
Design, Synthesis, and Antitumor Activity of Olmutinib Derivatives Containing Acrylamide Moiety



Abstract
:
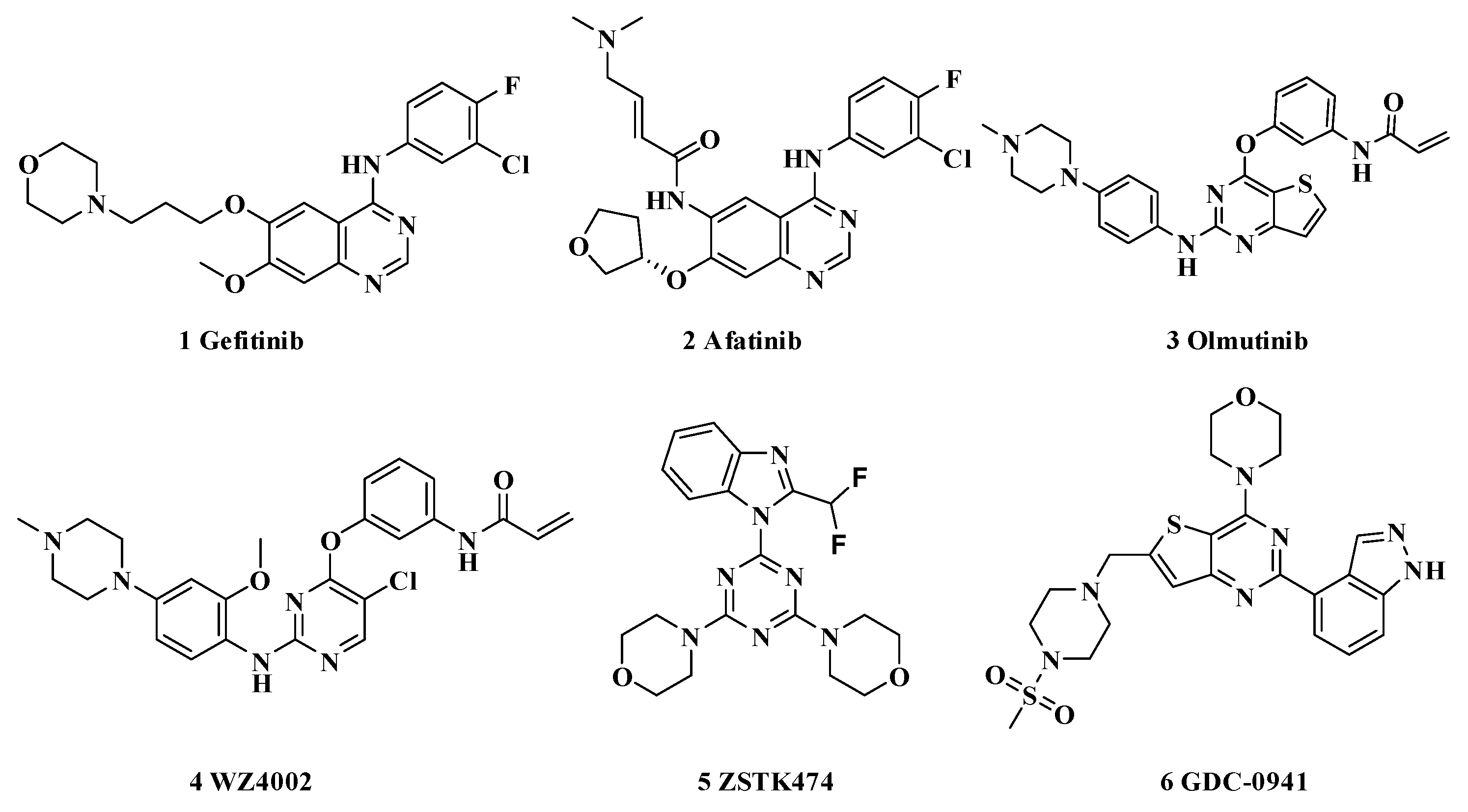
1. Introduction
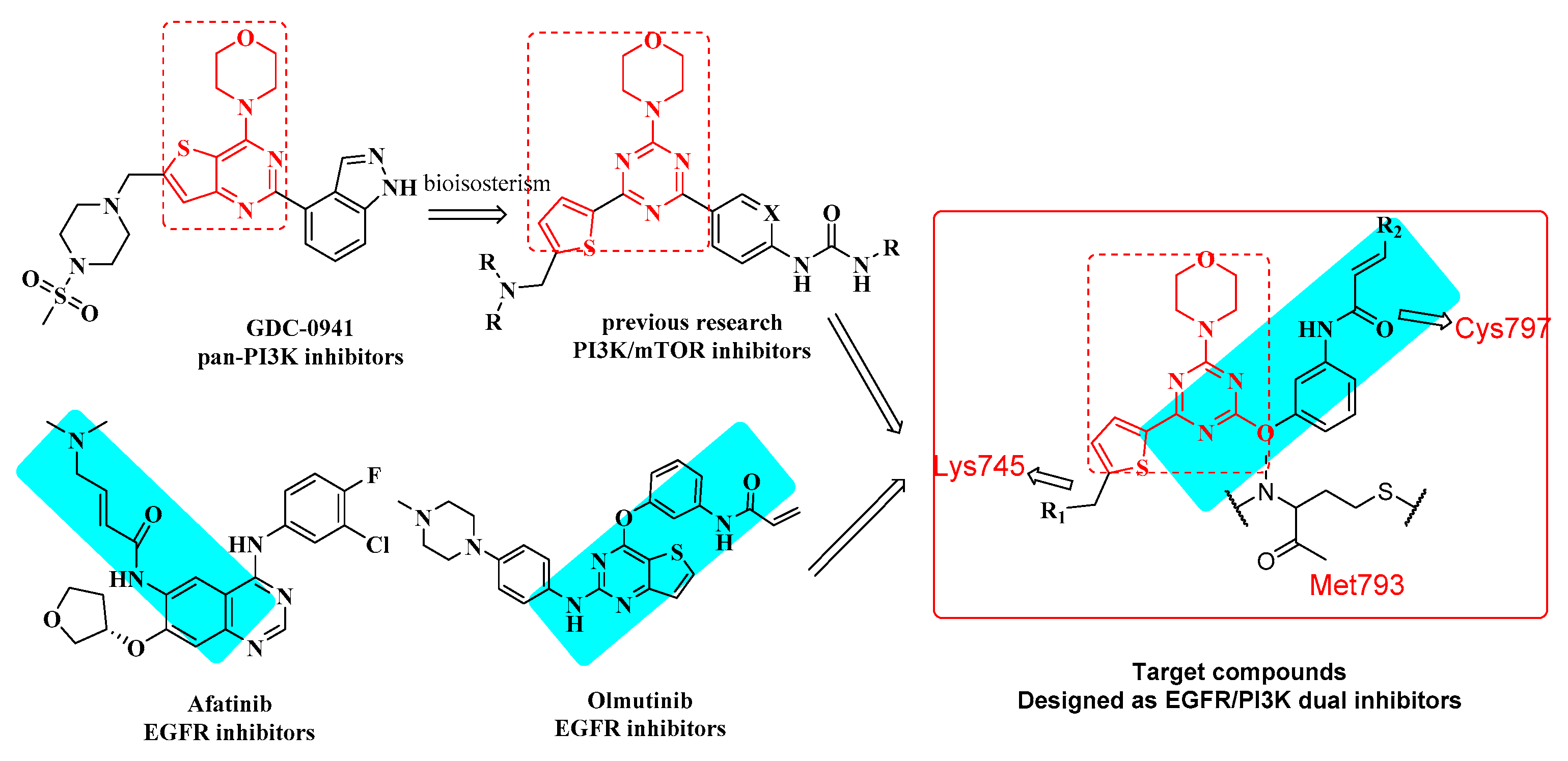
2. Results and Discussion
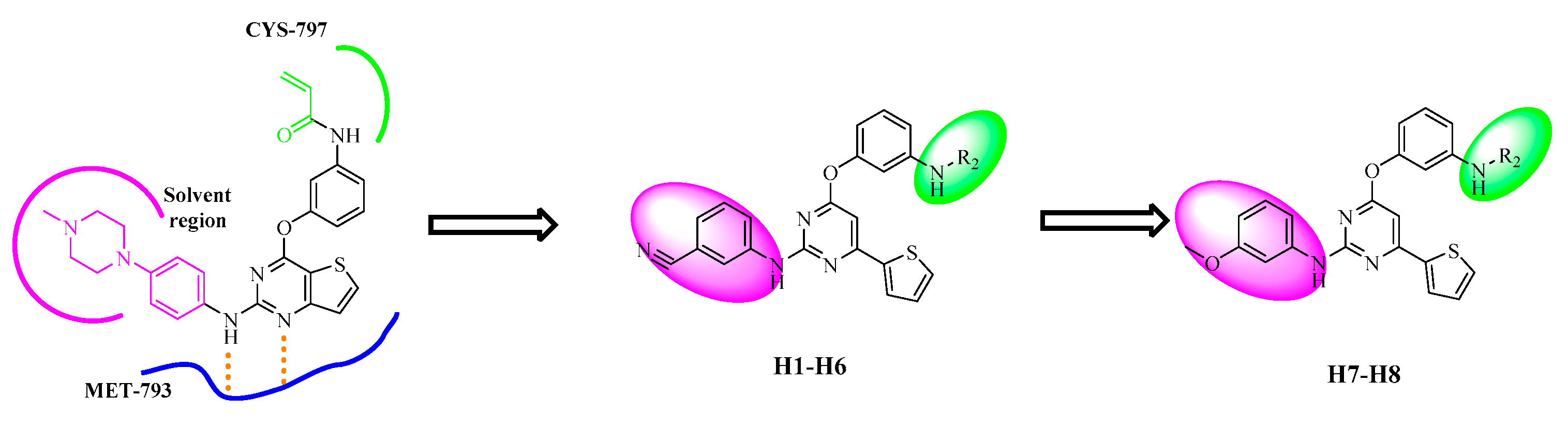
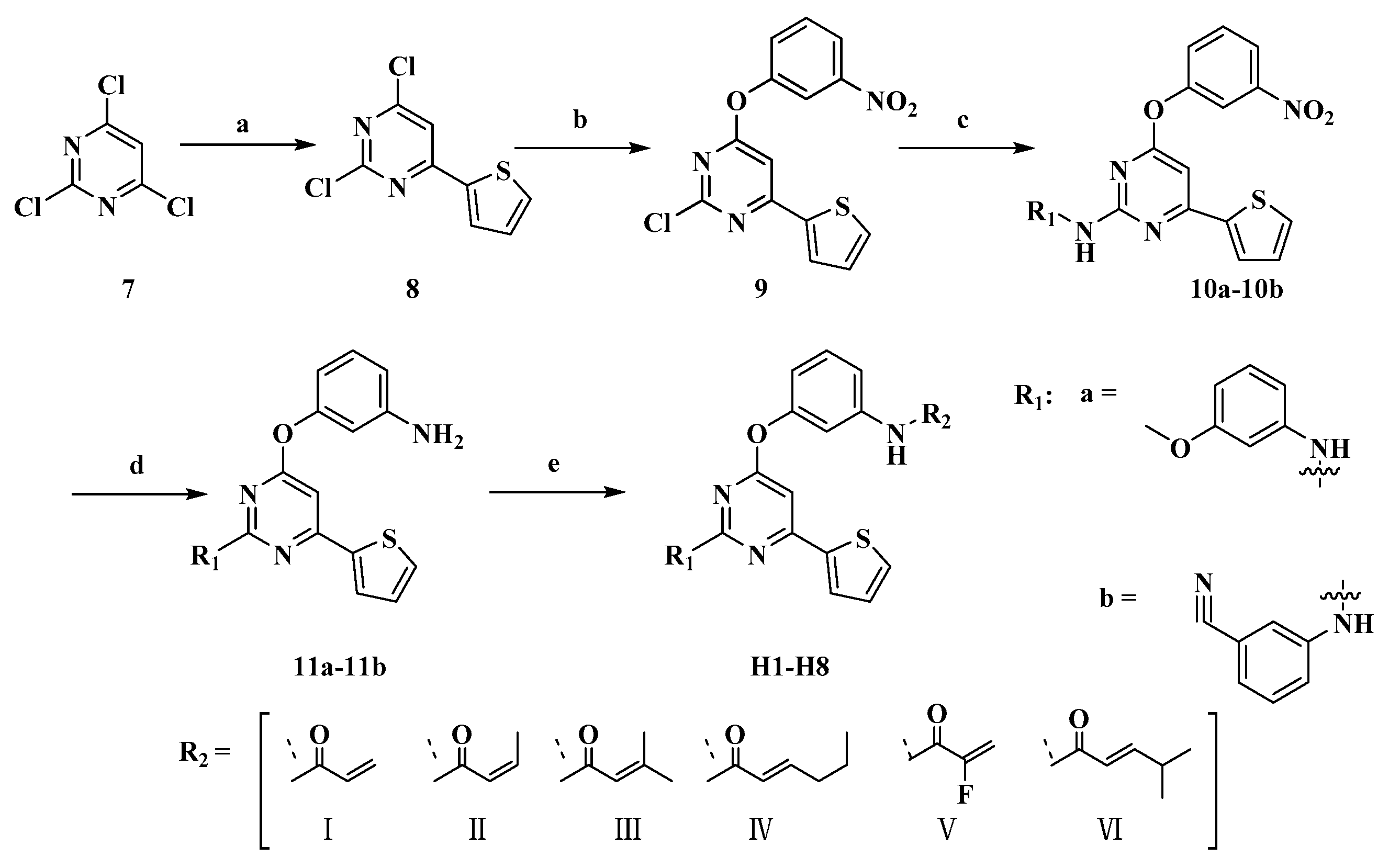
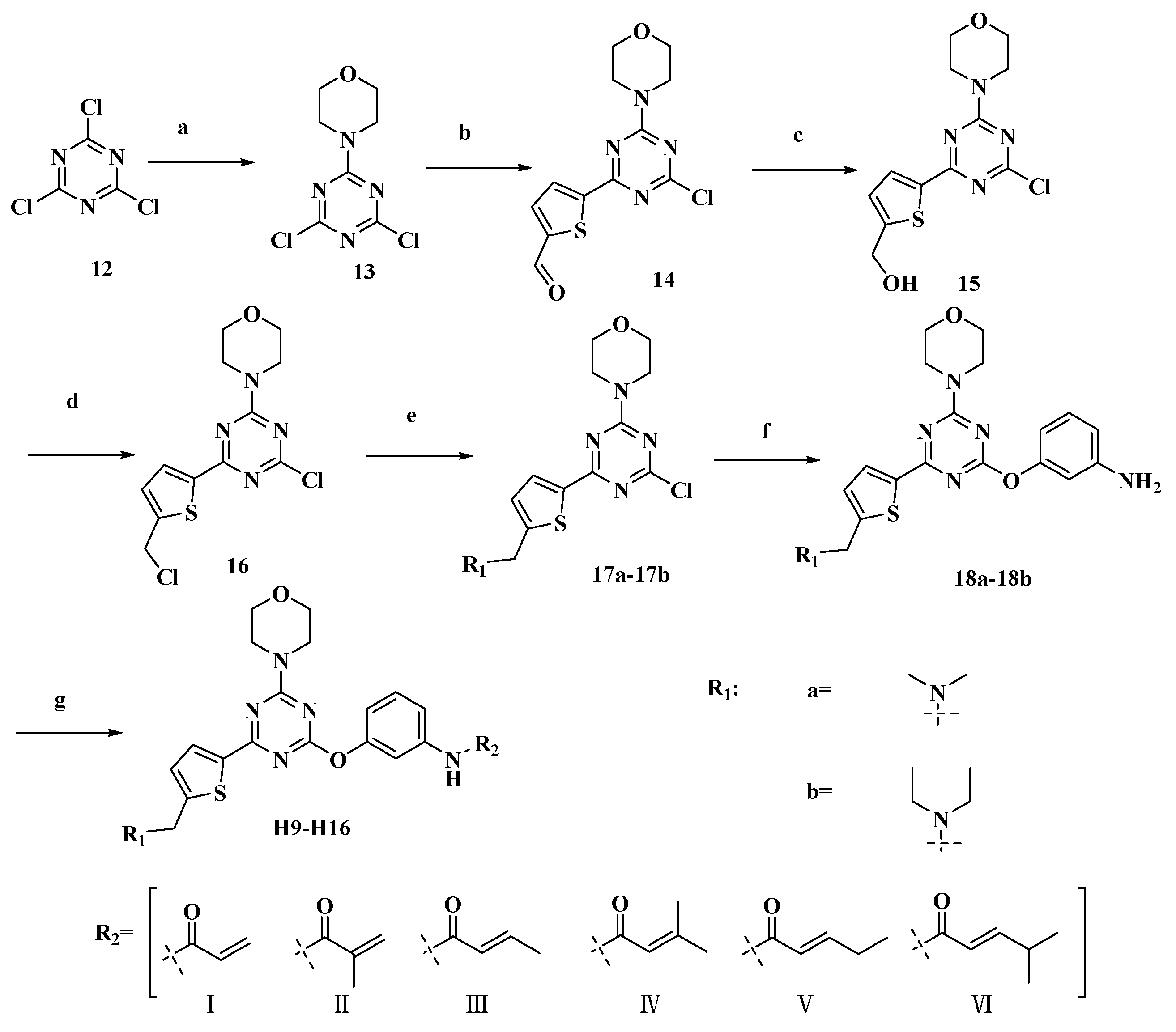
2.1. Chemistry
2.2. Biological Discussion
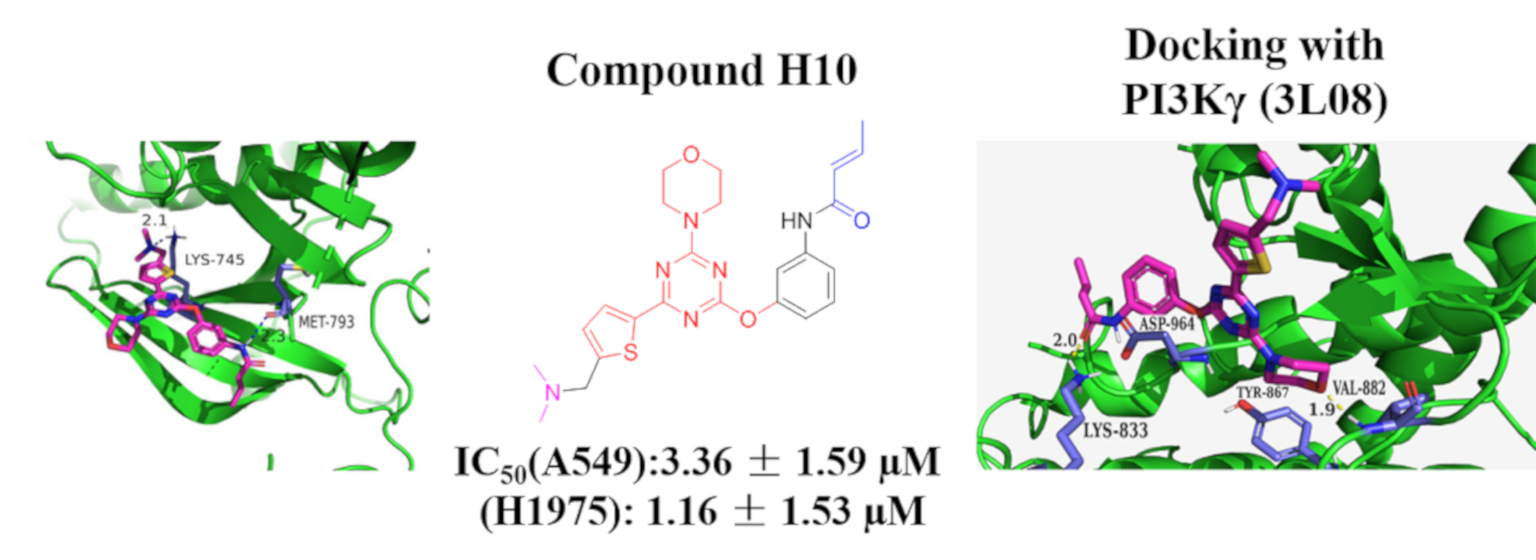
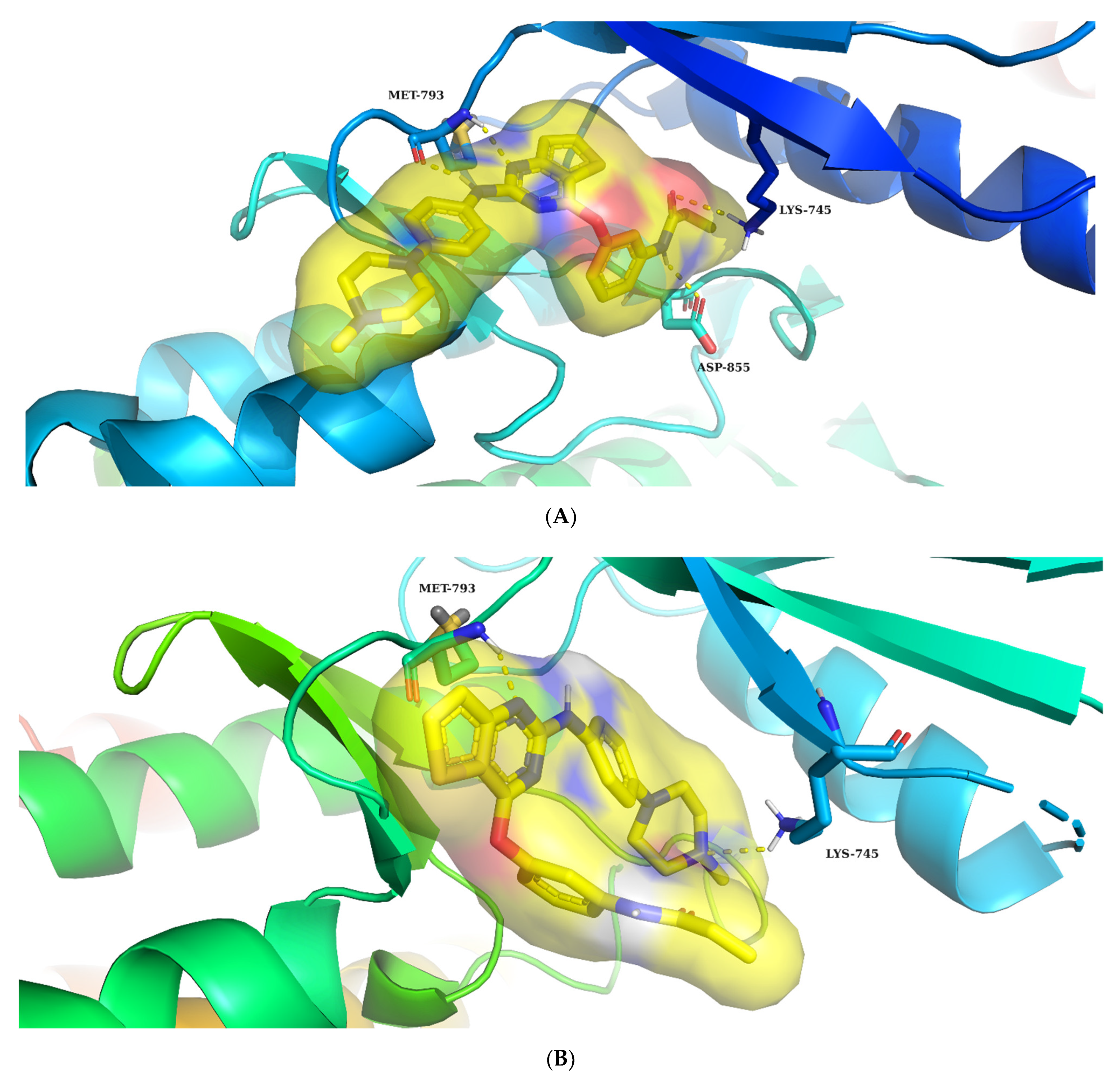
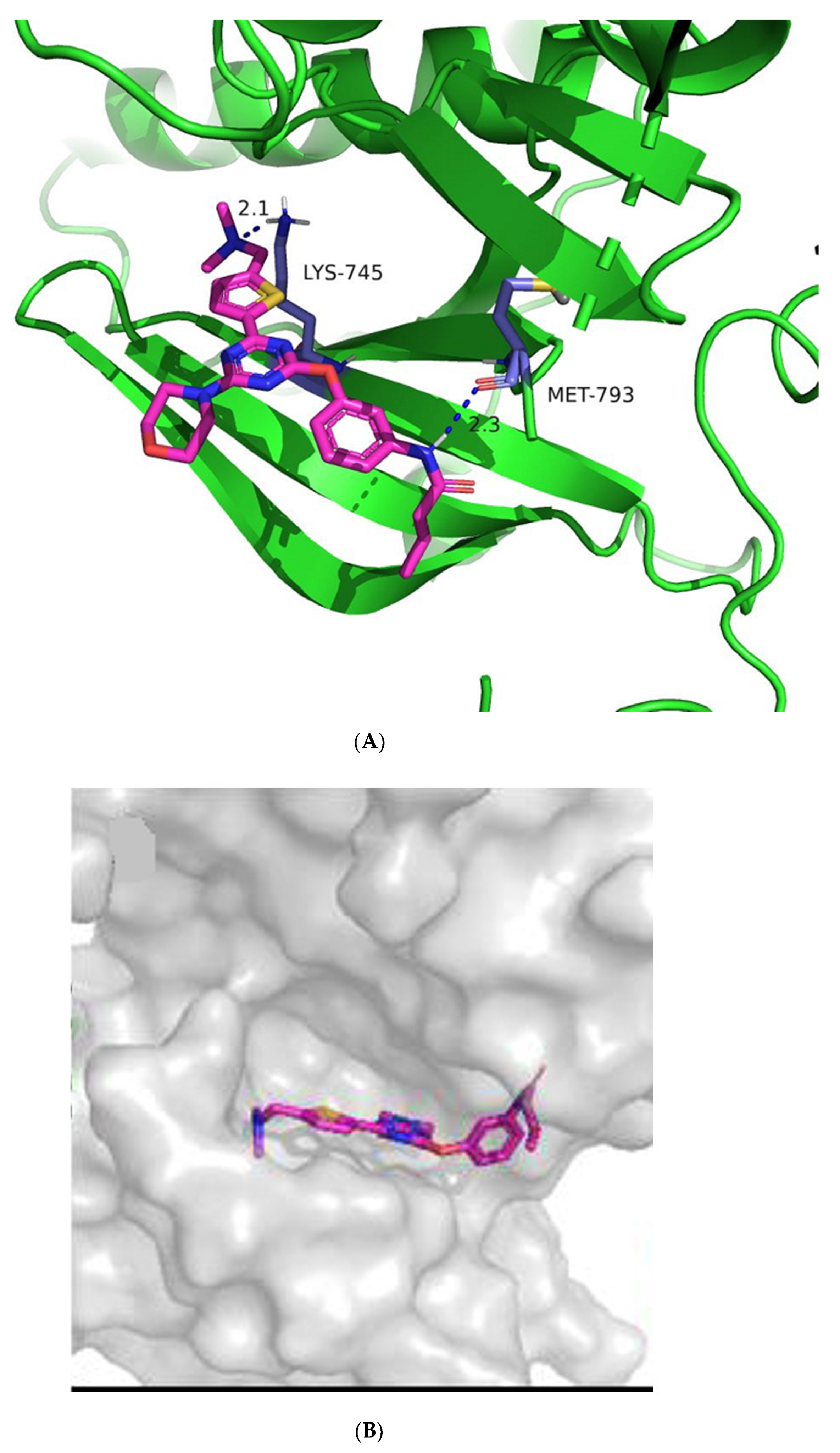
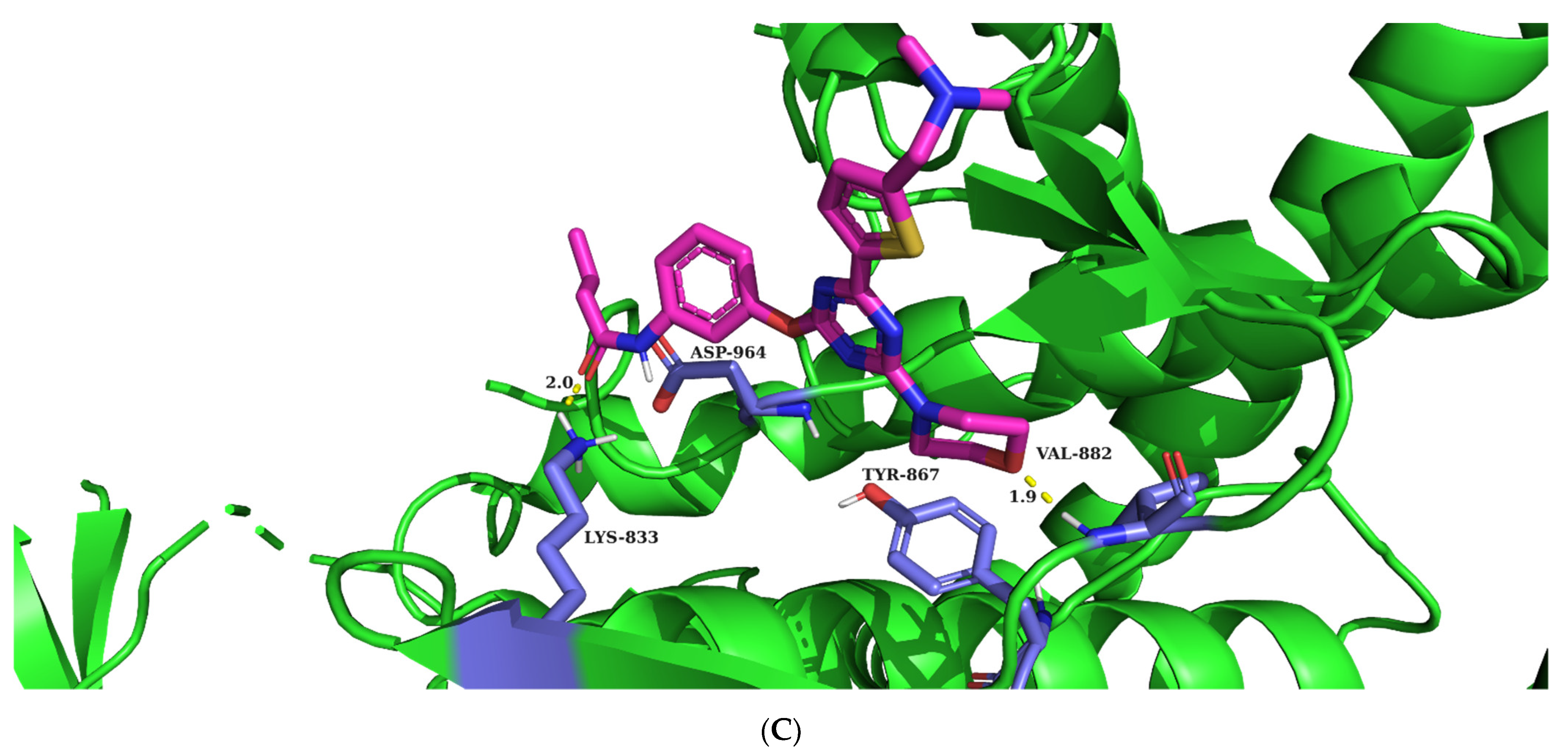
2.3. Molecular Docking Study
3. Experimental Section
3.1. General Information
3.2. Chemistry
3.2.1. Representative Procedure for the Synthesis of 2,4-dichloro-6-(thiophen-2-yl)pyrimidine (8)
3.2.2. Representative Procedure for the Synthesis of 2-chloro-4-(3-nitrophenoxy)-6-(thiophen-2-yl)pyrimidine (9)
3.2.3. Representative Procedure for the Synthesis of 10a–10b
- N-(3-methoxyphenyl)-4-(3-nitrophenoxy)-6-(thiophen-2-yl)pyrimidin-2-amine (10a)
- 3-((4-(3-nitrophenoxy)-6-(thiophen-2-yl)pyrimidin-2-yl)amino)benzonitrile (10b)
3.2.4. Representative Procedure for the Synthesis of 11a–11b
- 4-(3-aminophenoxy)-N-(3-methoxyphenyl)-6-(thiophen-2-yl)pyrimidin-2-amine (11a)
- 3-((4-(3-aminophenoxy)-6-(thiophen-2-yl)pyrimidin-2-yl)amino)benzonitrile (11b)
3.2.5. Representative Procedure for the Synthesis of Target Compounds H1–H8
3.2.6. Representative Procedure for the Synthesis of 12–16
3.2.7. Representative Procedure for the Synthesis of 17a–17b
- 1-(5-(4-chloro-6-morpholino-1,3,5-triazin-2-yl)thiophen-2-yl)-N,N-dimethylmethanamine (17a)
- N-((5-(4-chloro-6-morpholino-1,3,5-triazin-2-yl)thiophen-2-yl)methyl)-N-ethylethanamine (17b)
3.2.8. Representative Procedure for the Synthesis of 18a–18b
- 3-((4-(5-((dimethylamino)methyl)thiophen-2-yl)-6-morpholino-1,3,5-triazin-2-yl)oxy)aniline (18a)
- 3-((4-(5-((diethylamino)methyl)thiophen-2-yl)-6-morpholino-1,3,5-triazin-2-yl)oxy)aniline (18b)
3.2.9. Representative Procedure for the Synthesis of Target Compounds H9–H16
- N-(3-((2-((3-cyanophenyl)amino)-6-(thiophen-2-yl)pyrimidin-4-yl)oxy)phenyl)acrylamide (H1). Yield: 40.3%; color: yellow; m.p.: 197.2–199.4 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.34 (s, 1H), 9.95 (s, 1H), 8.07 (s, 1H), 7.85 (s, 2H), 7.70 (d, J = 6.3 Hz, 1H), 7.51 (d, J = 8.3 Hz, 1H), 7.46 (t, J = 8.1 Hz, 2H), 7.33 (s, 2H), 7.28–7.24 (m, 1H), 7.16 (s, 1H), 7.03–6.99 (m, 1H), 6.42 (dd, J = 16.9, 10.1 Hz, 1H), 6.27 (dd, J = 16.9, 2.0 Hz, 1H), 5.79–5.74 (m, 1H). TOF MS ES+ (m/z): [M + H]+, calcd for C25H19N5O2S: 456.3863, found, 456.3864.
- (E)-N-(3-((2-((3-cyanophenyl)amino)-6-(thiophen-2-yl)pyrimidin-4-yl)oxy)phenyl)but-2-enamide (H2). Yield: 58.3%; color: yellow; m.p.: 201.2–205.9 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.13 (s, 1H), 9.95 (s, 1H), 8.08 (d, J = 5.8 Hz, 2H), 7.85 (d, J = 5.1 Hz, 2H), 7.68 (d, J = 10.9 Hz, 1H), 7.47 (d, J = 8.2 Hz, 1H), 7.42 (d, J = 7.9 Hz, 1H), 7.34 (s, 2H), 7.27–7.24 (m, 1H), 7.16 (s, 1H), 6.99 (d, J = 8.1 Hz, 1H), 6.80 (dd, J = 15.3, 7.6 Hz, 1H), 6.14 (d, J = 15.3 Hz, 1H), 1.86 (d, J = 7.0 Hz, 3H). TOF MS ES+ (m/z): [M + H]+, calcd for C25H19N5O2S: 454.5743, found, 454.5746.
- N-(3-((2-((3-cyanophenyl)amino)-6-(thiophen-2-yl)pyrimidin-4-yl)oxy)phenyl)-3-methylbut-2-enamide (H3). Yield: 66.3%; color: yellow; m.p.: 215.5–218.7 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.01 (s, 1H), 9.95 (s, 1H), 8.07 (d, J = 3.8 Hz, 1H), 7.85 (d, J = 4.9 Hz, 2H), 7.67 (s, 1H), 7.42-7.40 (m, 3H), 7.34 (s, 2H), 7.27–7.24 (m, 1H), 7.15 (s, 1H), 6.96 (d, J = 8.1 Hz, 1H), 5.86 (s, 1H), 2.12 (s, 3H), 1.85 (s, 3H). TOF MS ES+ (m/z): [M + H]+, calcd for C25H19N5O2S: 468.5745, found, 468.5747.
- (E)-N-(3-((2-((3-cyanophenyl)amino)-6-(thiophen-2-yl)pyrimidin-4-yl)oxy)phenyl)hex-2-enamide (H4). Yield: 48.6%; color: yellow; m.p.: 209.6–211.4 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, 1H), 9.95 (s, 1H), 8.07 (d, J = 3.7 Hz, 1H), 7.85 (d, J = 5.0 Hz, 2H), 7.68 (s, 1H), 7.48 (d, J = 8.4 Hz, 1H), 7.42 (t, J = 8.0 Hz, 2H), 7.34 (s, 2H), 7.26 (dd, J = 5.0, 3.8 Hz, 1H), 7.16 (s, 1H), 6.98 (d, J = 7.9 Hz, 1H), 6.82–6.76 (m, 1H), 6.10 (d, J = 15.4 Hz, 1H), 2.18 (d, J = 7.0 Hz, 2H), 1.46 (d, J = 7.3 Hz, 2H), 0.90 (d, J = 3.5 Hz, 3H). TOF MS ES+ (m/z): [M + H]+, calcd for C25H19N5O2S: 482.3266, found, 482.3265.
- N-(3-((2-((3-cyanophenyl)amino)-6-(thiophen-2-yl)pyrimidin-4-yl)oxy)phenyl)-2-fluoroacrylamide (H5). Yield: 33.1%; color: yellow; m.p.: 222.9–226.8 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H), 9.97 (s, 1H), 8.08 (d, J = 4.0 Hz, 1H), 7.86 (d, J = 5.3 Hz, 1H), 7.70 (d, J = 10.7 Hz, 2H), 7.49 (t, J = 8.0 Hz, 2H), 7.34 (s, 2H), 7.27 (d, J = 4.7 Hz, 1H), 7.17 (s, 1H), 7.09 (d, J = 8.6 Hz, 1H), 5.78 (d, J = 3.7 Hz, 1H), 5.65 (s, 1H), 5.48–5.41 (m, 1H). TOF MS ES+ (m/z): [M + H]+, calcd for C24H16FN5O2S: 458.4987, found, 458.4989.
- (E)-N-(3-((2-((3-cyanophenyl)amino)-6-(thiophen-2-yl)pyrimidin-4-yl)oxy)phenyl)-4-methylpent-2-enamide (H6). Yield: 51.8%; color: yellow; m.p.: 223.1–225.4 °C 1H NMR (400 MHz, DMSO-d6) δ 10.19 (s, 1H), 9.94 (s, 1H), 8.06 (d, J = 3.8 Hz, 1H), 7.84 (d, J = 5.2 Hz, 2H), 7.68 (d, J = 4.5 Hz, 1H), 7.48 (d, J = 8.3 Hz, 1H), 7.42 (t, J = 8.1 Hz, 1H), 7.33 (s, 2H), 7.25 (t, J = 4.4 Hz, 1H), 7.15 (s, 1H), 6.98 (d, J = 8.0 Hz, 1H), 6.81 (d, J = 6.3 Hz, 1H), 6.79–6.74 (m, 1H), 6.06 (d, J = 15.4 Hz, 1H), 2.44 (dd, J = 13.1, 6.6 Hz, 1H), 1.03 (d, J = 6.5 Hz, 6H). TOF MS ES+ (m/z): [M + H]+, calcd for C27H23N5O2S: 482.5866, found, 482.5868.
- (E)-N-(3-((2-((3-methoxyphenyl)amino)-6-(thiophen-2-yl)pyrimidin-4-yl)oxy)phenyl)but-2-enamide (H7). Yield: 43.7%; color: yellow; m.p.: 231.7–234.3 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, 1H), 9.43 (s, 1H), 8.00 (s, 1H), 7.23 (s, 2H), 6.76 (d, J = 6.9 Hz, 5H), 6.08 (d, J = 15.2 Hz, 6H), 3.67 (s, 3H), 1.93 (d, J = 7.8 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.28, 163.61, 163.02, 159.31, 155.14, 154.13, 140.54, 140.28, 139.03, 133.31, 132.43, 130.17, 129.72, 128.50, 127.84, 126.10, 120.67, 116.37, 115.97, 113.36, 112.62, 55.11, 17.41. TOF MS ES+ (m/z): [M + H]+, calcd for C25H22N4O3S: 459.5356, found, 459.5359.
- (E)-N-(3-((2-((3-methoxyphenyl)amino)-6-(thiophen-2-yl)pyrimidin-4-yl)oxy)phenyl)-4-methylpent-2-enamide (H8). Yield: 22.6%; color: yellow; m.p.: 217.9–219.1 °C; 1H NMR 400 MHz, DMSO-d6) δ 9.83 (s, 2H), 7.55 (d, J = 8.8 Hz, 4H), 6.87 (d, J = 8.9 Hz, 5H), 6.75 (dd, J = 15.4, 6.4 Hz, 3H), 6.05 (s, 1H), 6.01 (s, 1H), 3.72 (s, 3H), 1.06 (s, 7H). TOF MS ES+ (m/z): [M + H]+, calcd for C27H26N4O3S: 487.6124, found, 487.6126.
- N-(3-((4-(5-((dimethylamino)methyl)thiophen-2-yl)-6-morpholino-1,3,5-triazin-2-yl)oxy)phenyl)acrylamide (H9). Yield: 76.8%; color: yellow; m.p.: 224.9–226.1 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.39 (s, 1H), 7.25 (s, 2H), 7.08 (s, 1H), 7.06 (s, 1H), 7.03 (s, 1H), 7.01 (s, 1H), 6.48–6.47 (m, 1H), 6.25–6.21 (m, 1H), 5.74 (s, 1H), 3.64 (s, 4H), 3.58 (s, 4H), 3.02 (s, 2H), 1.24 (s, 6H). TOF MS ES+ (m/z): [M + H]+, calcd for C23H26N6O3S: 466.5668, found, 466.5670.
- (E)-N-(3-((4-(5-((dimethylamino)methyl)thiophen-2-yl)-6-morpholino-1,3,5-triazin-2-yl)oxy)phenyl)but-2-enamide (H10). Yield: 63.5%; color: yellow; m.p.: 202.4–203.7 °C; 1H NMR ((400 MHz, DMSO-d6) δ 9.65 (s, 1H), 9.38 (s, 2H), 7.25 (s, 1H), 7.05 (s, 2H), 6.46 (s, 1H), 5.75 (s, 1H), 5.48 (s, 1H), 3.64 (s, 4H), 3.58 (s, 4H), 3.02 (s, 2H), 1.93 (s, 6H), 1.23 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.01, 169.89, 168.95, 163.76, 163.12, 157.33, 157.13, 146.17, 142.55, 142.09, 136.74, 136.45, 134.96, 128.48, 124.20, 121.58, 117.67, 114.48, 113.04, 58.14, 36.37, 35.08, 32.23, 24.75. TOF MS ES+ (m/z): [M + H]+, calcd for C24H28N6O3S: 480.5873, found, 480.5875.
- N-(3-((4-(5-((dimethylamino)methyl)thiophen-2-yl)-6-morpholino-1,3,5-triazin-2-yl)oxy)phenyl)-3-methylbut-2-enamide (H11). Yield: 84.5%; color: yellow; m.p.: 198.7–199.8 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.30 (s, 1H), 7.76 (s, 1H), 7.23 (s, 1H), 6.98 (s, 2H), 6.77 (s, 1H), 6.42 (s, 1H), 5.99 (s, 1H), 3.08-3.15 (m, 10H), 2.21 (s, 9H), 1.84 (s, 3H). TOF MS ES+ (m/z): [M + H]+, calcd for C24H28N6O3S: 482.6257, found, 482.6256.
- (E)-N-(3-((4-(5-((dimethylamino)methyl)thiophen-2-yl)-6-morpholino-1,3,5-triazin-2-yl)oxy)phenyl)-4-methylpent-2-enamide (H12). Yield: 78.5%; color: yellow; m.p.: 199.7–201.3 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.85 (s, 1H), 9.39 (s, 1H), 7.76 (s, 1H), 7.28 (s, 1H), 7.07 (m, 2H), 6.83 (m, 1H), 6.46 (s, 1H), 6.10 (m, 1H), 3.64 (s, 4H), 3.58 (s, 4H), 3.02 (s, 2H), 1.93(s, 7H), 1.06 (s, 6H). TOF MS ES+ (m/z): [M + H]+, calcd for C26H32N6O3S: 508.6263, found, 508.6266.
- N-(3-((4-(5-((diethylamino)methyl)thiophen-2-yl)-6-morpholino-1,3,5-triazin-2-yl)oxy)phenyl)methacrylamide (H13). Yield: 64.7%; color: yellow; m.p.: 240.7–243.5 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.71 (s, 1H), 7.78 (s, 1H), 7.35 (s, 1H), 6.97 (s, 1H), 6.54 (s, 1H), 6.02 (s, 1H), 5.82 (s, 1H), 5.60 (s, 1H), 5.51 (s, 1H), 3.75 (s, 6H), 1.98 (s, 4H), 1.89 (s, 4H), 1.16 (d, 3H), 1.00 (m, 6H). TOF MS ES+ (m/z): [M + H]+, calcd for C26H32N6O3S: 509.6956, found, 509.6953.
- (E)-N-(3-((4-(5-((diethylamino)methyl)thiophen-2-yl)-6-morpholino-1,3,5-triazin-2-yl)oxy)phenyl)but-2-enamide (H14). Yield: 35.8%; color: yellow; m.p.: 189.7–192.1 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.84 (s, 1H), 7.74 (s, 1H), 7.29 (s, 1H), 6.96 (s, 1H), 6.78 (d, J = 6.9 Hz, 1H), 6.15 (s, 1H), 6.11 (s, 1H), 5.82 (s, 1H), 5.78 (s, 1H), 3.74 (s, 6H), 1.83 (m, 4H), 1.14 (d, 4H), 0.98 (m, 9H). TOF MS ES+ (m/z): [M + H]+, calcd for C26H32N6O3S: 509.6933, found, 509.6931.
- N-(3-((4-(5-((diethylamino)methyl)thiophen-2-yl)-6-morpholino-1,3,5-triazin-2-yl)oxy)phenyl)-3-methylbut-2-enamide (H15). Yield: 41.5%; color: yellow; m.p.: 215.2–217.1 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.71 (s, 1H), 7.27 (s, 1H), 7.06 (s, 1H), 7.00 (s, 1H), 6.45 (s, 1H), 6.02 (s, 1H), 5.87 (s, 1H), 5.63 (s, 1H), 3.74 (s, 6H), 2.15 (s, 4H), 1.86 (s, 4H), 1.14 (s, 6H), 0.99 (s, 6H). TOF MS ES+ (m/z): [M + H]+, calcd for C26H32N6O3S: 523.8974, found, 523.8971.
- (E)-N-(3-((4-(5-((diethylamino)methyl)thiophen-2-yl)-6-morpholino-1,3,5-triazin-2-yl)oxy)phenyl)-4-methylpent-2-enamide (H16). Yield: 26.4%; color: yellow; m.p.: 222.9.2–226.4 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.88 (s, 1H), 7.75 (s, 1H), 7.30 (s, 1H), 7.05 (s, 1H), 6.79 (s, 1H), 6.48 (s, 1H), 6.10 (s, 1H), 6.07 (s, 1H), 3.72 (s, 6H), 1.14 (s, 10H), 1.05 (d, J = 6.7 Hz, 12H). TOF MS ES+ (m/z): [M + H]+, calcd for C26H32N6O3S: 536.6475, found, 536.6473.
3.3. EGFR and PI3Kα Kinase Assay
3.4. Cytotoxicity Assay In Vitro
3.5. Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Sample Availability
References
- Shepherd, F.A.; Rodrigues, P.J.; Ciuleanu, T.; Tan, E.H.; Hirsh, V.; Thongprasert, S.; Campos, D.; Maoleekoonpiroj, S.; Smylie, M.; Martins, R.; et al. Erlotinib in previously treated non-small-cell lung cancer. N. Engl. J. Med. 2005, 353, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.S.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.S.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Hiramatsu, M.; Ninomiya, H.; Inamura, K.; Okui, M.; Satoh, Y.; Fujiwara, M.; Yamori, T.; Kitagawa, T.; Ishikawa, Y. EGFR signal pathways selectively activated in lung adenocarcinoma. Cancer Res. 2007, 67, 5038. [Google Scholar]
- Cohen, M.H.; Williams, G.A.; Sridhara, R.; Chen, G.; McGuinn, W.D., Jr.; Morse, D.; Abraham, S.; Rahmans, A.; Liang, C.; Lostritto, R.; et al. United States Food and Drug Administration Drug Approval Summary: Gefitinib (ZD1839; Iressa) Tablets. Clin. Cancer Res. 2004, 10, 1212–1218. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J.; et al. MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Smit, E.F.; Groen, H.J.; Horn, L.; Gettinger, S.; Camidge, D.R.; Riely, G.J.; Wang, B.; Fu, Y.; Chand, V.K.; et al. Dual Inhibition of EGFR with Afatinib and Cetuximab in Kinase Inhibitor–Resistant EGFR-Mutant Lung Cancer with and without T790M Mutations. Cancer Discov. 2014, 4, 1036–1045. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.S. Olmutinib: First Global Approval. Drugs 2016, 76, 1153–1157. [Google Scholar] [CrossRef]
- Chen, K.L.; Cho, Y.T.; Yang, C.W.; Sheen, Y.S.; Liang, C.W.; Lacouture, M.E.; Chu, C.Y. Olmutinib-induced palmoplantar keratoderma. Br. J. Dermatol. 2018, 178, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-W.; Lee, D.H.; Han, J.-Y.; Lee, J.; Cho, B.C.; Kang, J.H.; Lee, K.H.; Cho, E.K.; Kim, J.-S.; Min, Y.J.; et al. Safety, tolerability, and anti-tumor activity of olmutinib in non-small cell lung cancer with T790M mutation: A single arm, open label, phase 1/2 trial. Lung Cancer 2019, 135, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, Y.; Yamazaki, Y.; Nakamura, Y.; Yoshihara, M.; Matsukuma, S.; Nakayama, H.; Yokose, T.; Kameda, Y.; Koizume, S.; Miyagi, Y. WZ4002, a third-generation EGFR inhibitor, can overcome anoikis resistance in EGFR-mutant lung adenocarcinomas more efficiently than Src inhibitors. Lab. Investig. 2012, 92, 371–383. [Google Scholar] [CrossRef] [Green Version]
- Dexin, K.T. ZSTK474 is an ATP-competitive inhibitor of class I phosphatidylinositol 3 kinase isoforms. Cancer Sci. 2007, 98, 1638–1642. [Google Scholar]
- Namatame, N.; Tamaki, N.; Yoshizawa, Y.; Okamura, M.; Nishimura, Y.; Yamazaki, K.; Tanaka, M.; Nakamura, T.; Semba, K.; Yamori, T.; et al. Antitumor profile of the PI3K inhibitor ZSTK474 in human sarcoma cell lines. Oncotarget 2018, 9, 35141–35161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkes, A.J.; Ahmadi, K.; Alderton, W.K.; Alix, S.; Baker, S.J.; Box, G.; Chickowree, I.S.; Clarke, P.A.; Depledge, P.; Eccles, S.A.; et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J. Med. Chem. 2008, 51, 5522–5532. [Google Scholar] [CrossRef] [PubMed]
- Yao, E.; Zhou, W.; Lee-Hoeflich, S.T.; Truong, T.; Haverty, P.M.; Eastham-Anderson, J.; Lewin-Koh, N.; Gunter, B.; Belvin, M.; Murray, L.J.; et al. Suppression of HER2/HER3-Mediated Growth of Breast Cancer Cells with Combinations of GDC-0941 PI3K Inhibitor, Trastuzumab, and Pertuzumab. Clin. Cancer Rese. 2009, 15, 4147–4156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, R.; Cantley, L. Ras, PI3K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Verbitski, S.M.; Mullally, J.E.; Fitzpatrick, F.A.; Ireland, C.M. Punaglandins, chlorinated prostaglandins, function as potent Michael receptors to inhibit ubiquitin isopeptidase activity. J. Med. Chem. 2004, 47, 2062–2070. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, Q.; Xiao, Z.; Sun, X.; Yang, Z.; Gu, Q.; Liu, Z.; Xie, T.; Jin, Q.; Zheng, P.; et al. Design, synthesis and biological evaluation of substituted 2-(thiophen-2-yl)-1,3,5-triazine derivatives as potential dual PI3Kα/mTOR inhibitors. Bioorg. Chem. 2020, 95, 103525. [Google Scholar] [CrossRef]
- Lei, F.; Sun, C.; Xu, S.; Wang, Q.; Ouyang, Y.; Chen, C. Design, synthesis, biological evaluation and docking studies of novel 2-substituted-4-morpholino-7,8-dihydro-5H-thiopyrano[4,3-d]- pyrimidine derivatives as dual PI3Kα/mTOR inhibitors. Eur. J. Med. Chem. 2016, 116, 27–35. [Google Scholar] [CrossRef]
- Zhao, B.; Lei, F.; Wang, C.; Zhang, B.; Yang, Z.; Li, W.; Zhu, W.; Xu, S. Design, Synthesis and Biological Evaluation of Novel Phenylsulfonylurea Derivatives as PI3K/mTOR Dual Inhibitors. Molecules 2018, 23, 1553. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Xu, S.; Liu, X.; Chen, X.; Xiong, H.; Hou, S. Discovery of thinopyrimidine-triazole conjugates as c-Met targeting and apoptosis inducing agents. Bioorg. Chem. 2018, 77, 370–380. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R1 | R2 | IC50(µM) a | Selectivity c | ||||
|---|---|---|---|---|---|---|---|---|
| A549 | H1975 | NCI-H460 | MCF-7 | LO2 | ||||
| H1 |  |  | 4.43 ± 0.50 | 9.64 ± 1.01 | 13.09 ± 1.17 | 18.99 ± 1.71 | >100 | >22.57 |
| H2 |  |  | 10.48 ± 1.17 | 12.45 ± 1.09 | 16.39 ± 0.98 | 14.56 ± 1.19 | 87.85 ± 1.67 | 8.38 |
| H3 |  |  | 27.69 ± 1.24 | 21.85 ± 1.33 | 25.79 ± 1.38 | 35.83 ± 1.64 | >100 | >3.61 |
| H4 |  |  | 37.10 ± 1.19 | 25.11 ± 1.30 | 23.38 ± 1.49 | 27.04 ± 1.63 | >100 | >2.70 |
| H5 |  |  | >100 | >100 | >100 | >100 | >100 | − |
| H6 |  |  | 15.56 ± 0.81 | 11.94 ± 1.11 | 18.09 ± 0.71 | 17.73 ± 1.01 | >100 | >6.43 |
| H7 |  |  | 4.37 ± 0.50 | 4.59 ± 0.46 | 13.48 ± 0.52 | 19.21 ± 1.12 | >100 | >22.88 |
| H8 |  |  | 19.01 ± 1.34 | 21.52 ± 1.21 | 24.12 ± 1.28 | 49.13 ± 2.13 | >100 | >5.26 |
| H9 |  |  | 23.46 ± 1.43 | 12.82 ± 1.25 | 28.24 ± 1.78 | 53.86 ± 1.73 | >100 | >4.26 |
| H10 |  |  | 3.36 ± 1.59 | 1.16 ± 1.53 | 10.65 ± 2.02 | 13.05 ± 1.36 | >100 | >29.76 |
| H11 |  |  | 13.54 ± 1.13 | 5.57 ± 1.53 | 19.72 ± 1.16 | 25.83 ± 1.31 | >100 | >7.39 |
| H12 |  |  | 21.86 ± 1.54 | 13.55 ± 1.94 | 23.81 ± 2.05 | 64.01 ± 1.80 | >100 | >4.57 |
| H13 |  |  | 23.45 ± 1.89 | 21.52 ± 1.21 | 54.53 ± 2.86 | 32.45 ± 1.73 | >100 | >4.26 |
| H14 |  |  | 20.56 ± 2.30 | 17.78 ± 1.34 | 46.22 ± 1.43 | 41.61 ± 1.61 | >100 | >4.86 |
| H15 |  |  | 21.35 ± 1.58 | 19.46 ± 1.55 | 62.43 ± 2.94 | 38.54 ± 1.78 | >100 | >4.68 |
| H16 |  |  | 36.14 ± 1.61 | 15.57 ± 2.55 | 30.28 ± 1.68 | 51.82 ± 1.27 | >100 | >2.77 |
| Olmutinib b | - | - | 4.29 ± 0.21 | 0.52 ± 0.10 | 5.29 ± 0.59 | 26.90 ± 0.93 | 25.76 ± 1.31 | 6.00 |
| Compound | IC50 (µM) a | |
|---|---|---|
| EGFRT790M/L858R | PI3Kα | |
| H7 | 0.63 | >10 |
| H10 | 0.25 | 8.56 |
| Olmutinib b | 0.01 | >10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, X.; Tang, S.; Yang, F.; Zheng, P.; Xu, S.; Pan, Q.; Zhu, W. Design, Synthesis, and Antitumor Activity of Olmutinib Derivatives Containing Acrylamide Moiety. Molecules 2021, 26, 3041. https://doi.org/10.3390/molecules26103041
Hu X, Tang S, Yang F, Zheng P, Xu S, Pan Q, Zhu W. Design, Synthesis, and Antitumor Activity of Olmutinib Derivatives Containing Acrylamide Moiety. Molecules. 2021; 26(10):3041. https://doi.org/10.3390/molecules26103041
Chicago/Turabian StyleHu, Xiaohan, Sheng Tang, Feiyi Yang, Pengwu Zheng, Shan Xu, Qingshan Pan, and Wufu Zhu. 2021. "Design, Synthesis, and Antitumor Activity of Olmutinib Derivatives Containing Acrylamide Moiety" Molecules 26, no. 10: 3041. https://doi.org/10.3390/molecules26103041