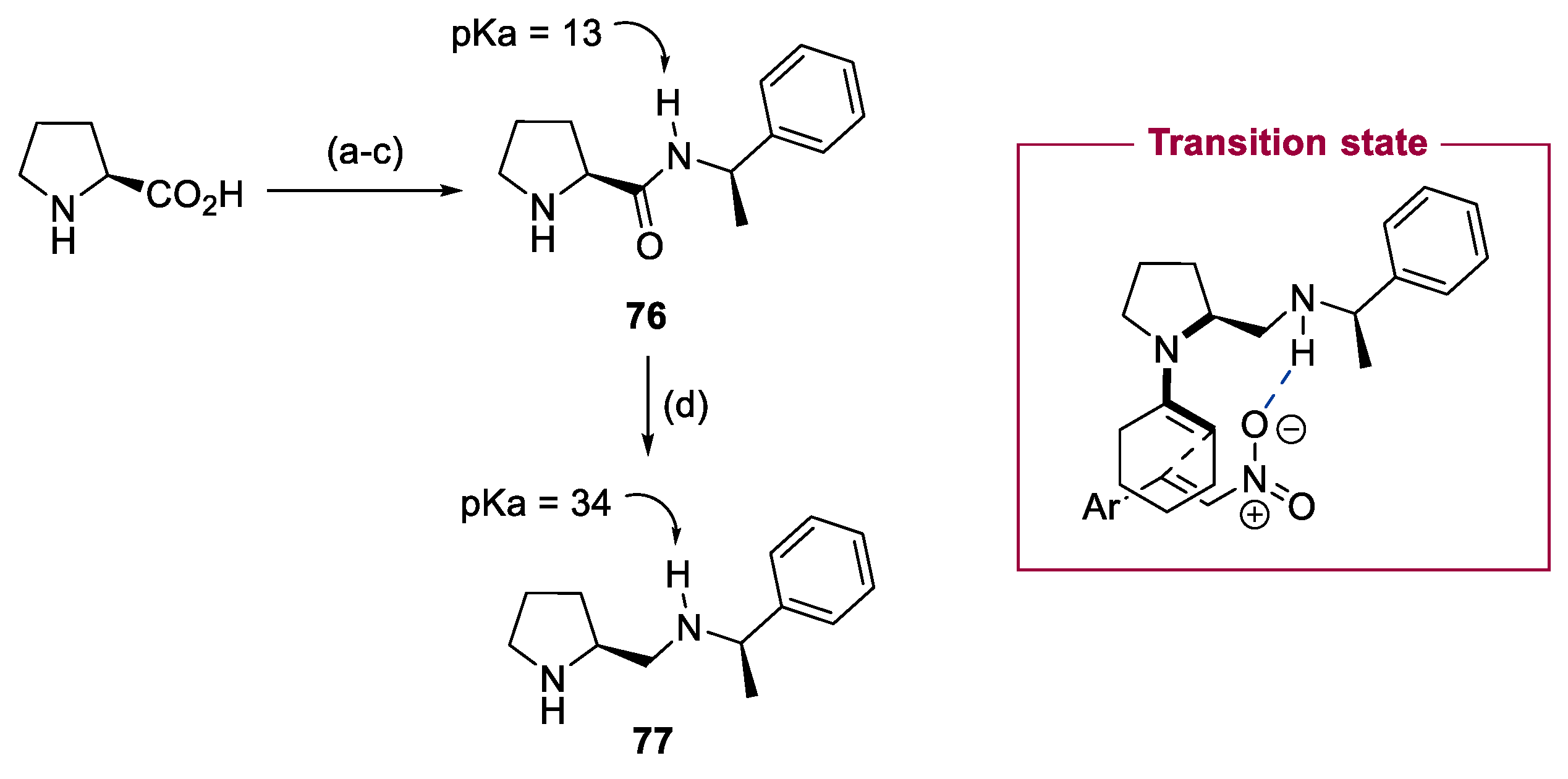
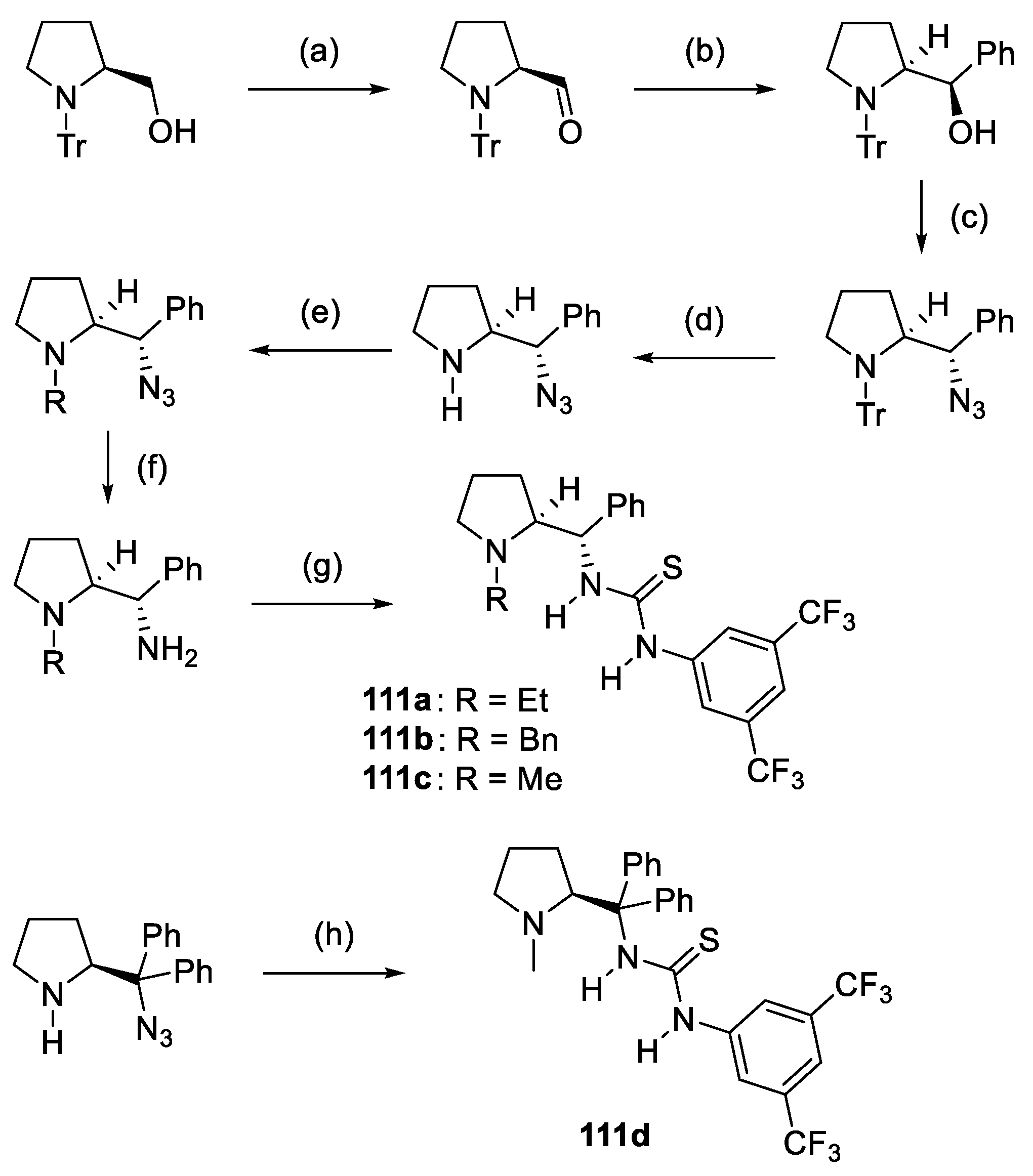
3. Prolinol-Related Organocatalysts
4. Diarylprolinol-Related Organocatalysts
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li Petri, G.; Raimondi, M.V.; Spanò, V.; Holl, R.; Barraja, P.; Montalbano, A. Pyrrolidine in Drug Discovery: A Versatile Scaffold for Novel Biologically Active Compounds. Top. Curr. Chem. 2021, 379, 34. [Google Scholar] [CrossRef] [PubMed]
- Stocker, B.L.; Dangerfield, E.M.; Win-Mason, A.L.; Haslett, G.W.; Timmer, M.S.M. Recent Developments in the Synthesis of Pyrrolidine-Containing Iminosugars. Eur. J. Org. Chem. 2010, 2010, 1615–1637. [Google Scholar] [CrossRef]
- Martineau, D.; Beley, M.; Gros, P.C. Pyrrolidine-Containing Polypyridines: New Ligands for Improved Visible Light Absorption by Ruthenium Complexes. J. Org. Chem. 2006, 71, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; El-Sayed, E.S.M.; Ju, Z.; Wang, W.; Yuan, D. The Synthesis and Applications of Chiral Pyrrolidine Functionalized Metal-Organic Frameworks and Covalent-Organic Frameworks. Inorg. Chem. Front. 2020, 7, 1319–1333. [Google Scholar] [CrossRef]
- Vega-Peñaloza, A.; Paria, S.; Bonchio, M.; Dell’Amico, L.; Companyó, X. Profiling the Privileges of Pyrrolidine-Based Catalysts in Asymmetric Synthesis: From Polar to Light-Driven Radical Chemistry. ACS Catal. 2019, 9, 6058–6072. [Google Scholar] [CrossRef]
- Oliveira, V.D.G.; Cardoso, M.F.D.C.; Forezi, L.D.S.M. Organocatalysis: A Brief Overview on Its Evolution and Applications. Catalysts 2018, 8, 605. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, L. Recent Advances in Asymmetric Reactions Catalyzed by Proline and Its Derivatives. Synthesis 2017, 49, 960–972. [Google Scholar] [CrossRef]
- Thorat, B.R.; Mali, S.N.; Wavhal, S.S.; Bhagat, D.S.; Borade, R.M.; Chapolikar, A.; Gandhi, A.; Shinde, P. L-Proline: A Versatile Organo-Catalyst in Organic Chemistry. Comb. Chem. High Throughput Screen. 2023, 26, 1108–1140. [Google Scholar] [CrossRef]
- Cozzi, P.G.; Gualandi, A.; Mengozzi, L.; Wilson, C.M. Imidazolidinones as Asymmetric Organocatalysts. In Sustainable Catalysis: Without Metals or Other Endangered Elements, Part 2; North, M., Ed.; Royal Society of Chemistry: London, UK, 2015; pp. 164–195. [Google Scholar]
- Jensen, K.L.; Dickmeiss, G.; Jiang, H.; Albrecht, Ł.; Jørgensen, K.A. The Diarylprolinol Silyl Ether System: A General Organocatalyst. Acc. Chem. Res. 2012, 45, 248–264. [Google Scholar] [CrossRef]
- Gotoh, H.; Hayashi, Y. Diarylprolinol silyl ethers: Development and application as organocatalysts. In Sustainable Catalysis: Challenges and Practices for the Pharmaceutical and Fine Chemical Industries; Dunn, P.J., Hii, K.K., Krische, M.J., Williams, M.T., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 287–316. [Google Scholar]
- Moyano, A. Activation modes in asymmetric organocatalysis. In Stereoselective Organocatalysis; Torres, R.R., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2013; ISBN 9781118604755. [Google Scholar]
- Chen, Z.; Yang, Q.-Q.; Du, W.; Chen, Y.-C. Asymmetric Organocatalysis Involving Double Activation. Tetrahedron Chem 2022, 2, 100017. [Google Scholar] [CrossRef]
- Han, M.Y.; Jia, J.Y.; Wang, W. Recent Advances in Organocatalytic Asymmetric Synthesis of Polysubstituted Pyrrolidines. Tetrahedron Lett. 2014, 55, 784–794. [Google Scholar] [CrossRef]
- Pandey, G.; Banerjee, P.; Gadre, S.R. Construction of Enantiopure Pyrrolidine Ring System via Asymmetric [3 + 2]-Cycloaddition of Azomethine Ylides. Chem. Rev. 2006, 106, 4484–4517. [Google Scholar] [CrossRef]
- Adrio, J.; Carretero, J.C. Stereochemical Diversity in Pyrrolidine Synthesis by Catalytic Asymmetric 1,3-Dipolar Cycloaddition of Azomethine Ylides. Chem. Commun. 2019, 55, 11979–11991. [Google Scholar] [CrossRef]
- Philip, R.M.; Treesa, G.S.S.; Saranya, S.; Anilkumar, G. Applications of Aryl-Sulfinamides in the Synthesis of N-Heterocycles. RSC Adv. 2021, 11, 20591–20600. [Google Scholar] [CrossRef]
- Smolobochkin, A.V.; Gazizov, A.S.; Turmanov, R.A.; Abdullaeva, D.S.; Burilov, A.R.; Pudovik, M.A. N-Phosphorylated Pyrrolidines: An Overview of Synthetic Approaches. Synthesis 2020, 52, 2162–2170. [Google Scholar] [CrossRef]
- Li, J.; Ye, Y.; Zhang, Y. Cycloaddition/Annulation Strategies for the Construction of Multisubstituted Pyrrolidines and Their Applications in Natural Product Synthesis. Org. Chem. Front. 2018, 5, 864–892. [Google Scholar] [CrossRef]
- Schultz, D.M.; Wolfe, J.P. Recent Developments in Palladium-Catalyzed Alkene Aminoarylation Reactions for the Synthesis of Nitrogen Heterocycles. Synthesis 2012, 44, 351–361. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Giacalone, F.; Noto, R. Supported Proline and Proline-Derivatives as Recyclable Organocatalysts. Chem. Soc. Rev. 2008, 37, 1666–1688. [Google Scholar] [CrossRef]
- Singh, S. Recent Development of Recoverable MacMillan Catalyst in Asymmetric Organic Transformations. Adv. Synth. Catal. 2021, 363, 629–656. [Google Scholar] [CrossRef]
- Susam, Z.D.; Tanyeli, C. Recyclable Organocatalysts in Asymmetric Synthesis. Asian J. Org. Chem. 2021, 10, 1251–1266. [Google Scholar] [CrossRef]
- Ferré, M.; Pleixats, R.; Wong Chi Man, M.; Cattoën, X. Recyclable Organocatalysts Based on Hybrid Silicas. Green Chem. 2016, 18, 881–922. [Google Scholar] [CrossRef] [Green Version]
- Fulgheri, T.; Della Penna, F.; Baschieri, A.; Carlone, A. Advancements in the Recycling of Organocatalysts: From Classical to Alternative Approaches. Curr. Opin. Green Sustain. Chem. 2020, 25, 100387. [Google Scholar] [CrossRef]
- Shajahan, R.; Sarang, R.; Saithalavir, A. Polymer Supported Proline-Based Organocatalysts in Asymmetric Aldol Reactions: A Review. Curr. Organocatalysis 2022, 9, 124–146. [Google Scholar] [CrossRef]
- Bui, T.; Barbas, C.F. A Proline-Catalyzed Asymmetric Robinson Annulation Reaction. Tetrahedron Lett. 2000, 41, 6951–6954. [Google Scholar] [CrossRef]
- List, B.; Lerner, R.A.; Barbas III, C.F. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Bahmanyar, S.; Houk, K.N.; Martin, H.J.; List, B. Quantum Mechanical Predictions of the Stereoselectivities of Proline-Catalyzed Asymmetric Intermolecular Aldol Reactions. J. Am. Chem. Soc. 2003, 125, 2475–2479. [Google Scholar] [CrossRef]
- Yang, H.; Carter, R.G. Synthesis of All-Carbon, Quaternary Center-Containing Cyclohexenones through an Organocatalyzed, Multicomponent Coupling. Org. Lett. 2010, 12, 3108–3111. [Google Scholar] [CrossRef]
- Naziroglu, H.N.; Durmaz, M.; Bozkurt, S.; Demir, A.S.; Sirit, A. Application of L-Prolinamides as Highly Efficient Organocatalysts for the Asymmetric Michael Addition of Unmodified Aldehydes to Nitroalkenes. Tetrahedron Asymmetry 2012, 23, 164–169. [Google Scholar] [CrossRef]
- Watts, J.; Luu, L.; McKee, V.; Carey, E.; Kelleher, F. Structure-Reactivity Studies of Simple 4-Hydroxyprolinamide Organocatalysts in the Asymmetric Michael Addition Reaction of Aldehydes to Nitroolefins. Adv. Synth. Catal. 2012, 354, 1035–1042. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lin, J.; Wei, K. Aromatic L-Prolinamide-Catalyzed Asymmetric Michael Addition of Aldehydes to Nitroalkenes. Tetrahedron Asymmetry 2014, 25, 1599–1604. [Google Scholar] [CrossRef]
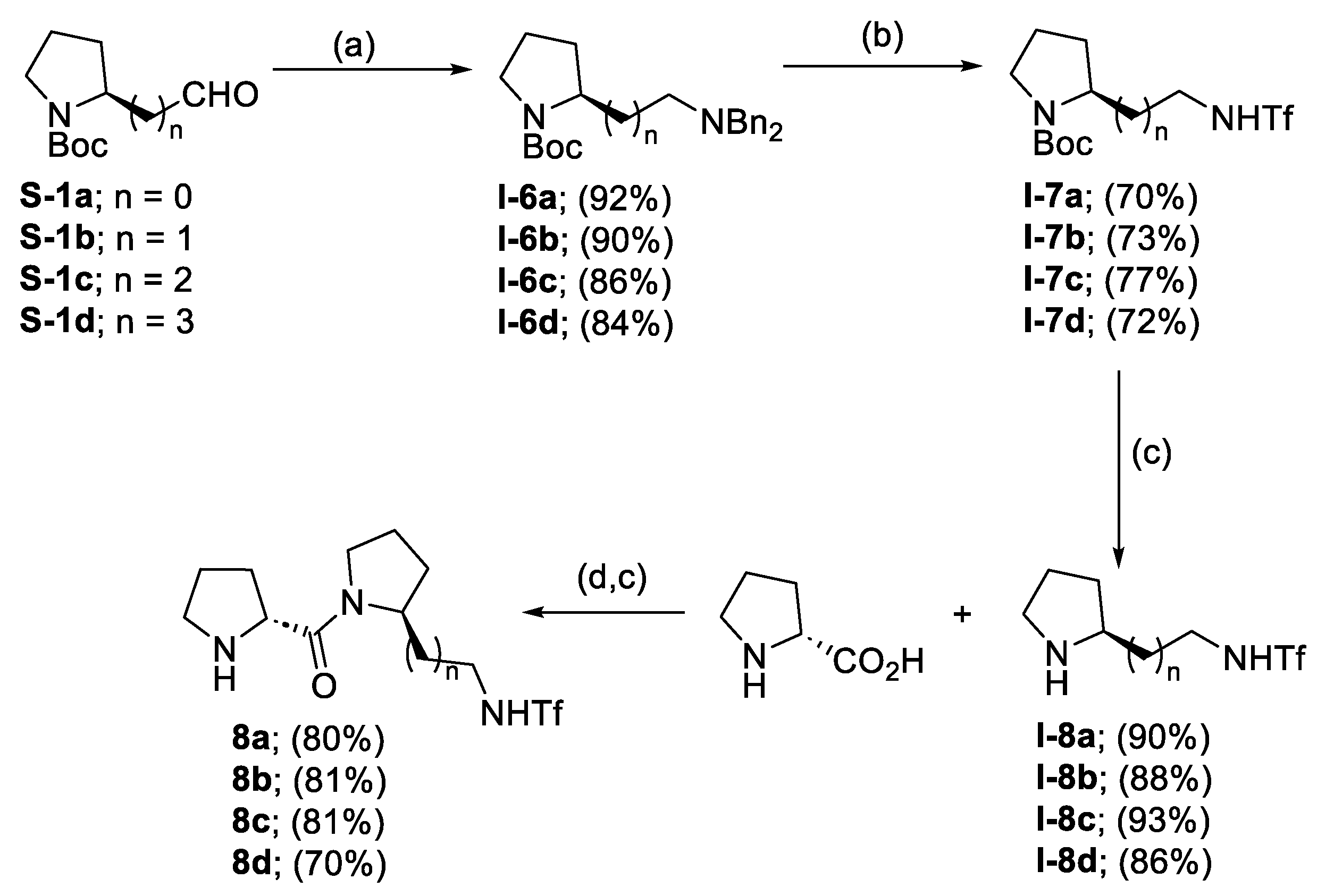
- Gorde, A.B.; Ramapanicker, R. D-Prolyl-2-(Trifluoromethylsulfonamidopropyl)Pyrrolidine: An Organocatalyst for Asymmetric Michael Addition of Aldehydes to β-Nitroalkenes at Ambient Conditions. J. Org. Chem. 2019, 84, 1523–1533. [Google Scholar] [CrossRef]
- Wiesner, M.; Neuburger, M.; Wennemers, H. Tripeptides of the Type H-D-Pro-Pro-Xaa-NH2 as Catalysts for Asymmetric 1,4-Addition Reactions: Structural Requirements for High Catalytic Efficiency. Chem.–A Eur. J. 2009, 15, 10103–10109. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Clerget, M.; Gager, O.; Monteil, M.; Pirat, J.L.; Migianu-Griffoni, E.; Deschamp, J.; Lecouvey, M. Novel Easily Recyclable Bifunctional Phosphonic Acid Carrying Tripeptides for the Stereoselective Michael Addition of Aldehydes with Nitroalkenes. Adv. Synth. Catal. 2016, 358, 34–40. [Google Scholar] [CrossRef]
- Almaşi, D.; Alonso, D.A.; Nájera, C. Prolinamides versus Prolinethioamides as Recyclable Catalysts in the Enantioselective Solvent-Free Inter- and Intramolecular Aldol Reactions. Adv. Synth. Catal. 2008, 350, 2467–2472. [Google Scholar] [CrossRef]
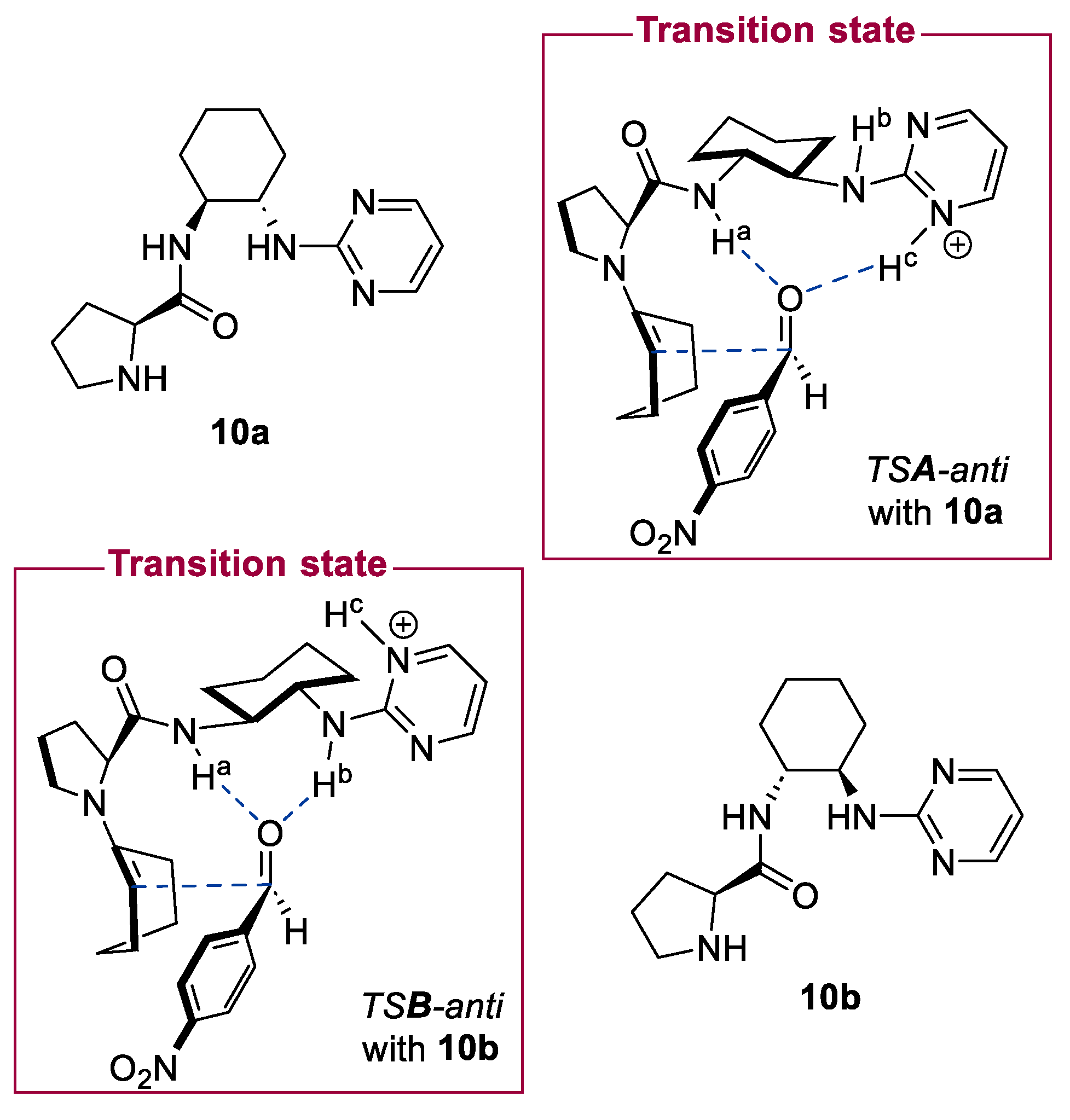
- Vizcaíno-Milla, P.; Sansano, J.M.; Nájera, C.; Fiser, B.; Gómez-Bengoa, E. Pyrimidine-Derived Prolinamides as Recoverable Bifunctional Organocatalysts for Enantioselective Inter- and Intramolecular Aldol Reactions under Solvent-Free Conditions. Eur. J. Org. Chem. 2015, 2015, 2614–2621. [Google Scholar] [CrossRef] [Green Version]
- Tzeng, Z.H.; Chen, H.Y.; Reddy, R.J.; Huang, C.T.; Chen, K. Highly Diastereo- and Enantioselective Direct Aldol Reactions Promoted by Water-Compatible Organocatalysts Bearing a Pyrrolidinyl-Camphor Structural Scaffold. Tetrahedron 2009, 65, 2879–2888. [Google Scholar] [CrossRef]
- Xu, J.; Fu, X.; Wu, C.; Hu, X. Simple, Inexpensive, and Facile L-Prolinamide Used as a Recyclable Organocatalyst for Highly Efficient Large-Scale Asymmetric Direct Aldol Reactions. Tetrahedron Asymmetry 2011, 22, 840–850. [Google Scholar] [CrossRef]
- Saha, S.; Moorthy, J.N. Enantioselective Organocatalytic Biginelli Reaction: Dependence of the Catalyst on Sterics, Hydrogen Bonding, and Reinforced Chirality. J. Org. Chem. 2011, 76, 396–402. [Google Scholar] [CrossRef] [PubMed]
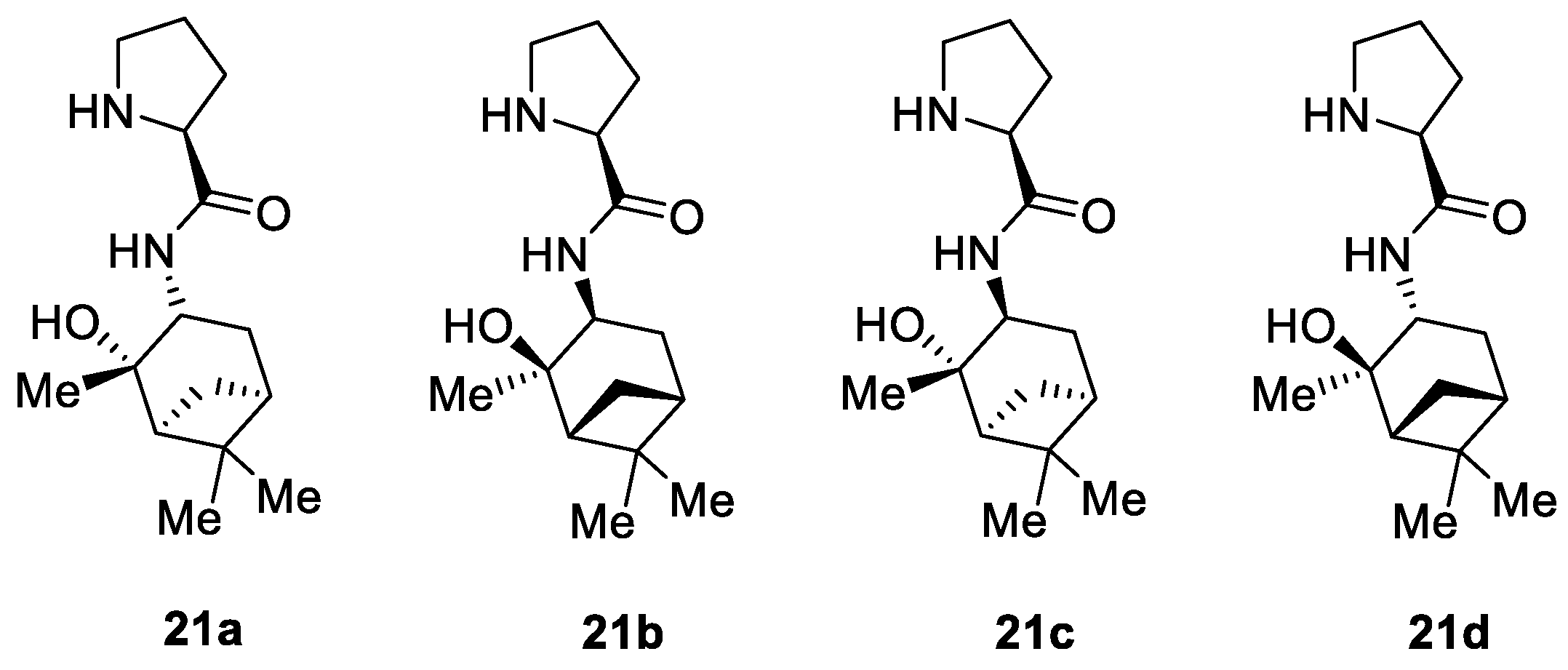
- Siyutkin, D.E.; Kucherenko, A.S.; Frolova, L.L.; Kuchin, A.V.; Zlotin, S.G. 2-Hydroxy-3-[(S)-Prolinamido]Pinanes as Novel Bifunctional Organocatalysts for Asymmetric Aldol Reactions in Aqueous Media. Tetrahedron Asymmetry 2011, 22, 1320–1324. [Google Scholar] [CrossRef]
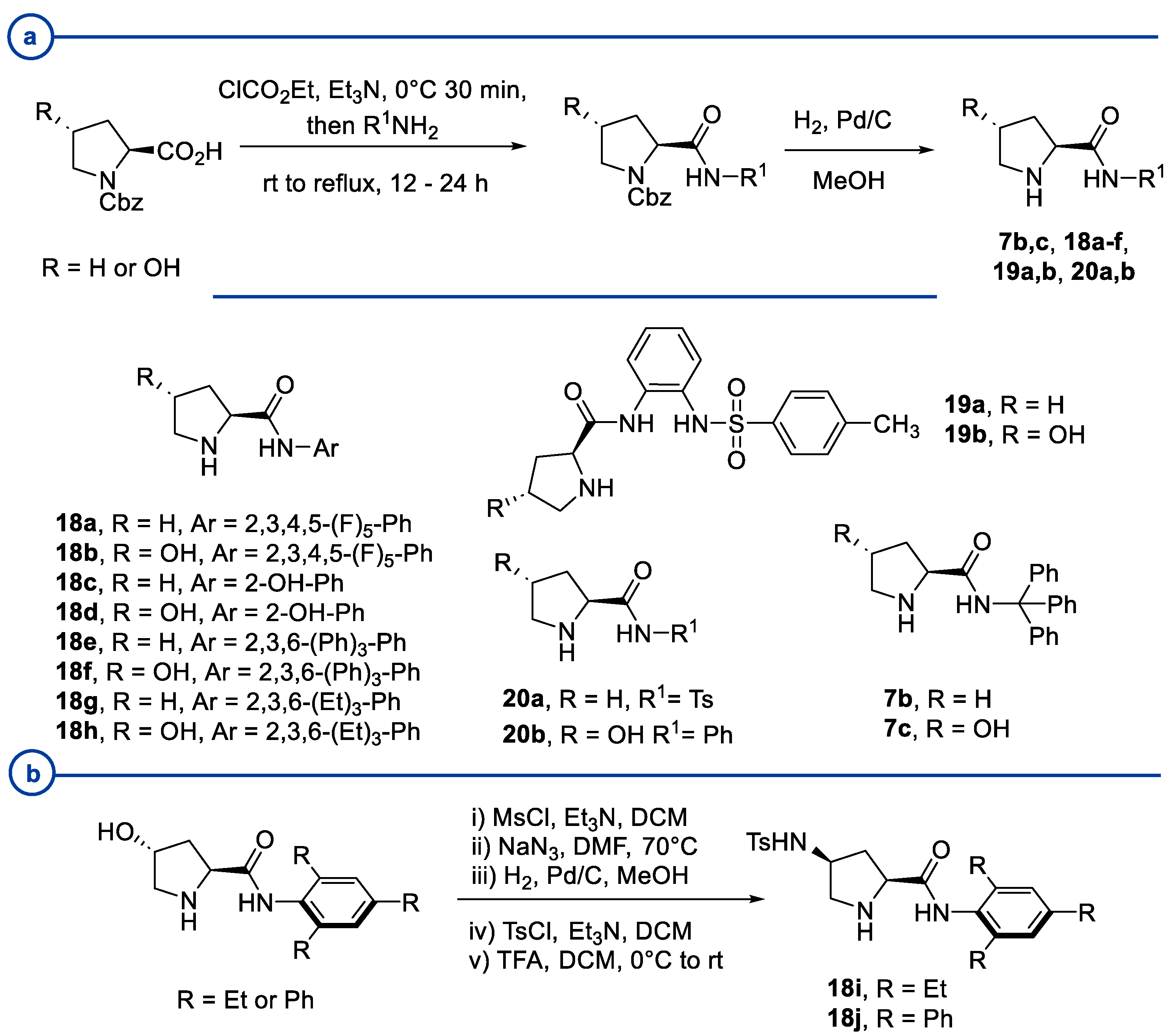
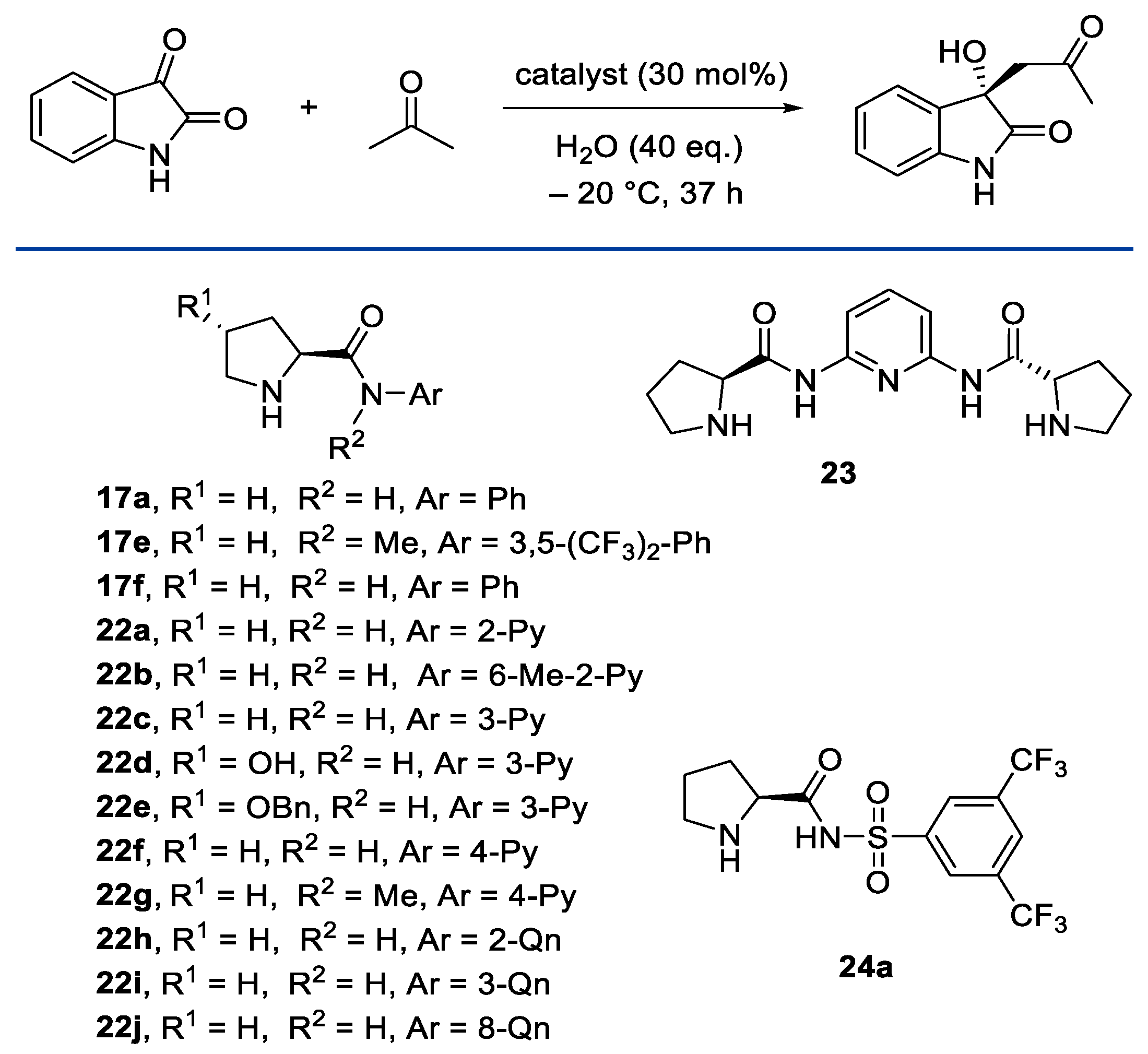
- Kinsella, M.; Duggan, P.G.; Lennon, C.M. Screening of Simple N-Aryl and N-Heteroaryl Pyrrolidine Amide Organocatalysts for the Enantioselective Aldol Reaction of Acetone with Isatin. Tetrahedron Asymmetry 2011, 22, 1423–1433. [Google Scholar] [CrossRef]
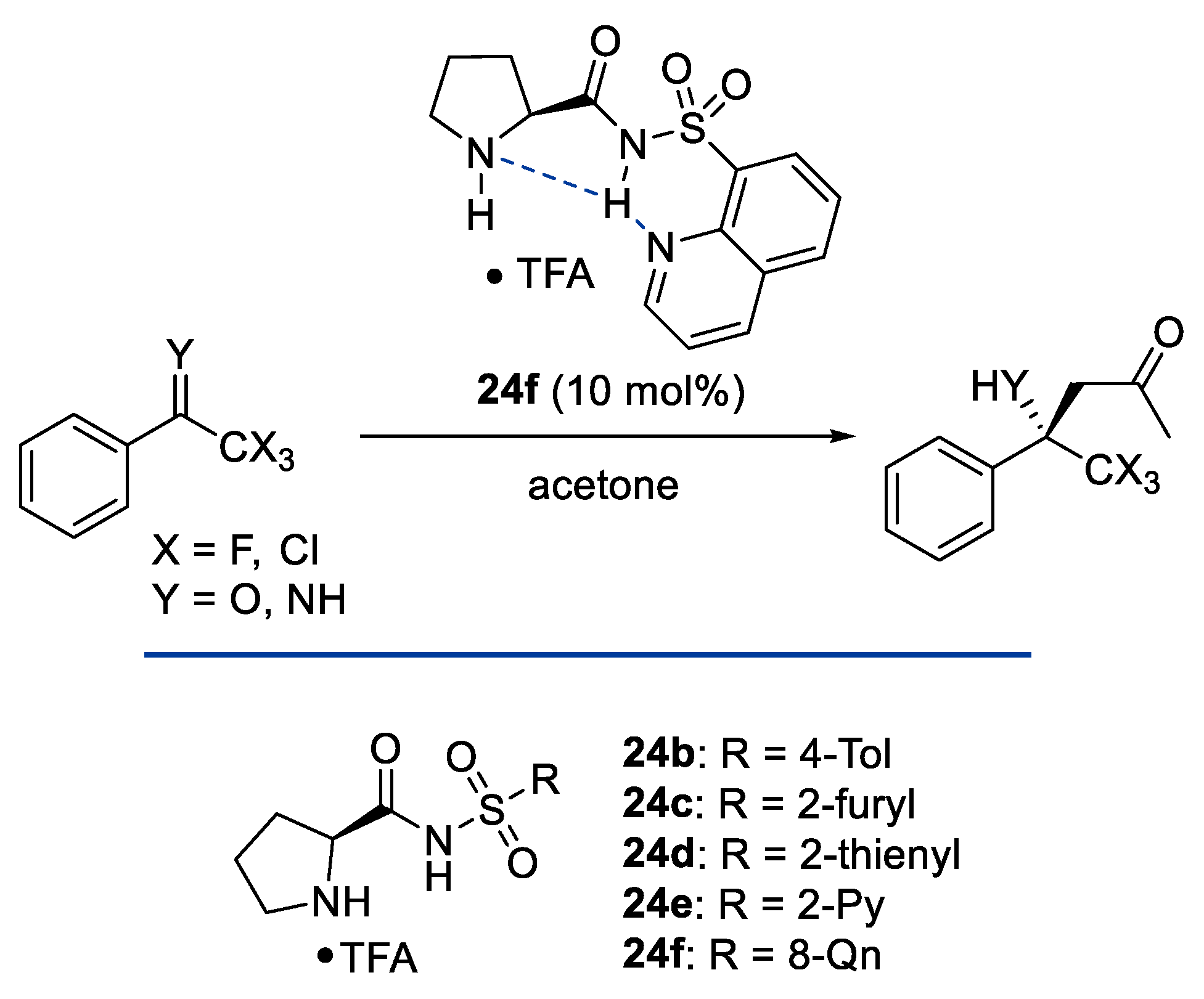
- Hara, N.; Tamura, R.; Funahashi, Y.; Nakamura, S. N-(Heteroarenesulfonyl)Prolinamides-Catalyzed Aldol Reaction between Acetone and Aryl Trihalomethyl Ketones. Org. Lett. 2011, 13, 1662–1665. [Google Scholar] [CrossRef]
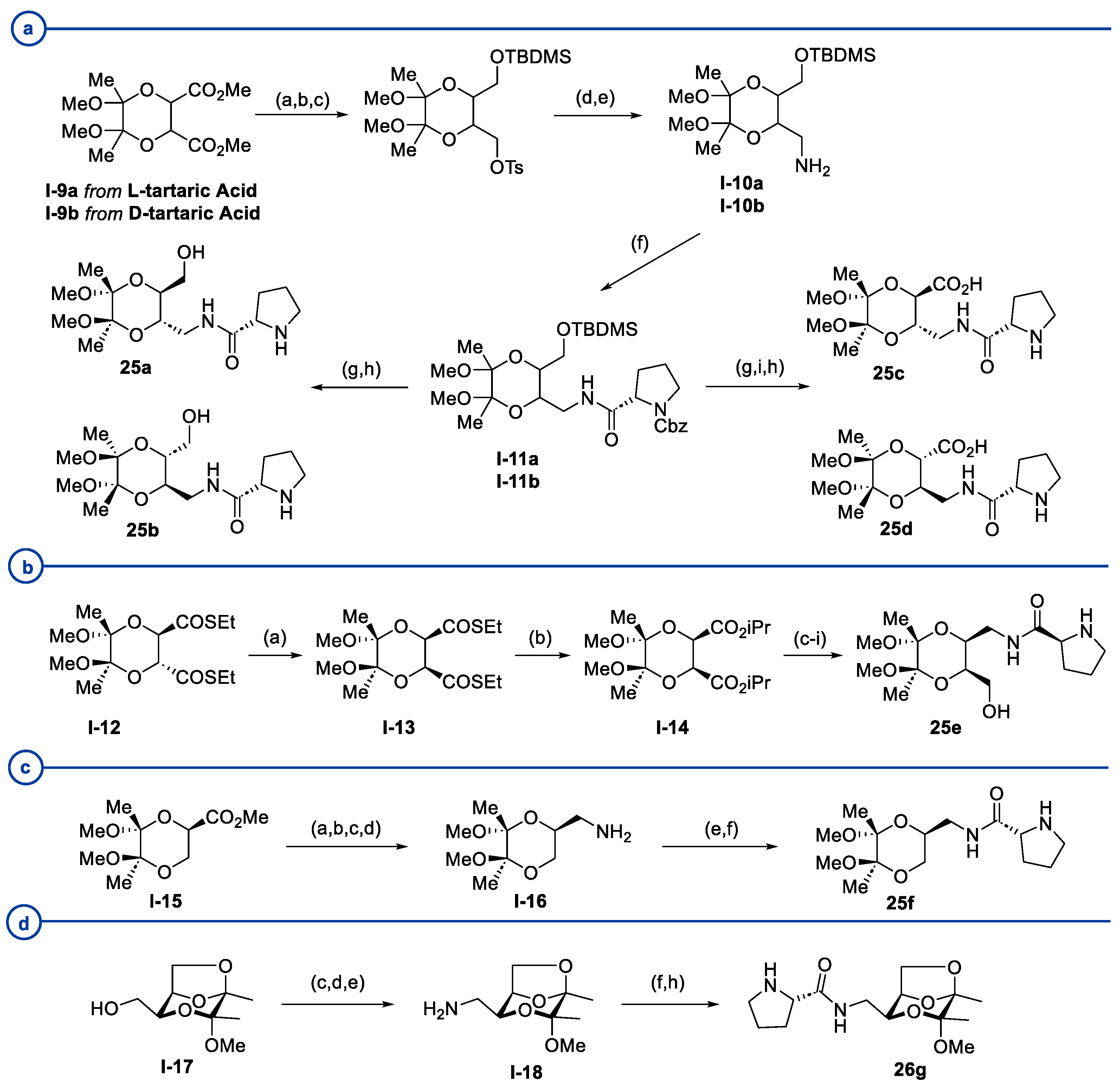
- Maycock, C.D.; Rita Ventura, M. New Organocatalysts Derived from Tartaric and Glyceric Acids for Direct Aldol Reactions. Tetrahedron Asymmetry 2012, 23, 1262–1271. [Google Scholar] [CrossRef]
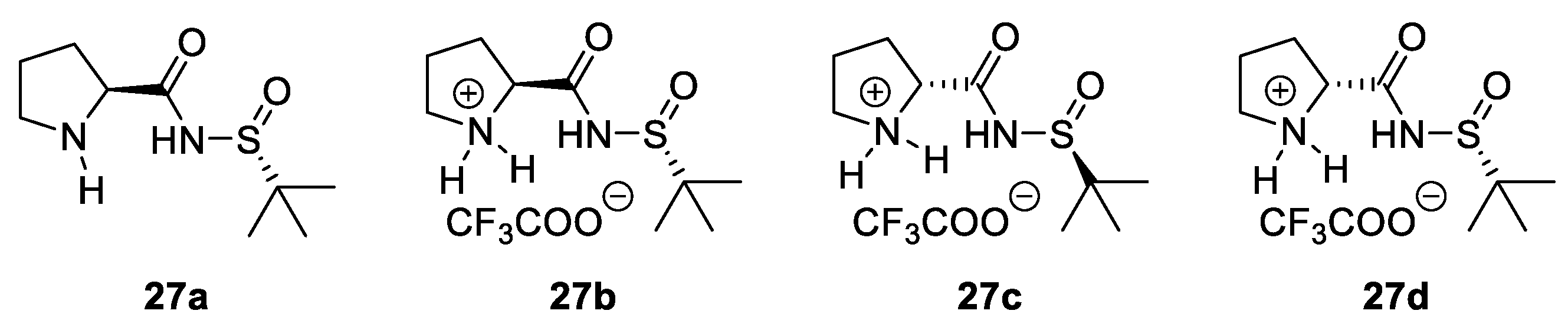
- Wan, W.; Gao, W.; Ma, G.; Ma, L.; Wang, F.; Wang, J.; Jiang, H.; Zhu, S.; Hao, J. Asymmetric Aldol Reaction Organocatalyzed by Bifunctional N-Prolyl Sulfinamides under Solvent-Free Conditions. RSC Adv. 2014, 4, 26563–26568. [Google Scholar] [CrossRef]
- Kumar, T.P.; Vavle, N.C.; Patro, V.; Haribabu, K. Phthalimido-Prolinamide: A New Chiral Catalyst for Solvent Free Enantioselective Aldol Reactions. Tetrahedron Asymmetry 2014, 25, 457–461. [Google Scholar] [CrossRef]
- Naresh, T.; Kumar, T.P.; Haribabu, K.; Chandrasekhar, S. AZT-Prolinamide: The Nucleoside Derived Pyrrolidine Catalysts for Asymmetric Aldol Reactions Using Water as Solvent. Tetrahedron Asymmetry 2014, 25, 1340–1345. [Google Scholar] [CrossRef]
- Yadav, G.D.; Singh, S. Direct Asymmetric Aldol Reactions Catalysed by Trans-4-Hydroxy-(S)-Prolinamide in Solvent-Free Conditions. Tetrahedron Asymmetry 2015, 26, 1156–1166. [Google Scholar] [CrossRef]
- Yadav, G.D.; Singh, S. Trans-4-Hydroxy-L-Prolinamide as an Efficient Catalyst for Direct Asymmetric Aldol Reaction of Acetone with Isatins. Tetrahedron Asymmetry 2016, 27, 463–466. [Google Scholar] [CrossRef]
- Rojas Cabrera, H.; Huelgas, G.; Hernández Pérez, J.M.; Walsh, P.J.; Somanathan, R.; Anaya De Parrodi, C. Homochiral L-Prolinamido-Sulfonamides and Their Use as Organocatalysts in Aldol Reactions. Tetrahedron Asymmetry 2015, 26, 163–172. [Google Scholar] [CrossRef]
- Davie, E.A.C.; Mennen, S.M.; Xu, Y.; Miller, S.J. Asymmetric Catalysis Mediated by Synthetic Peptides. Chem. Rev. 2007, 107, 5759–5812. [Google Scholar] [CrossRef]
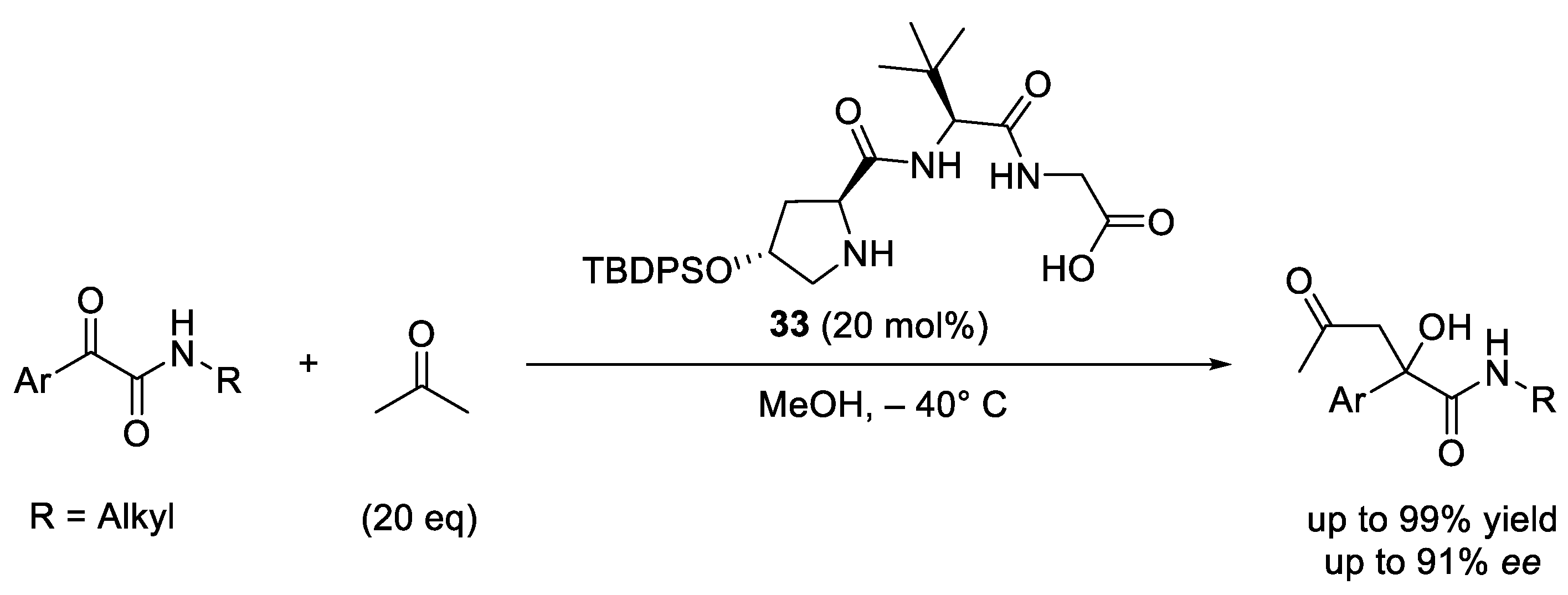
- Revell, J.D.; Wennemers, H. Investigating Sequence Space: How Important Is the Spatial Arrangement of Functional Groups in the Asymmetric Aldol Reaction Catalyst H-Pro-Pro-Asp- NH2? Adv. Synth. Catal. 2008, 350, 1046–1052. [Google Scholar] [CrossRef]
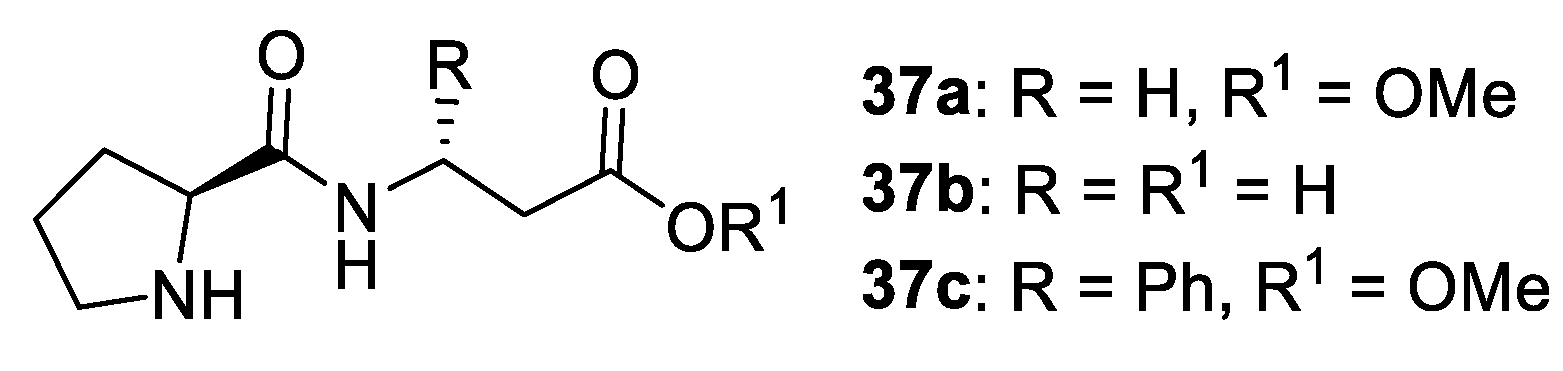
- Psarra, A.; Kokotos, C.G.; Moutevelis-Minakakis, P. Tert-Butyl Esters of Tripeptides Based on Pro-Phe as Organocatalysts for the Asymmetric Aldol Reaction in Aqueous or Organic Medium. Tetrahedron 2014, 70, 608–615. [Google Scholar] [CrossRef]
- Kon, K.; Kohari, Y.; Murata, M. Unnatural Tripeptide as Highly Enantioselective Organocatalyst for Asymmetric Aldol Reaction of Isatins. Tetrahedron Lett. 2019, 60, 415–418. [Google Scholar] [CrossRef]
- Kon, K.; Takai, H.; Kohari, Y.; Murata, M. Tripeptide-Catalyzed Asymmetric Aldol Reaction Between α-Ketoesters and Acetone Under Acidic Cocatalyst-Free Conditions. Catalysts 2019, 9, 514. [Google Scholar] [CrossRef] [Green Version]
- Kon, K.; Takai, H.; Kobayashi, T.; Kohari, Y.; Murata, M. Organocatalyzed Asymmetric Aldol Reaction of α-Keto Amides with A Tripeptide Catalyst. Synlett 2021, 32, 829–832. [Google Scholar] [CrossRef]
- Chandrasekhar, S.; Johny, K.; Reddy, C.R. Proline-Threonine Dipeptide as an Organocatalyst for the Direct Asymmetric Aldol Reaction. Tetrahedron Asymmetry 2009, 20, 1742–1745. [Google Scholar] [CrossRef]
- Tang, Z.; Jiang, F.; Cui, X.; Gong, L.Z.; Mi, A.Q.; Jiang, Y.Z.; Wu, Y.D. Enantioselective Direct Aldol Reactions Catalyzed by L-Prolinamide Derivatives. Proc. Natl. Acad. Sci. USA 2004, 101, 5755–5760. [Google Scholar] [CrossRef] [Green Version]
- Panov, I.; Drabina, P.; Hanusek, J.; Sedlák, M. The Synthesis and Characterisation of N-(1-Carbamoyl-1,1-Dialkyl-Methyl)-(S)-Prolinamides and Related Pyrrolidin-2-yl-4,5-Dihydro-1H-Imidazol-5-Ones as Potential Enantioselective Organocatalysts. Tetrahedron Asymmetry 2011, 22, 215–221. [Google Scholar] [CrossRef]
- Hu, X.M.; Zhang, D.X.; Zhang, S.Y.; Wang, P.A. Highly Modular Dipeptide-like Organocatalysts for Direct Asymmetric Aldol Reactions in Brine. RSC Adv. 2015, 5, 39557–39564. [Google Scholar] [CrossRef]
- Machuca, E.; Rojas, Y.; Juaristi, E. Synthesis and Evaluation of (S)-Proline-Containing α,β-Dipeptides as Organocatalysts in Solvent-Free Asymmetric Aldol Reactions under Ball-Milling Conditions. Asian J. Org. Chem. 2015, 4, 46–53. [Google Scholar] [CrossRef]
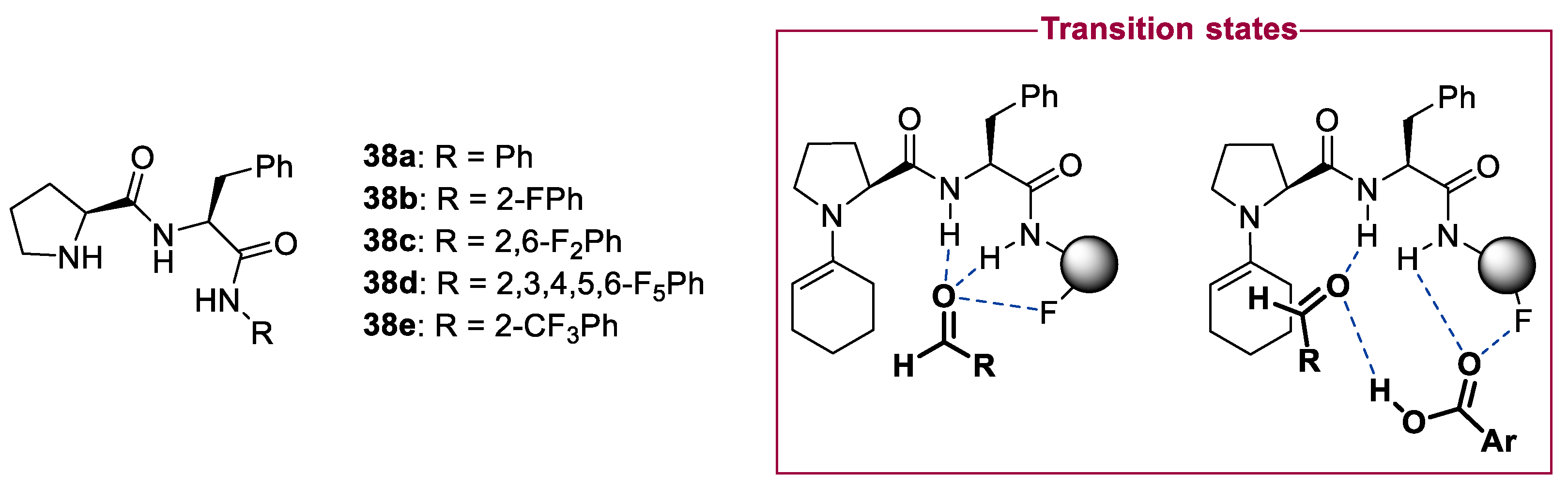
- Ahmetlli, A.; Spiliopoulou, N.; Magi-Oikonomopoulou, A.; Gerokonstantis, D.T.; Moutevelis-Minakakis, P.; Kokotos, C.G. Proline Dipeptides Containing Fluorine Moieties as Oganocatalysts for the Asymmetric Aldol Reaction. Tetrahedron 2018, 74, 5987–5995. [Google Scholar] [CrossRef]
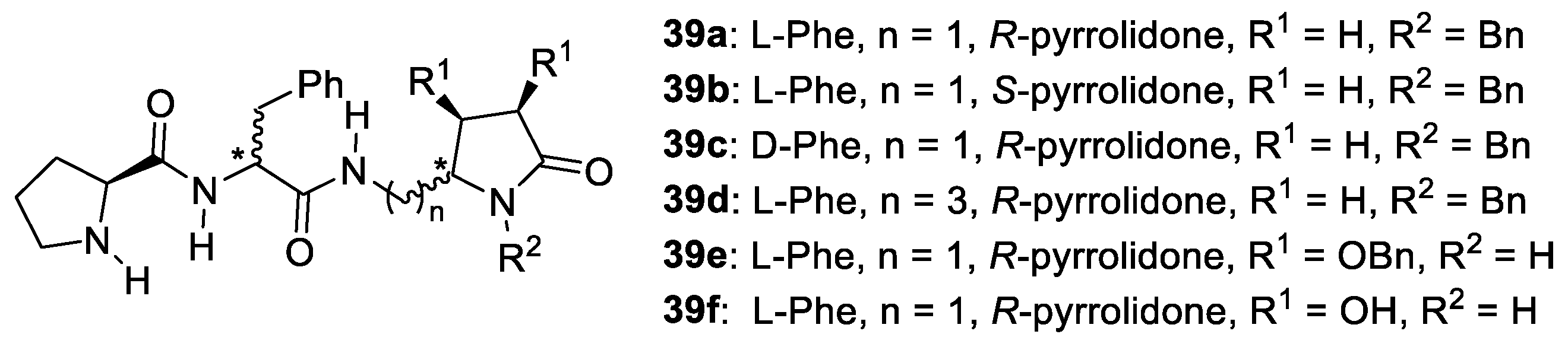
- Vlasserou, I.; Sfetsa, M.; Gerokonstantis, D.T.; Kokotos, C.G.; Moutevelis-Minakakis, P. Combining Prolinamides with 2-Pyrrolidinone: Novel Organocatalysts for the Asymmetric Aldol Reaction. Tetrahedron 2018, 74, 2338–2349. [Google Scholar] [CrossRef]
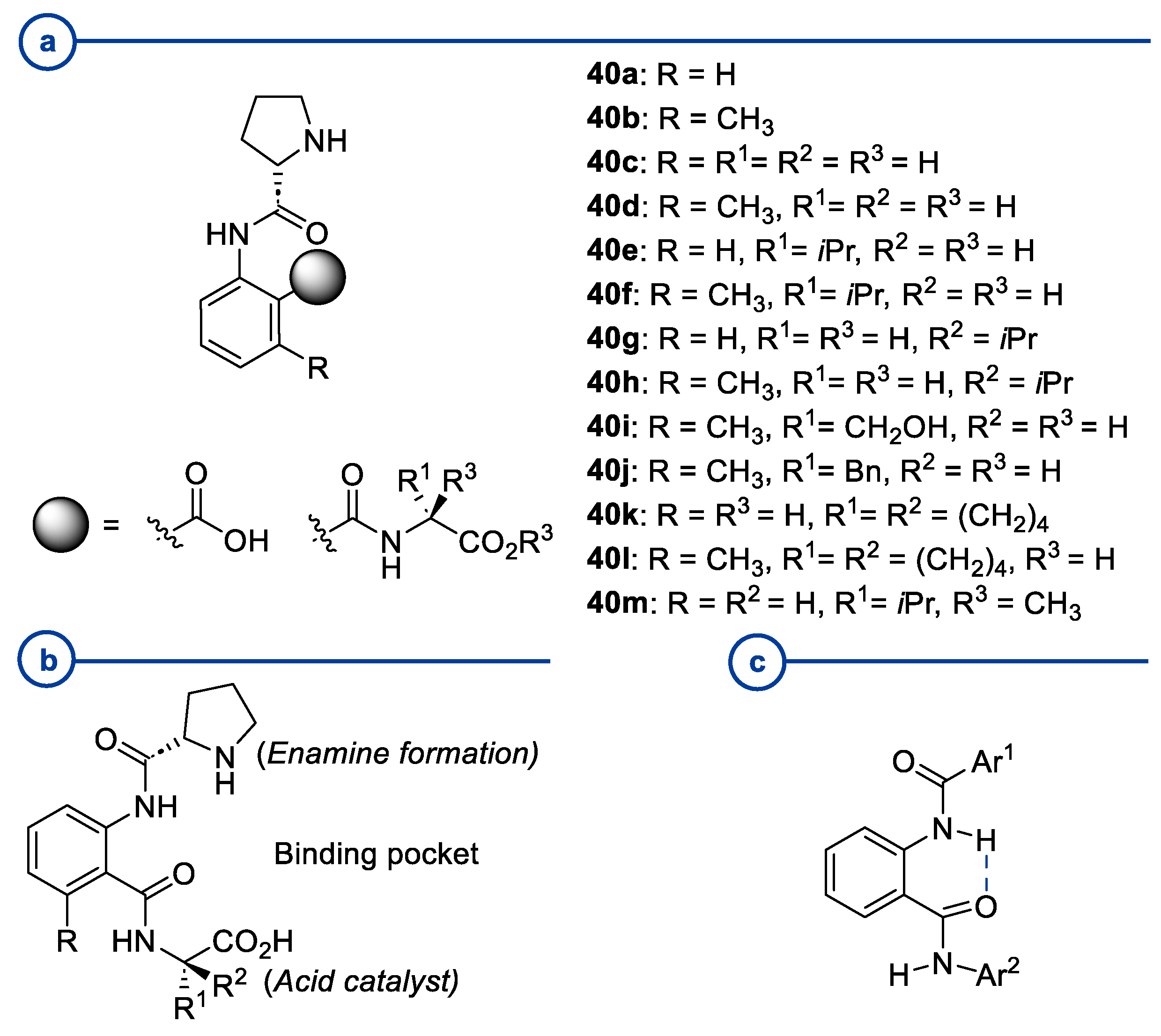
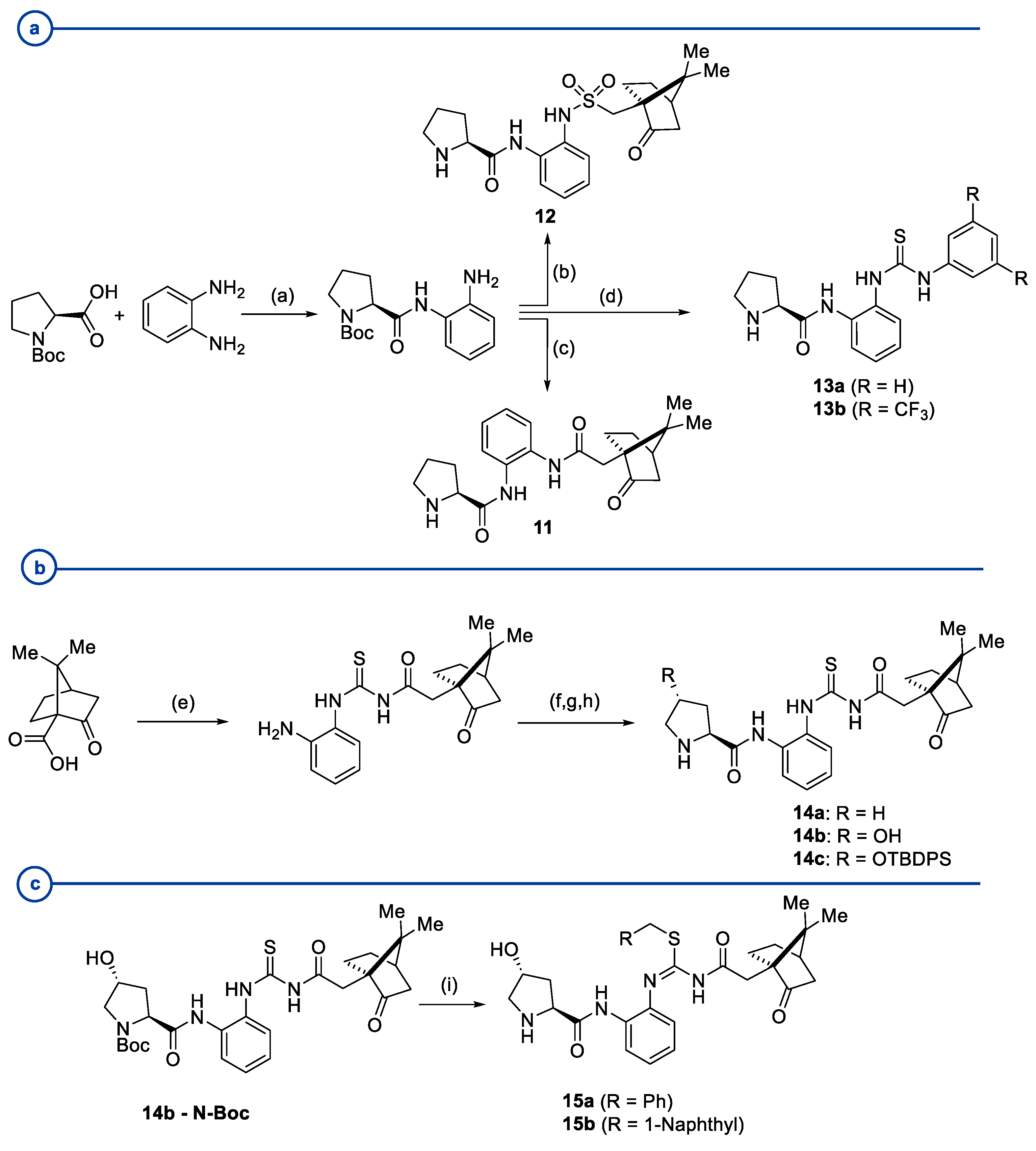
- Pearson, A.J.; Panda, S. N-Prolinylanthranilamide Pseudopeptides as Bifunctional Organocatalysts for Asymmetric Aldol Reactions. Org. Lett. 2011, 13, 5548–5551. [Google Scholar] [CrossRef] [PubMed]
- Kokotos, C.G. Construction of Tertiary Alcohols Bearing Perfluoroalkyl Chains Catalyzed by Prolinamide-Thioureas. J. Org. Chem. 2012, 77, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Freund, M.; Schenker, S.; Tsogoeva, S.B. Enantioselective Nitro-Michael Reactions Catalyzed by Short Peptides on Water. Org. Biomol. Chem. 2009, 7, 4279–4284. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, M.; Upert, G.; Angelici, G.; Wennemers, H. Enamine Catalysis with Low Catalyst Loadings—High Efficiency via Kinetic Studies. J. Am. Chem. Soc. 2010, 132, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Schnitzer, T.; Wennemers, H. Influence of the Trans/Cis Conformer Ratio on the Stereoselectivity of Peptidic Catalysts. J. Am. Chem. Soc. 2017, 139, 15356–15362. [Google Scholar] [CrossRef] [PubMed]
- Duschmalé, J.; Wennemers, H. Adapting to Substrate Challenges: Peptides as Catalysts for Conjugate Addition Reactions of Aldehydes to α,β-Disubstituted Nitroolefins. Chem.–A Eur. J. 2012, 18, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Kastl, R.; Wennemers, H. Peptide-Catalyzed Stereoselective Conjugate Addition Reactions Generating All-Carbon Quaternary Stereogenic Centers. Angew. Chem.—Int. Ed. 2013, 52, 7228–7232. [Google Scholar] [CrossRef]
- Xu, D.; Wang, J.; Yan, L.; Yuan, M.; Xie, X.; Wang, Y. Novel Bifunctional L-Prolinamide Derivatives as Highly Efficient Organocatalysts for Asymmetric Nitro-Michael Reactions. Tetrahedron Asymmetry 2016, 27, 1121–1132. [Google Scholar] [CrossRef]
- Cortes-Clerget, M.; Jover, J.; Dussart, J.; Kolodziej, E.; Monteil, M.; Migianu-Griffoni, E.; Gager, O.; Deschamp, J.; Lecouvey, M. Bifunctional Tripeptide with a Phosphonic Acid as a Brønsted Acid for Michael Addition: Mechanistic Insights. Chem.—A Eur. J. 2017, 23, 6654–6662. [Google Scholar] [CrossRef] [Green Version]
- Borges-González, J.; Feher-Voelger, A.; Crisóstomo, F.P.; Morales, E.Q.; Martín, T. Tetrahydropyran-Based Hybrid Dipeptides as Asymmetric Catalysts for Michael Addition of Aldehydes to β-Nitrostyrenes. Adv. Synth. Catal. 2017, 359, 576–583. [Google Scholar] [CrossRef]
- Schnitzer, T.; Budinská, A.; Wennemers, H. Organocatalysed Conjugate Addition Reactions of Aldehydes to Nitroolefins with Anti Selectivity. Nat. Catal. 2020, 3, 143–147. [Google Scholar] [CrossRef]
- Grünenfelder, C.E.; Kisunzu, J.K.; Wennemers, H. Peptide-Catalyzed Stereoselective Conjugate Addition Reactions of Aldehydes to Maleimide. Angew. Chem.—Int. Ed. 2016, 55, 8571–8574. [Google Scholar] [CrossRef] [PubMed]
- Panday, S.K. Advances in the Chemistry of Proline and Its Derivatives: An Excellent Amino Acid with Versatile Applications in Asymmetric Synthesis. Tetrahedron Asymmetry 2011, 22, 1817–1847. [Google Scholar] [CrossRef]
- Xiao, J.; Xu, F.X.; Lu, Y.P.; Loh, T.P. Chemzymes: A New Class of Structurally Rigid Tricyclic Amphibian Organocatalyst Inspired by Natural Product. Org. Lett. 2010, 12, 1220–1223. [Google Scholar] [CrossRef] [PubMed]
- Montroni, E.; Sanap, S.P.; Lombardo, M.; Quintavalla, A.; Trombini, C.; Dhavale, D.D. A New Robust and Efficient Ion-Tagged Proline Catalyst Carrying an Amide Spacer for the Asymmetric Aldol Reaction. Adv. Synth. Catal. 2011, 353, 3234–3240. [Google Scholar] [CrossRef]
- Lombardo, M.; Easwar, S.; Pasi, F.; Trombini, C. The Ion Tag Strategy as a Route to Highly Efficient Organocatalysts for the Direct Asymmetric Aldol Reaction. Adv. Synth. Catal. 2009, 351, 276–282. [Google Scholar] [CrossRef]
- Bottoni, A.; Lombardo, M.; Miscione, G.P.; Montroni, E.; Quintavalla, A.; Trombini, C. Electrosteric Activation by Using Ion-Tagged Prolines: A Combined Experimental and Computational Investigation. ChemCatChem 2013, 5, 2913–2924. [Google Scholar] [CrossRef]
- Montroni, E.; Lombardo, M.; Quintavalla, A.; Trombini, C.; Gruttadauria, M.; Giacalone, F. A Liquid-Liquid Biphasic Homogeneous Organocatalytic Aldol Protocol Based on the Use of a Silica Gel Bound Multilayered Ionic Liquid Phase. ChemCatChem 2012, 4, 1000–1006. [Google Scholar] [CrossRef]
- Bhowmick, S.; Kunte, S.S.; Bhowmick, K.C. A New Organocatalyst Derived from Abietic Acid and 4-Hydroxy-L-Proline for Direct Asymmetric Aldol Reactions in Aqueous Media. Tetrahedron Asymmetry 2014, 25, 1292–1297. [Google Scholar] [CrossRef]
- Mase, N.; Tanaka, F.; Barbas, C.F. Rapid Fluorescent Screening for Bifunctional Amine-Acid Catalysts: Efficient Syntheses of Quaternary Carbon-Containing Aldols under Organocatalysis. Org. Lett. 2003, 5, 4369–4372. [Google Scholar] [CrossRef] [PubMed]
- Mase, N.; Tanaka, F.; Barbas, C.F. Synthesis of β-Hydroxyaldehydes with Stereogenic Quaternary Carbon Centers by Direct Organocatalytic Asymmetric Aldol Reactions. Angew. Chem.—Int. Ed. 2004, 43, 2420–2423. [Google Scholar] [CrossRef]
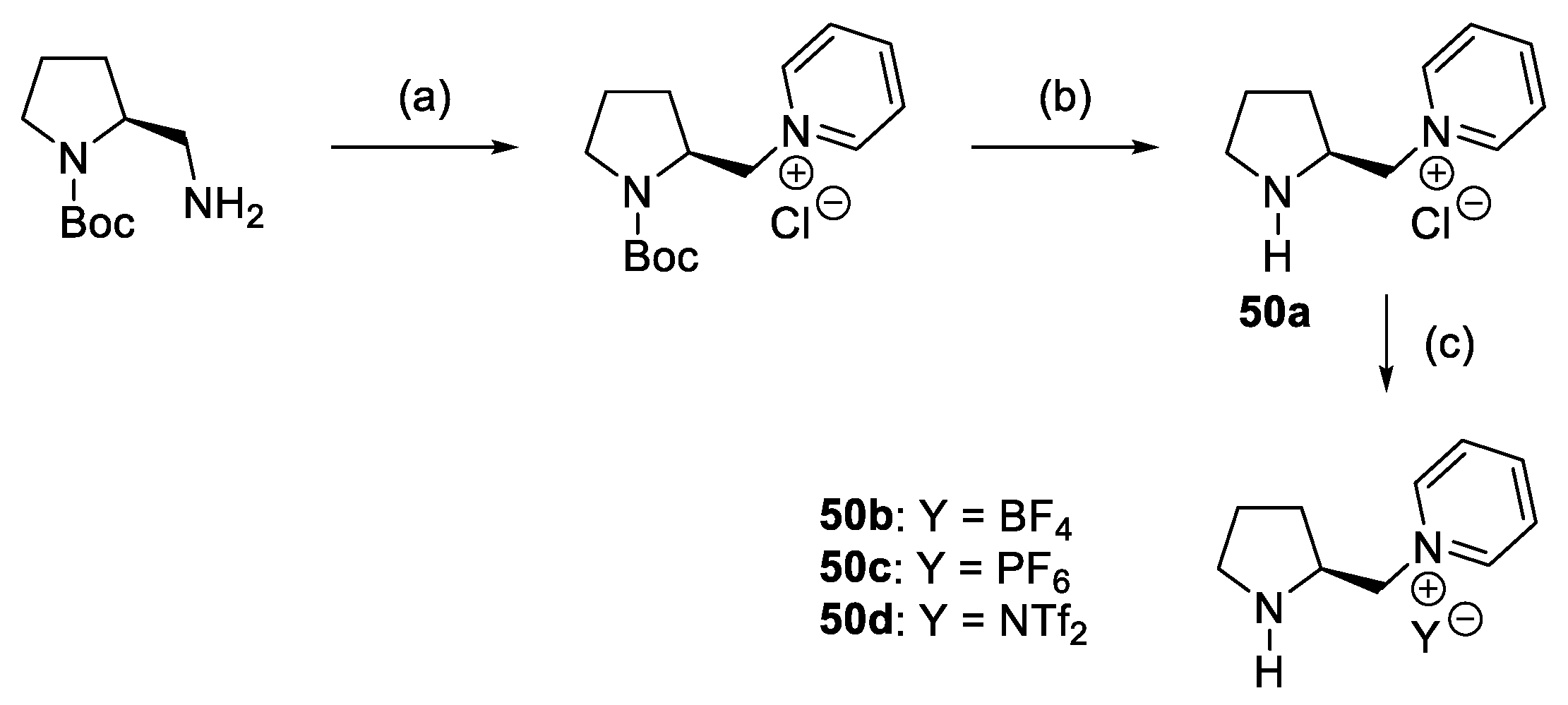
- Ni, B.; Zhang, Q.; Headley, A.D. Pyrrolidine-Based Chiral Pyridinium Ionic Liquids (ILs) as Recyclable and Highly Efficient Organocatalysts for the Asymmetric Michael Addition Reactions. Tetrahedron Lett. 2008, 49, 1249–1252. [Google Scholar] [CrossRef]
- Barbier, D.; Marazano, C.; Das, B.C.; Potier, P. New Chiral Isoquinolinium Salt Derivatives from Chiral Primary Amines via Zincke Reaction. J. Org. Chem. 1996, 61, 9596–9598. [Google Scholar] [CrossRef]
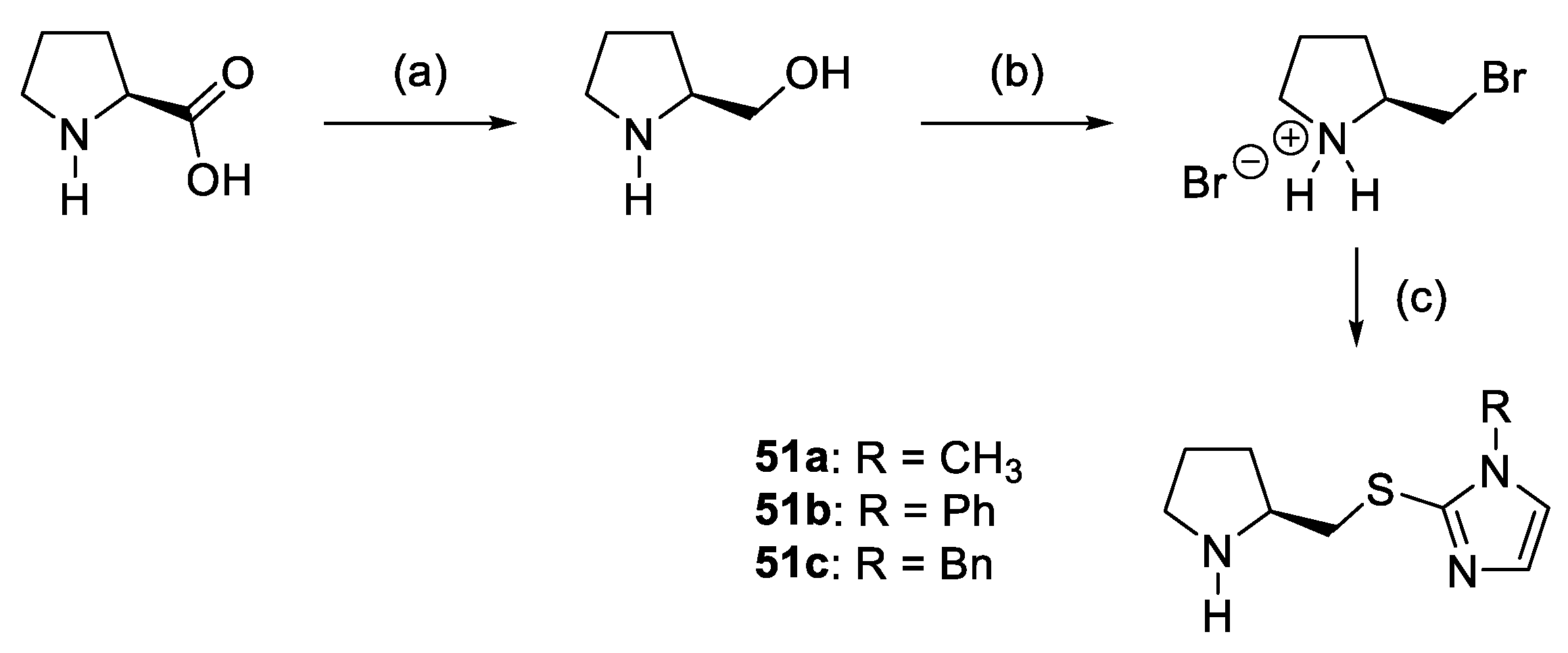
- Xu, D.Q.; Wang, L.P.; Luo, S.P.; Wang, Y.F.; Zhang, S.; Xu, Z.Y. 2-[(Imidazolylthio)Methyl]Pyrrolidine as a Trifunctional Organocatalyst for the Highly Asymmetric Michael Addition of Ketones to Nitroolefins. Eur. J. Org. Chem. 2008, 2008, 1049–1053. [Google Scholar] [CrossRef]
- Dahlin, N.; Bøgevig, A.; Adolfsson, H. N-Arenesulfonyl-2-Aminomethylpyrrolidmes—Novel Modular Ligands and Organocatalysts for Asymmetric Catalysis. Adv. Synth. Catal. 2004, 346, 1101–1105. [Google Scholar] [CrossRef]
- Miao, T.; Wang, L.; Li, P.; Yan, J. A Highly Efficient and Recyclable Ionic Liquid Anchored Pyrrolidine Catalyst for Enantioselective Michael Additions. Synthesis 2008, 23, 3828–3834. [Google Scholar] [CrossRef]
- Gaitor, J.C.; Paul, L.M.; Reardon, M.M.; Hmissa, T.; Minkowicz, S.; Regner, M.; Sheng, Y.; Michael, S.F.; Isern, S.; Mirjafari, A. Ionic Liquids with Thioether Motifs as Synthetic Cationic Lipids for Gene Delivery. Chem. Commun. 2017, 53, 8328–8331. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ni, B.; Headley, A.D. Asymmetric Michael Addition Reactions of Aldehydes with Nitrostyrenes Catalyzed by Functionalized Chiral Ionic Liquids. Tetrahedron 2008, 64, 5091–5097. [Google Scholar] [CrossRef]
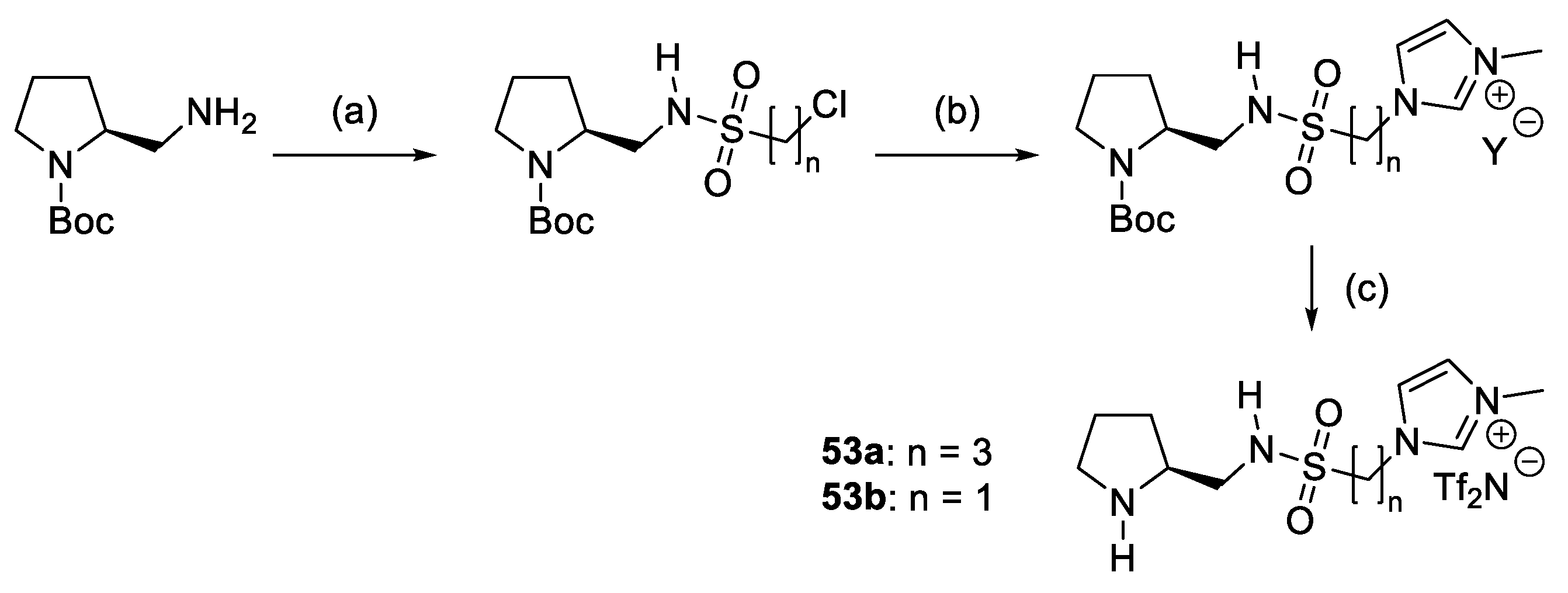
- Ni, B.; Zhang, Q.; Dhungana, K.; Headley, A.D. Ionic Liquid-Supported (ILS) (S)-Pyrrolidine Sulfonamide, a Recyclable Organocatalyst for the Highly Enantioselective Michael Addition to Nitroolefins. Org. Lett. 2009, 11, 1037–1040. [Google Scholar] [CrossRef]
- Carley, A.P.; Dixon, S.; Kilburn, J.D. Pyrrolidine-Based Organocatalysts for Enantioselective Michael Addition of Cyclohexanone to Trans-β-Nitrostyrene. Synthesis 2009, 15, 2509–2516. [Google Scholar] [CrossRef]
- Tan, B.; Zeng, X.; Lu, Y.; Chua, P.J.; Zhong, G. Rational Design of Organocatalyst: Highly Stereoselective Michael Addition of Cyclic Ketones to Nitroolefins. Org. Lett. 2009, 11, 1927–1930. [Google Scholar] [CrossRef]
- Chang, C.; Li, S.H.; Reddy, R.J.; Chen, K. Pyrrolidine-Camphor Derivative as an Organocatalyst for Asymmetic Michael Additions of α,α-Disubstituted Aldehydes to β-Nitroalkenes: Construction of Quaternary Carbon-Bearing Aldehydes under Solvent-Free Conditions. Adv. Synth. Catal. 2009, 351, 1273–1278. [Google Scholar] [CrossRef]
- Reddy, R.J.; Kuan, H.H.; Chou, T.Y.; Chen, K. Novel Prolinamide-Camphor-Containing Organocatalysts for Direct Asymmetric Michael Addition of Unmodified Aldehydes to Nitroalkenes. Chem.—A Eur. J. 2009, 15, 9294–9298. [Google Scholar] [CrossRef]
- Liu, P.M.; Chang, C.; Reddy, R.J.; Ting, Y.F.; Kuan, H.H.; Chen, K. Remarkable Reaction Rate and Excellent Enantioselective Direct α-Amination of Aldehydes with Azodicarboxylates Catalyzed by Pyrrolidinylcamphor-Derived Organocatalysts. Eur. J. Org. Chem. 2010, 2010, 42–46. [Google Scholar] [CrossRef]
- Magar, D.R.; Chang, C.; Ting, Y.F.; Chen, K. Highly Enantioselective Conjugate Addition of Ketones to Alkylidene Malonates Catalyzed by a Pyrrolidinyl-Camphor-Derived Organocatalyst. Eur. J. Org. Chem. 2010, 2010, 2062–2066. [Google Scholar] [CrossRef]
- Ting, Y.F.; Chang, C.; Reddy, R.J.; Magar, D.R.; Chen, K. Pyrrolidinyl-Camphor Derivatives as a New Class of Organocatalyst for Direct Asymmetric Michael Addition of Aldehydes and Ketones to β-Nitroalkenes. Chem.—A Eur. J. 2010, 16, 7030–7038. [Google Scholar] [CrossRef]
- Liu, P.M.; Magar, D.R.; Chen, K. Highly Efficient and Practical Pyrrolidine-Camphor-Derived Organocatalysts for the Direct α-Amination of Aldehydes. Eur. J. Org. Chem. 2010, 2010, 5705–5713. [Google Scholar] [CrossRef]
- Vermote, A.; Brackman, G.; Risseeuw, M.D.P.; Coenye, T.; Van Calenbergh, S. Novel Hamamelitannin Analogues for the Treatment of Biofilm Related MRSA Infections–A Scaffold Hopping Approach. Eur. J. Med. Chem. 2017, 127, 757–770. [Google Scholar] [CrossRef] [Green Version]
- Anwar, S.; Lee, P.H.; Chou, T.Y.; Chang, C.; Chen, K. Pyrrolidine-Linker-Camphor Assembly: Bifunctional Organocatalysts for Efficient Michael Addition of Cyclohexanone to Nitroolefins under Neat Conditions. Tetrahedron 2011, 67, 1171–1177. [Google Scholar] [CrossRef]
- Xu, D.Z.; Shi, S.; Wang, Y. Simple Chiral Pyrrolidine-Pyridine-Based Catalysts for Highly Enantioselective Michael Addition to Nitro Olefins. Eur. J. Org. Chem. 2009, 2009, 4848–4853. [Google Scholar] [CrossRef]
- Wang, C.; Yu, C.; Liu, C.; Peng, Y. 4-Trifluoromethanesulfonamidyl Prolinol Tert-Butyldiphenylsilyl Ether as a Highly Efficient Bifunctional Organocatalyst for Michael Addition of Ketones and Aldehydes to Nitroolefins. Tetrahedron Lett. 2009, 50, 2363–2366. [Google Scholar] [CrossRef]
- Conrow, R.E.; Dean, W.D. Diazidomethane Explosion. Org. Process Res. Dev. 2008, 12, 1285–1286. [Google Scholar] [CrossRef]
- Zheng, B.; Wang, H.; Han, Y.; Liu, C.; Peng, Y. A Novel Method to Access Chiral Nonnatural 2,4-Disubstituted Pyrrolidines from Aldehydes and Nitroolefins Only with an α-Substituent. Chem. Commun. 2013, 49, 4561–4563. [Google Scholar] [CrossRef]
- Ban, S.; Du, D.M.; Liu, H.; Yang, W. Synthesis of Binaphthyl Sulfonimides and Their Application in the Enantioselective Michael Addition of Ketones to Nitroalkenes. Eur. J. Org. Chem. 2010, 2010, 5160–5164. [Google Scholar] [CrossRef]
- Wang, L.; Liu, J.; Miao, T.; Zhou, W.; Li, P.; Ren, K.; Zhang, X. Sugar-Based Pyrrolidine as a Highly Enantioselective Organocatalyst for Asymmetric Michael Addition of Ketones to Nitrostyrenes. Adv. Synth. Catal. 2010, 352, 2571–2578. [Google Scholar] [CrossRef]
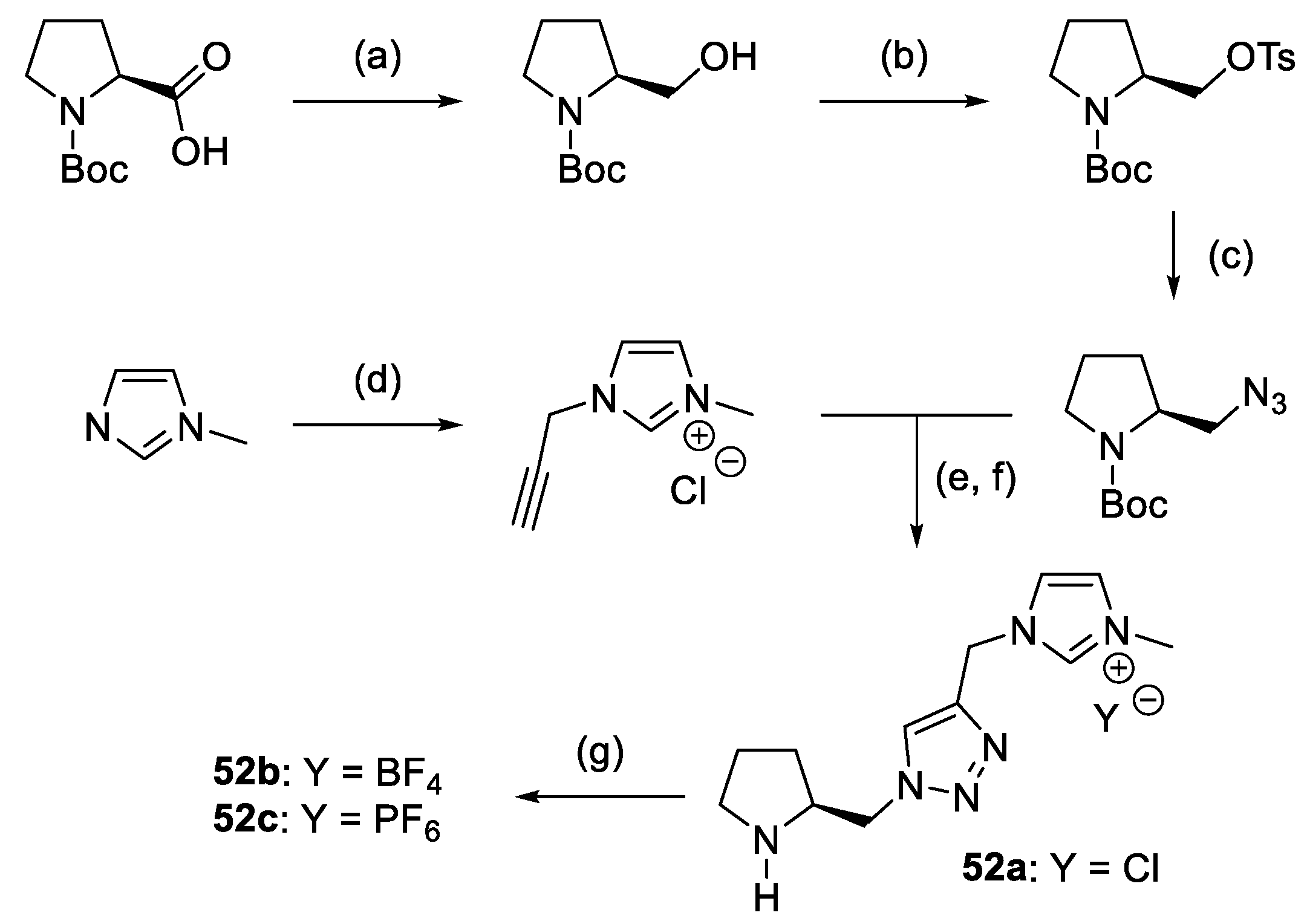
- Chandrasekhar, S.; Kumar, T.P.; Haribabu, K.; Reddy, C.R. Hydroxyphthalimide Allied Triazole-Pyrrolidine Catalyst for Asymmetric Michael Additions in Water. Tetrahedron Asymmetry 2010, 21, 2372–2375. [Google Scholar] [CrossRef]
- Widianti, T.; Hiraga, Y.; Kojima, S.; Abe, M. Novel Cyclic β-Aminophosphonate Derivatives as Efficient Organocatalysts for the Asymmetric Michael Addition Reactions of Ketones to Nitrostyrenes. Tetrahedron Asymmetry 2010, 21, 1861–1868. [Google Scholar] [CrossRef]
- Syu, S.-en; Kao, T.T.; Lin, W. A New Type of Organocatalyst for Highly Stereoselective Michael Addition of Ketones to Nitroolefins on Water. Tetrahedron 2010, 66, 891–897. [Google Scholar] [CrossRef]
- Chen, J.R.; Cao, Y.J.; Zou, Y.Q.; Tan, F.; Fu, L.; Zhu, X.Y.; Xiao, W.J. Novel Thiourea-Amine Bifunctional Catalysts for Asymmetric Conjugate Addition of Ketones/Aldehydes to Nitroalkenes: Rational Structural Combination for High Catalytic Efficiency. Org. Biomol. Chem. 2010, 8, 1275–1279. [Google Scholar] [CrossRef]
- Cao, X.Y.; Zheng, J.C.; Li, Y.X.; Shu, Z.C.; Sun, X.L.; Wang, B.Q.; Tang, Y. Pyrrolidine-Ureas as Bifunctional Organocatalysts for Asymmetric Michael Addition of Ketone to Nitroalkenes: Unexpected Hydrogen Bonding Effect. Tetrahedron 2010, 66, 9703–9707. [Google Scholar] [CrossRef]
- Yu, C.; Qiu, J.; Zheng, F.; Zhong, W. Highly Efficient Bifunctional Organocatalysts for the Asymmetric Michael Addition of Ketones to Nitroolefins. Tetrahedron Lett. 2011, 52, 3298–3302. [Google Scholar] [CrossRef]
- Lin, J.; Tian, H.; Jiang, Y.J.; Huang, W.B.; Zheng, L.Y.; Zhang, S.Q. Novel Pyrrolidine-Aminobenzimidazole Bifunctional Organocatalysts for Asymmetric Nitro-Michael Reactions in Brine. Tetrahedron Asymmetry 2011, 22, 1434–1440. [Google Scholar] [CrossRef]
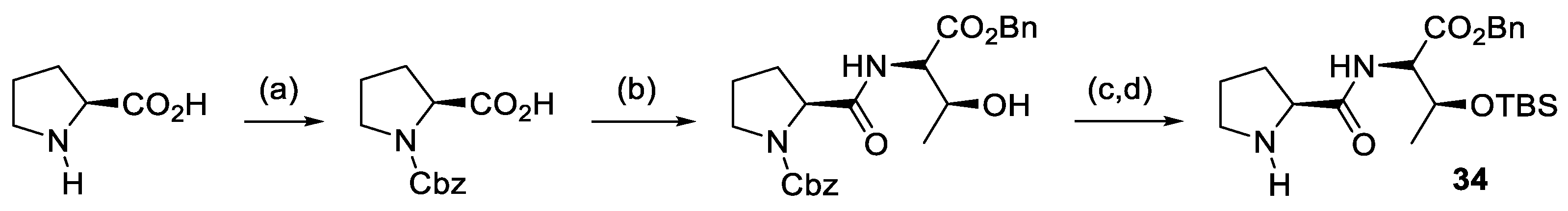
- Zhang, H.; Chuan, Y.; Li, Z.; Peng, Y. 4-Aminothiourea Prolinol Tert-Butyldiphenylsilyl Ether: A Chiral Secondary Amine-Thiourea as Organocatalyst for Enantioselective Anti-Mannich Reactions. Adv. Synth. Catal. 2009, 351, 2288–2294. [Google Scholar] [CrossRef]
- Wang, L.; Cai, C.; Curran, D.P.; Zhang, W. Enantioselective α-Chlorination of Aldehydes with Recyclable Fluorous (S)-Pyrrolidine-Thiourea Bifunctional Organocatalyst. Synlett 2010, 3, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Kokotos, C.G.; Limnios, D.; Triggidou, D.; Trifonidou, M.; Kokotos, G. Novel Pyrrolidine-Thiohydantoins/Thioxotetrahydropyrimidinones as Highly Effective Catalysts for the Asymmetric Michael Addition. Org. Biomol. Chem. 2011, 9, 3386–3395. [Google Scholar] [CrossRef]
- Chen, J.R.; Fu, L.; Zou, Y.Q.; Chang, N.J.; Rong, J.; Xiao, W.J. Pyrrolidinyl-Sulfamide Derivatives as a New Class of Bifunctional Organocatalysts for Direct Asymmetric Michael Addition of Cyclohexanone to Nitroalkenes. Org. Biomol. Chem. 2011, 9, 5280–5287. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Liu, Y.; Lu, S.M.; Li, J.; Li, C. Recyclable Enamine Catalysts for Asymmetric Direct Cross-Aldol Reaction of Aldehydes in Emulsion Media. Green Chem. 2011, 13, 1983–1985. [Google Scholar] [CrossRef]
- Albrecht, Ł.; Dickmeiss, G.; Acosta, F.C.; Rodríguez-Escrich, C.; Davis, R.L.; Jørgensen, K.A. Asymmetric Organocatalytic Formal [2 + 2]-Cycloadditions via Bifunctional H-Bond Directing Dienamine Catalysis. J. Am. Chem. Soc. 2012, 134, 2543–2546. [Google Scholar] [CrossRef]
- Jiang, H.; Rodríguez-Escrich, C.; Johansen, T.K.; Davis, R.L.; Jørgensen, K.A. Organocatalytic Activation of Polycyclic Aromatic Compounds for Asymmetric Diels-Alder Reactions. Angew. Chem.—Int. Ed. 2012, 51, 10271–10274. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.N.; Singh, P.; Singh, P.; Lal, N.; Sharma, S.K. Pyrrolidine Based Chiral Organocatalyst for Efficient Asymmetric Michael Addition of Cyclic Ketones to β-Nitrostyrenes. Bioorganic Med. Chem. Lett. 2012, 22, 4225–4228. [Google Scholar] [CrossRef]
- Siyutkin, D.E.; Kucherenko, A.S.; Frolova, L.L.; Kuchin, A.V.; Zlotin, S.G. N-Pyrrolidine-2-ylmethyl)-2-Hydroxy-3-Aminopinanes as Novel Organocatalysts for Asymmetric Conjugate Additions of Ketones to α-Nitroalkenes. Tetrahedron Asymmetry 2013, 24, 776–779. [Google Scholar] [CrossRef]
- Singh, K.; Singh, P.; Kaur, A.; Singh, P.; Sharma, S.; Khullar, S.; Mandal, S.K. (2 S)-2-[(Phenylsulfinyl)Methyl]Pyrrolidine-Catalyzed Efficient Stereoselective Michael Addition of Cyclohexanone and Cyclopentanone to Nitroolefins. Synthesis 2013, 45, 1406–1413. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, K.; Hirashima, S.I.; Kawada, M.; Koseki, Y.; Tada, N.; Itoh, A.; Miura, T. Pyrrolidine-Diaminomethylenemalononitrile Organocatalyst for Michael Additions of Carbonyl Compounds to Nitroalkenes under Solvent-Free Conditions. Tetrahedron Lett. 2014, 55, 2703–2706. [Google Scholar] [CrossRef]
- Hirashima, S.I.; Narushima, T.; Kawada, M.; Nakashima, K.; Hanai, K.; Koseki, Y.; Miura, T. Asymmetric Conjugate Additions of Carbonyl Compounds to Nitroalkenes under Solvent-Free Conditions Using Fluorous Diaminomethylenemalononitrile Organocatalyst. Chem. Pharm. Bull. 2017, 85, 1185–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Jiang, M.; Liu, J.T. Enantioselective Michael Addition of Cyclic Ketones to Nitroolefins Catalyzed by a Novel Fluorine-Insertion Organocatalyst. Tetrahedron Asymmetry 2014, 25, 212–218. [Google Scholar] [CrossRef]
- Kaplaneris, N.; Koutoulogenis, G.; Raftopoulou, M.; Kokotos, C.G. 4-Fluoro and 4-Hydroxy Pyrrolidine-Thioxotetrahydropyrimidinones: Organocatalysts for Green Asymmetric Transformations in Brine. J. Org. Chem. 2015, 80, 5464–5473. [Google Scholar] [CrossRef]
- Kumar, T.P.; Balaji, S.V. Sugar Amide-Pyrrolidine Catalyst for the Asymmetric Michael Addition of Ketones to Nitroolefins. Tetrahedron Asymmetry 2014, 25, 473–477. [Google Scholar] [CrossRef]
- Kumar, T.P.; Radhika, L.; Haribabu, K.; Kumar, V.N. Pyrrolidine-Oxyimides: New Chiral Catalysts for Enantioselective Michael Addition of Ketones to Nitroolefins in Water. Tetrahedron Asymmetry 2014, 25, 1555–1560. [Google Scholar] [CrossRef]
- Kumar, T.P. Pyrrolidine-Oxyimide Catalyzed Asymmetric Michael Addition of α,α-Disubstituted Aldehydes to Nitroolefins. Tetrahedron Asymmetry 2015, 26, 907–911. [Google Scholar] [CrossRef]
- Kumar, T.P.; Prasad, S.S.; Haribabu, K.; Kumar, V.N.; Reddy, C.S. Pyrrolidine-HOBt: An Oxytriazole Catalyst for the Enantioselective Michael Addition of Cyclohexanone to Nitroolefins in Water. Tetrahedron Asymmetry 2016, 27, 1133–1138. [Google Scholar] [CrossRef]
- Zhao, H.W.; Yang, Z.; Yue, Y.Y.; Li, H.L.; Song, X.Q.; Sheng, Z.H.; Meng, W.; Guo, X.Y. Asymmetric Direct Michael Reactions of Cyclohexanone with Aromatic Nitroolefins in Water Catalyzed by Novel Axially Unfixed Biaryl-Based Bifunctional Organocatalysts. Synlett 2014, 25, 293–297. [Google Scholar] [CrossRef]
- Hu, X.; Wei, Y.F.; Wu, N.; Jiang, Z.; Liu, C.; Luo, R.S. Indolinol-Catalyzed Asymmetric Michael Reaction of Aldehydes to Nitroalkenes in Brine. Tetrahedron Asymmetry 2016, 27, 420–427. [Google Scholar] [CrossRef]
- Reyes-Rangel, G.; Vargas-Caporali, J.; Juaristi, E. Asymmetric Michael Addition Reaction Organocatalyzed by Stereoisomeric Pyrrolidine Sulfinamides under Neat Conditions. A Brief Study of Self-Disproportionation of Enantiomers. Tetrahedron 2017, 73, 4707–4718. [Google Scholar] [CrossRef]
- Kaur, A.; Singh, K.N.; Sharma, E.; Rani, P.; Sharma, S.K. Pyrrolidine-Carbamate Based New and Efficient Chiral Organocatalyst for Asymmetric Michael Addition of Ketones to Nitroolefins. Tetrahedron 2018, 74, 6137–6143. [Google Scholar] [CrossRef]
- Mahato, C.K.; Mukherjee, S.; Kundu, M.; Pramanik, A. Pyrrolidine-Oxadiazolone Conjugates as Organocatalysts in Asymmetric Michael Reaction. J. Org. Chem. 2019, 84, 1053–1063. [Google Scholar] [CrossRef]
- Ormandyová, K.; Bilka, S.; Mečiarová, M.; Šebesta, R. Bifunctional Thio/Squaramide Catalyzed Stereoselective Michael Additions of Aldehydes to Nitroalkenes towards Synthesis of Chiral Pyrrolidines. ChemistrySelect 2019, 4, 8870–8875. [Google Scholar] [CrossRef]
- Polácková, V.; Krištofíková, D.; Némethová, B.; Górová, R.; Meciarová, M.; Šebesta, R. N-Sulfinylpyrrolidine-Containing Ureas and Thioureas as Bifunctional Organocatalysts. Beilstein J. Org. Chem. 2021, 17, 2629–2641. [Google Scholar] [CrossRef] [PubMed]
- Dargó, G.; Nagy, S.; Kis, D.; Bagi, P.; Mátravölgyi, B.; Tóth, B.; Huszthy, P.; Drahos, L.; Kupai, J. Application of Proline-Derived (Thio)Squaramide Organocatalysts in Asymmetric Diels-Alder and Conjugate Addition Reactions. Synthesis 2022, 54, 3823–3830. [Google Scholar] [CrossRef]
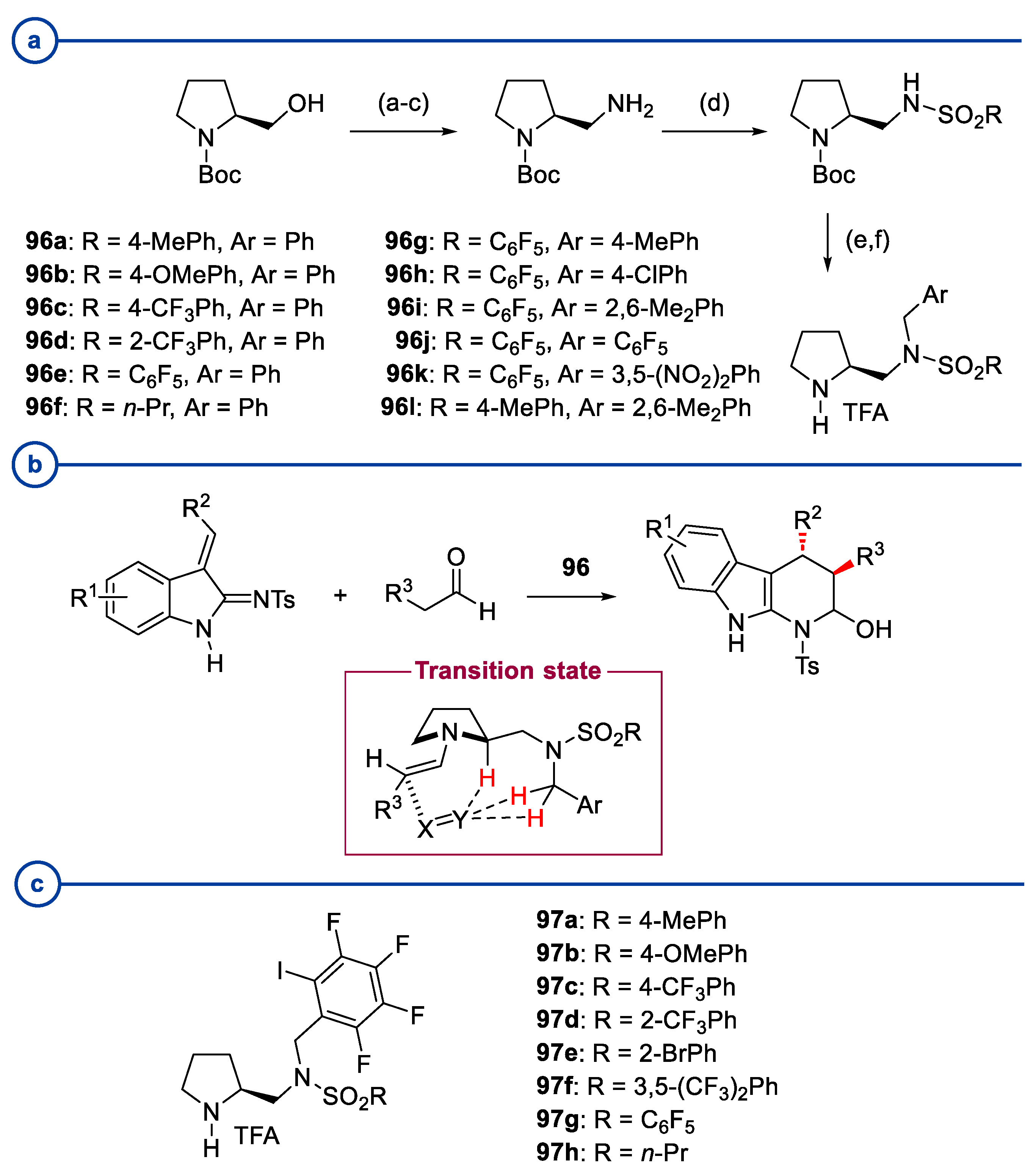
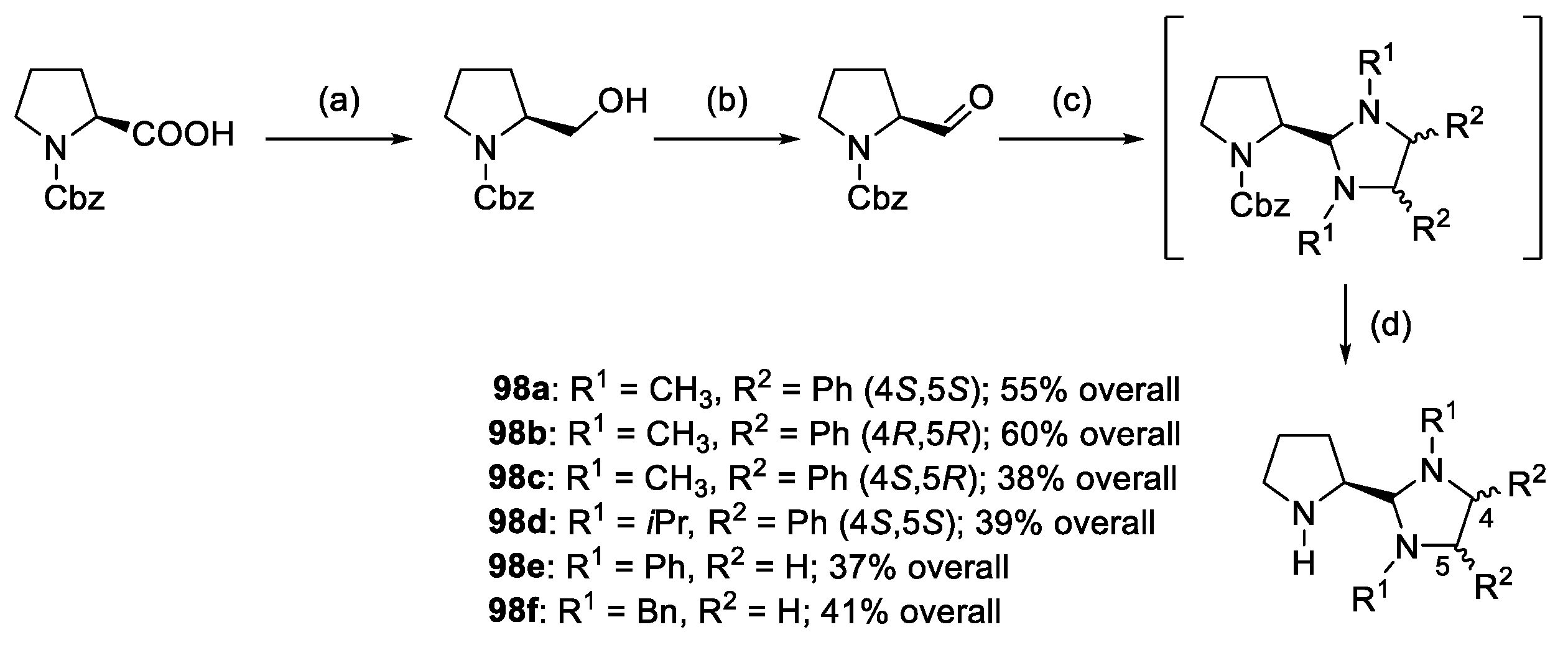
- Moriyama, K.; Oka, Y. Enantioselective Cascade Michael/Hemiaminal Formation of α,β-Unsaturated Iminoindoles with Aldehydes Using a Chiral Aminomethylpyrrolidine Catalyst Bearing a SO2C6F5 Group as a Strongly Electron Withdrawing Arylsulfonyl Group. ACS Catal. 2022, 12, 7436–7442. [Google Scholar] [CrossRef]
- Moriyama, K.; Oka, Y.; Kaiho, T. A Chiral N-Tetrafluoroiodobenzyl-N-Sulfonyl Aminomethylpyrrolidine Catalyst for the Enantioselective Michael/Hemiaminal Formation Cascade Reaction of α,β-Unsaturated Iminoindoles with Aldehydes. Synlett 2022, 33, 1763–1769. [Google Scholar] [CrossRef]
- Marigo, M.; Wabnitz, T.C.; Fielenbach, D.; Jørgensen, K.A. Enantioselective Organocatalyzed Alpha-Sulfenylation of Aldehydes. Angew. Chem.—Int. Ed. 2005, 44, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Gotoh, H.; Hayashi, T.; Shoji, M. Diphenylprolinol Silyl Ethers as Efficient Organocatalysts for the Asymmetric Michael Reaction of Aldehydes and Nitroalkenes. Angew. Chem.—Int. Ed. 2005, 44, 4212–4215. [Google Scholar] [CrossRef]
- Hayashi, Y.; Hatano, Y.; Mori, N. Asymmetric Michael Reaction of Malononitrile and α,β-Unsaturated Aldehydes Catalyzed by Diarylprolinol Silyl Ether. Synlett 2022, 33, 1831–1836. [Google Scholar] [CrossRef]
- Reyes-Rodríguez, G.J.; Rezayee, N.M.; Vidal-Albalat, A.; Jørgensen, K.A. Prevalence of Diarylprolinol Silyl Ethers as Catalysts in Total Synthesis and Patents. Chem. Rev. 2019, 119, 4221–4260. [Google Scholar] [CrossRef]
- Quintard, A.; Bournaud, C.; Alexakis, A. Diversity-Oriented Synthesis towards Conceptually New Highly Modular Aminal-Pyrrolidine Organocatalysts. Chem.—A Eur. J. 2008, 14, 7504–7507. [Google Scholar] [CrossRef]
- Luis Olivares-Romero, J.; Juaristi, E. Synthesis of Three Novel Chiral Diamines Derived from (S)-Proline and Their Evaluation as Precursors of Diazaborolidines for the Catalytic Borane-Mediated Enantioselective Reduction of Prochiral Ketones. Tetrahedron 2008, 64, 9992–9998. [Google Scholar] [CrossRef]
- Shi, Z.; Tan, B.; Leong, W.W.Y.; Zeng, X.; Lu, M.; Zhong, G. Catalytic Asymmetric Formal [4 + 1] Annulation Leading to Optically Active Cis-Isoxazoline N-Oxides. Org. Lett. 2010, 12, 5402–5405. [Google Scholar] [CrossRef]
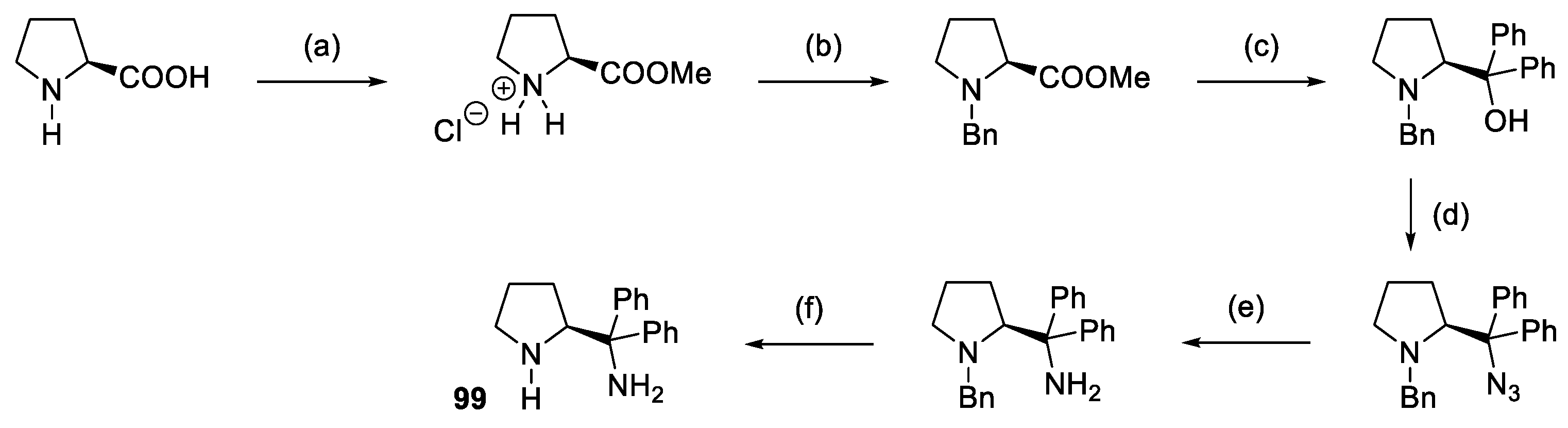
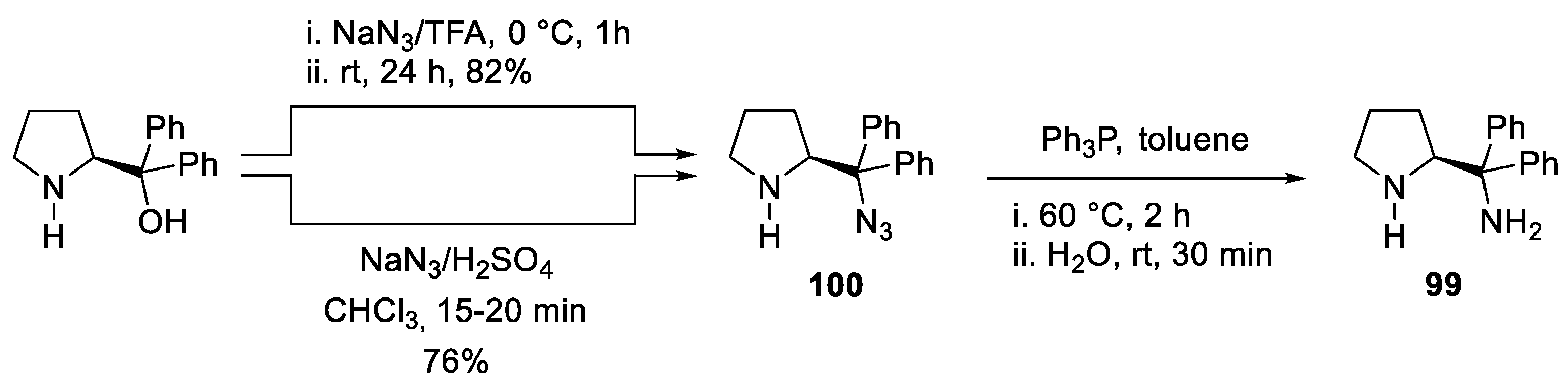
- Roy, H.N.; Pitchaiah, A.; Kim, M.; Hwang, I.T.; Lee, K.I. Protective Group-Free Synthesis of New Chiral Diamines via Direct Azidation of 1,1-Diaryl-2-Aminoethanols. RSC Adv. 2013, 3, 3526–3530. [Google Scholar] [CrossRef]
- Reyes-Rangel, G.; Vargas-Caporali, J.; Juaristi, E. In Search of Diamine Analogs of the α,α-Diphenyl Prolinol Privileged Chiral Organocatalyst. Synthesis of Diamine Derivatives of α,α-Diphenyl-(S)-Prolinol and Their Application as Organocatalysts in the Asymmetric Michael and Mannich Reactions. Tetrahedron 2016, 72, 379–391. [Google Scholar] [CrossRef]
- Vargas-Caporali, J.; Juaristi, E. The Diamino Analogues of Privileged Corey-Bakshi-Shibata and Jorgensen-Hayashi Catalysts: A Comparison of Their Performance. Synthesis 2016, 48, 3890–3906. [Google Scholar] [CrossRef]
- Kano, T.; Mii, H.; Maruoka, K. Direct Asymmetric Benzoyloxylation of Aldehydes Catalyzed by 2-Tritylpyrrolidine. J. Am. Chem. Soc. 2009, 131, 3450–3451. [Google Scholar] [CrossRef]
- Shimogaki, M.; Maruyama, H.; Tsuji, S.; Homma, C.; Kano, T.; Maruoka, K. Synthesis of Chiral Tritylpyrrolidine Derivatives and Their Application to Asymmetric Benzoyloxylation. J. Org. Chem. 2017, 82, 12928–12932. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.S.; Weng, J.; Ai, H.B.; Lu, G.; Chan, A.S.C. Highly Efficient Asymmetric Michael Reaction of Aldehydes to Nitroalkenes with Diphenylperhydroindolinol Silyl Ethers as Organocatalysts. Adv. Synth. Catal. 2009, 351, 2449–2459. [Google Scholar] [CrossRef]
- Wu, J.; Ni, B.; Headley, A.D. Di(Methylimidazole)Prolinol Silyl Ether Catalyzed Highly Michael Addition of Aldehydes to Nitroolefins in Water. Org. Lett. 2009, 11, 3354–3356. [Google Scholar] [CrossRef]
- Lombardo, M.; Chiarucci, M.; Quintavalla, A.; Trombini, C. Highly Efficient Ion-Tagged Catalyst for the Enantioselective Michael Addition of Aldehydes to Nitroalkenes. Adv. Synth. Catal. 2009, 351, 2801–2806. [Google Scholar] [CrossRef]
- Quintard, A.; Belot, S.; Marchai, E.; Alexakis, A. Aminal-Pyrrolidine Organocatalysts—Highly Efficient and Modular Catalysts for α-Functionalization of Carbonyl Compounds. Eur. J. Org. Chem. 2010, 2010, 927–936. [Google Scholar] [CrossRef]
- Husmann, R.; Jörres, M.; Raabe, G.; Bolm, C. Silylated Pyrrolidines as Catalysts for Asymmetric Michael Additions of Aldehydes to Nitroolefins. Chem.—A Eur. J. 2010, 16, 12549–12552. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.O.; Stiller, J.; Marqués-López, E.; Strohfeldt, K.; Christmann, M.; Strohmann, C. Silyl-Modified Analogues of 2-Tritylpyrrolidine: Synthesis and Applications in Asymmetric Organocatalysis. Chem.—A Eur. J. 2010, 16, 12553–12558. [Google Scholar] [CrossRef]
- Kerrick, S.T.; Beak, P. Asymmetric Deprotonations: Enantioselective Syntheses of 2-Substituted (Tert-Butoxycarbonyl)Pyrrolidines. J. Am. Chem. Soc. 1991, 113, 9708–9710. [Google Scholar] [CrossRef]
- Eaborn, C. Organosilicon Compounds. Part III. Some Sterically Hindered Compounds. J. Chem. Soc. 1952, 0, 2840–2846. [Google Scholar] [CrossRef]
- Jentzsch, K.I.; Min, T.; Etcheson, J.I.; Fettinger, J.C.; Franz, A.K. Silyl Fluoride Electrophiles for the Enantioselective Synthesis of Silylated Pyrrolidine Catalysts. J. Org. Chem. 2011, 76, 7065–7075. [Google Scholar] [CrossRef] [PubMed]
- Sparr, C.; Schweizer, W.B.; Senn, H.M.; Gilmour, R. The Fluorine-Iminium Ion Gauche Effect: Proof of Principle and Application to Asymmetric Organocatalysis. Angew. Chem.—Int. Ed. 2009, 48, 3065–3068. [Google Scholar] [CrossRef] [PubMed]
- Sparr, C.; Tanzer, E.M.; Bachmann, J.; Gilmour, R. A Concise Synthesis of (S)-2-(Fluorodiphenylmethyl)Pyrrolidine: A Novel Organocatalyst for the Stereoselective Epoxidation of α,β-Unsaturated Aldehydes. Synthesis 2010, 8, 1394–1397. [Google Scholar] [CrossRef]
- Tanzer, E.M.; Zimmer, L.E.; Schweizer, W.B.; Gilmour, R. Fluorinated Organocatalysts for the Enantioselective Epoxidation of Enals: Molecular Preorganisation by the Fluorine-Iminium Ion Gauche Effect. Chem.—A Eur. J. 2012, 18, 11334–11342. [Google Scholar] [CrossRef]
- Quintard, A.; Langlois, J.B.; Emery, D.; Mareda, J.; Guénée, L.; Alexakis, A. Conformationally Stabilized Catalysts by Fluorine Insertion: Tool for Enantioselectivity Improvement. Chem.—A Eur. J. 2011, 17, 13433–13437. [Google Scholar] [CrossRef]
- Conde, E.; Bello, D.; De Cózar, A.; Sánchez, M.; Vázquez, M.A.; Cossío, F.P. Densely Substituted Unnatural L- and D-Prolines as Catalysts for Highly Enantioselective Stereodivergent (3 + 2) Cycloadditions and Aldol Reactions. Chem. Sci. 2012, 3, 1486–1491. [Google Scholar] [CrossRef]
- De Gracia Retamosa, M.; de Cózar, A.; Sánchez, M.; Miranda, J.I.; Sansano, J.M.; Castelló, L.M.; Nájera, C.; Jiménez, A.I.; Sayago, F.J.; Cativiela, C.; et al. Remote Substituent Effects on the Stereoselectivity and Organocatalytic Activity of Densely Substituted Unnatural Proline Esters in Aldol Reactions. Eur. J. Org. Chem. 2015, 2015, 2503–2516. [Google Scholar] [CrossRef] [Green Version]
- Arrastia, I.; Arrieta, A.; Cossío, F.P. Application of 1,3-Dipolar Reactions between Azomethine Ylides and Alkenes to the Synthesis of Catalysts and Biologically Active Compounds. Eur. J. Org. Chem. 2018, 2018, 5889–5904. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, M.; Montroni, E.; Quintavalla, A.; Trombini, C. A New Family of Conformationally Constrained Bicyclic Diarylprolinol Silyl Ethers as Organocatalysts. Adv. Synth. Catal. 2012, 354, 3428–3434. [Google Scholar] [CrossRef]
- Lombardo, M.; Cerisoli, L.; Manoni, E.; Montroni, E.; Quintavalla, A.; Trombini, C. Properties and Reactivity of Conformationally Constrained Bicyclic Diarylprolinol Silyl Ethers as Organocatalysts. Eur. J. Org. Chem. 2014, 2014, 5946–5953. [Google Scholar] [CrossRef]
- Vinayagam, P.; Vishwanath, M.; Kesavan, V. New Class of Bifunctional Thioureas from L-Proline: Highly Enantioselective Michael Addition of 1,3-Dicarbonyls to Nitroolefins. Tetrahedron Asymmetry 2014, 25, 568–577. [Google Scholar] [CrossRef]
- Tian, J.M.; Yuan, Y.H.; Tu, Y.Q.; Zhang, F.M.; Zhang, X.B.; Zhang, S.H.; Wang, S.H.; Zhang, X.M. The Design of a Spiro-Pyrrolidine Organocatalyst and Its Application to Catalytic Asymmetric Michael Addition for the Construction of All-Carbon Quaternary Centers. Chem. Commun. 2015, 51, 9979–9982. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.M.; Yuan, Y.H.; Xie, Y.Y.; Zhang, S.Y.; Ma, W.Q.; Zhang, F.M.; Wang, S.H.; Zhang, X.M.; Tu, Y.Q. Catalytic Asymmetric Cascade Using Spiro-Pyrrolidine Organocatalyst: Efficient Construction of Hydrophenanthridine Derivatives. Org. Lett. 2017, 19, 6618–6621. [Google Scholar] [CrossRef]
- Zou, Y.; Li, C.Y.; Xiang, M.; Li, W.S.; Zhang, J.; Wan, W.J.; Wang, L.X. New Scaffold Organocatalysts of Chiral 3,2′-Pyrrolidinyl Spirooxindoles Promoted Enantioselective Aldol Condensation between Isatins and Acetone. Tetrahedron Lett. 2022, 97, 153780. [Google Scholar] [CrossRef]
- Silvi, M.; Verrier, C.; Rey, Y.P.; Buzzetti, L.; Melchiorre, P. Visible-Light Excitation of Iminium Ions Enables the Enantioselective Catalytic β-Alkylation of Enals. Nat. Chem. 2017, 9, 868–873. [Google Scholar] [CrossRef]
- Berger, M.; Carboni, D.; Melchiorre, P. Photochemical Organocatalytic Regio- and Enantioselective Conjugate Addition of Allyl Groups to Enals. Angew. Chem.—Int. Ed. 2021, 60, 26373–26377. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.H.F.; Ma, D.; Di Sanza, R.; Melchiorre, P. Photoredox Organocatalysis for the Enantioselective Synthesis of 1,7-Dicarbonyl Compounds. Org. Lett. 2022, 24, 1695–1699. [Google Scholar] [CrossRef]
- Rodríguez, R.I.; Sicignano, M.; Alemán, J. Fluorinated Sulfinates as Source of Alkyl Radicals in the Photo-Enantiocontrolled β-Functionalization of Enals. Angew. Chem.—Int. Ed. 2022, 61, e202112632. [Google Scholar] [CrossRef]
- Brinner, K.M.; Ellman, J.A. A Rapid and General Method for the Asymmetric Synthesis of 2-Substituted Pyrrolidines Using Tert-Butanesulfinamide. Org. Biomol. Chem. 2005, 3, 2109–2113. [Google Scholar] [CrossRef]
- Ruano, J.L.G.; Alemán, J.; Cid, M.B. Quick Access to Optically Pure 2-(1-Hydroxybenzyl)Piperidine and Pyrrolidine. Synthesis 2006, 4, 687–691. [Google Scholar] [CrossRef]
- Leemans, E.; Mangelinckx, S.; De Kimpe, N. Asymmetric Synthesis of 2-Arylpyrrolidines Starting from γ-Chloro N-(Tert-Butanesulfinyl)Ketimines. Chem. Commun. 2010, 46, 3122–3124. [Google Scholar] [CrossRef] [PubMed]
- Reddy, L.R.; Das, S.G.; Liu, Y.; Prashad, M. A Facile Asymmetric Synthesis of Either Enantiomer of 2-Substituted Pyrrolidines. J. Org. Chem. 2010, 75, 2236–2246. [Google Scholar] [CrossRef]
- Reddy, L.R.; Prashad, M. Asymmetric Synthesis of 2-Substituted Pyrrolidines by Addition of Grignard Reagents to γ-Chlorinated N-Tert-Butanesulfinyl Imine. Chem. Commun. 2010, 46, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.A.; Prasad, K.R. Addition of the Lithium Anion of Diphenylmethanol Methyl/Methoxymethyl Ether to Nonracemic Sulfinimines: Two-Step Asymmetric Synthesis of Diphenylprolinol Methyl Ether and Chiral (Diphenylmethoxymethyl)Amines. J. Org. Chem. 2018, 83, 10776–10785. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, X.; Shen, H.; Liu, Q.; Pan, J.; Hu, W.; Xiong, Y.; Chen, C. Boron Trifluoride•Diethyl Ether-Catalyzed Etherification of Alcohols: A Metal-Free Pathway to Diphenylmethyl Ethers. Adv. Synth. Catal. 2015, 357, 3115–3120. [Google Scholar] [CrossRef]
- Enders, D.; Kipphardt, H.; Gerdes, P.; Brena-Valle, L.J.; Bhushan, V. Large Scale Preparation of Versatile Chiral Auxiliaries Derived from (S)-Proline. Bull. Soc. Chin. Belg. 1988, 97, 691–704. [Google Scholar] [CrossRef]
- Ho, C.Y.; Chen, Y.C.; Wong, M.K.; Yang, D. Fluorinated Chiral Secondary Amines as Catalysts for Epoxidation of Olefins with Oxone. J. Org. Chem. 2005, 70, 898–906. [Google Scholar] [CrossRef]
- Yuan, Y.H.; Han, X.; Zhu, F.P.; Tian, J.M.; Zhang, F.M.; Zhang, X.M.; Tu, Y.Q.; Wang, S.H.; Guo, X. Development of Bifunctional Organocatalysts and Application to Asymmetric Total Synthesis of Naucleofficine I and II. Nat. Commun. 2019, 10, 3394. [Google Scholar] [CrossRef] [Green Version]
- Aukland, M.H.; List, B. Organocatalysis Emerging as a Technology. Pure Appl. Chem. 2021, 93, 1371–1381. [Google Scholar] [CrossRef]
- Han, B.; He, X.H.; Liu, Y.Q.; He, G.; Peng, C.; Li, J.L. Asymmetric Organocatalysis: An Enabling Technology for Medicinal Chemistry. Chem. Soc. Rev. 2021, 50, 1522–1586. [Google Scholar] [CrossRef] [PubMed]








































































































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quintavalla, A.; Carboni, D.; Lombardo, M. Recent Advances in Asymmetric Synthesis of Pyrrolidine-Based Organocatalysts and Their Application: A 15-Year Update. Molecules 2023, 28, 2234. https://doi.org/10.3390/molecules28052234
Quintavalla A, Carboni D, Lombardo M. Recent Advances in Asymmetric Synthesis of Pyrrolidine-Based Organocatalysts and Their Application: A 15-Year Update. Molecules. 2023; 28(5):2234. https://doi.org/10.3390/molecules28052234
Chicago/Turabian StyleQuintavalla, Arianna, Davide Carboni, and Marco Lombardo. 2023. "Recent Advances in Asymmetric Synthesis of Pyrrolidine-Based Organocatalysts and Their Application: A 15-Year Update" Molecules 28, no. 5: 2234. https://doi.org/10.3390/molecules28052234