
Methylated Xanthones from the Rootlets of Metaxya rostrata Display Cytotoxic Activity in Colorectal Cancer Cells


 , and
, and
Abstract
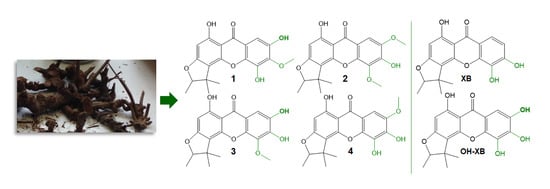
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Chemicals and Reagents
3.3. Plant Material
3.4. Extraction and Isolation
3.5. Cell Lines
3.6. Cytotoxic Activity
3.7. Caspase Activity
3.8. Western Blot
3.9. Topoisomerase I Assay
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cárdenas, G.G.; Tuomisto, H.; Lehtonen, S. Newly discovered diversity in the tropical fern genus Metaxya based on morphology and molecular phylogenetic analyses. Kew Bull. 2016, 71, 5. [Google Scholar] [CrossRef]
- Smith, A.; Tuomisto, H.; Pryer, K.; Hunt, J.; Wolf, P. Metaxya lanosa, a Second Species in the Genus and Fern Family Metaxyaceae. Syst. Bot. 2009, 26, 480–486. [Google Scholar] [CrossRef]
- Kainz, K.P.; Krenn, L.; Erdem, Z.; Kählig, H.; Zehl, M.; Bursch, W.; Berger, W.; Marian, B. 2-Deprenyl-Rheediaxanthone B Isolated from Metaxya rostrata Induces Active Cell Death in Colorectal Tumor Cells. PLoS ONE 2013, 8, e65745. [Google Scholar] [CrossRef]
- Waltenberger, B.; Atanasov, A.G.; Heiss, E.H.; Bernhard, D.; Rollinger, J.M.; Breuss, J.M.; Schuster, D.; Bauer, R.; Kopp, B.; Franz, C.; et al. Drugs from nature targeting inflammation (DNTI): A successful Austrian interdisciplinary network project. Mon. für Chem.-Chem. Mon. 2016, 147, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Khazir, J.; Riley, D.L.; Pilcher, L.A.; De Maayer, P.; Mir, B.A. Anticancer Agents from Diverse Natural Sources. Nat. Prod. Commun. 2014, 9, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Ye, J.; Zhang, R.; Wu, F.; Zhai, L.; Wang, K.; Xiao, M.; Xie, T.; Sui, X. Non-apoptotic cell death in malignant tumor cells and natural compounds. Cancer Lett. 2018, 420, 210–227. [Google Scholar] [CrossRef]
- Krajarng, A.; Nakamura, Y.; Suksamrarn, S.; Watanapokasin, R. α-Mangostin Induces Apoptosis in Human Chondrosarcoma Cells through Downregulation of ERK/JNK and Akt Signaling Pathway. J. Agric. Food Chem. 2011, 59, 5746–5754. [Google Scholar] [CrossRef]
- Krajarng, A.; Nilwarankoon, S.; Suksamrarn, S.; Watanapokasin, R. Antiproliferative effect of α-mangostin on canine osteosarcoma cells. Res. Veter. Sci. 2012, 93, 788–794. [Google Scholar] [CrossRef]
- Watanapokasin, R.; Jarinthanan, F.; Nakamura, Y.; Sawasjirakij, N.; Jaratrungtawee, A.; Suksamrarn, S. Effects of α-mangostin on apoptosis induction of human colon cancer. World J. Gastroenterol. 2011, 17, 2086–2095. [Google Scholar] [CrossRef]
- Mittermair, E.; Krenn, L.; Marian, B. Prenylated xanthones from Metaxya rostrata suppress FoxM1 and induce active cell death by distinct mechanisms. Phytomedicine 2019, 60, 152912. [Google Scholar] [CrossRef] [PubMed]
- Mittermair, E.; Schueffl, H.; Heffeter, P.; Krenn, L.; Marian, B. Destabilization of FoxM1 and Inhibition of Topoisomerase I Contribute to Cytotoxicity of Prenylated Xanthones Isolated from Metaxya rostrata. Planta Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Klein-Júnior, L.C.; Campos, A.; Niero, R.; Corrêa, R.; Heyden, Y.V.; Filho, V.C. Xanthones and Cancer: From Natural Sources to Mechanisms of Action. Chem. Biodivers. 2020, 17, e1900499. [Google Scholar] [CrossRef] [PubMed]
- Akao, Y.; Nakagawa, Y.; Nozawa, Y.; Iinuma, M. Anti-Cancer Effects of Xanthones from Pericarps of Mangosteen. Int. J. Mol. Sci. 2008, 9, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Akao, Y.; Yi, H.; Ohguchi, K.; Ito, T.; Tanaka, T.; Kobayashi, E.; Iinuma, M.; Nozawa, Y. Preferential target is mitochondria in α-mangostin-induced apoptosis in human leukemia HL60 cells. Bioorg. Med. Chem. 2004, 12, 5799–5806. [Google Scholar] [CrossRef]
- Gong, D.; Ferrell, J.E. The Roles of Cyclin A2, B1, and B2 in Early and Late Mitotic Events. Mol. Boil. Cell 2010, 21, 3149–3161. [Google Scholar] [CrossRef]
- Khongkow, P.; Gomes, A.R.; Gong, C.; Man, E.P.S.; Tsang, J.W.-H.; Zhao, F.; Monteiro, L.J.; Coombes, R.C.; Medema, R.; Khoo, U.-S.; et al. Paclitaxel targets FOXM1 to regulate KIF20A in mitotic catastrophe and breast cancer paclitaxel resistance. Oncogene 2015, 35, 990–1002. [Google Scholar] [CrossRef]
- Wierstra, I. FOXM1 (Forkhead box M1) in Tumorigenesis: Overexpression in human cancer, implication in tumorigenesis, oncogenic functions, tumor-suppressive properties, and target of anticancer therapy. In Advances in Cancer Research; Academic Press: Cambridge, UK, 2013; pp. 191–419. [Google Scholar] [CrossRef]
- Wierstra, I. The Transcription Factor FOXM1 (Forkhead box M1): Proliferation-specific expression, transcription factor function, target genes, mouse models, and normal biological roles. In Advances in Cancer Research; Academic Press: New York, NY, USA, 2013; pp. 97–398. [Google Scholar] [CrossRef]
- Yao, S.; Fan, L.Y.-N.; Lam, E.W.-F. The FOXO3-FOXM1 axis: A key cancer drug target and a modulator of cancer drug resistance. Semin. Cancer Boil. 2018, 50, 77–89. [Google Scholar] [CrossRef]
- Li, H.; Huang, J.; Yang, B.; Xiang, T.; Yin, X.; Peng, W.; Cheng, W.; Wan, J.; Luo, F.; Li, H.; et al. Mangiferin exerts antitumor activity in breast cancer cells by regulating matrix metalloproteinases, epithelial to mesenchymal transition, and β-catenin signaling pathway. Toxicol. Appl. Pharmacol. 2013, 272, 180–190. [Google Scholar] [CrossRef]
- Halasi, M.; Gartel, A.L. Targeting FOXM1 in cancer. Biochem. Pharmacol. 2013, 85, 644–652. [Google Scholar] [CrossRef]
- Kainz, K. Phytochemical Investigation and Bioactivity-Guided Isolation of Cytotoxic Compounds from Metaxya rostrata. Ph.D. Thesis, University of Vienna, Vienna, Austria, 2011. [Google Scholar]
- Erdem, Z.N.; Schwarz, S.; Drev, D.; Heinzle, C.; Reti, A.; Heffeter, P.; Hudec, X.; Holzmann, K.; Grasl-Kraupp, B.; Berger, W.; et al. Irinotecan Upregulates Fibroblast Growth Factor Receptor 3 Expression in Colorectal Cancer Cells, Which Mitigates Irinotecan-Induced Apoptosis. Transl. Oncol. 2017, 10, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Dornetshuber, R.; Heffeter, P.; Lemmens-Gruber, R.; Elbling, L.; Marko, D.; Micksche, M.; Berger, W. Oxidative stress and DNA interactions are not involved in Enniatin- and Beauvericin-mediated apoptosis induction. Mol. Nutr. Food Res. 2009, 53, 1112–1122. [Google Scholar] [CrossRef] [PubMed]
- Evdokimov, N.M.; Van Slambrouck, S.; Heffeter, P.; Tu, L.; Le Calvé, B.; Lamoral-Theys, D.; Hooten, C.J.; Uglinskii, P.Y.; Rogelj, S.; Kiss, R.; et al. Structural Simplification of Bioactive Natural Products with Multicomponent Synthesis. 3. Fused Uracil-Containing Heterocycles as Novel Topoisomerase-Targeting Agents. J. Med. Chem. 2011, 54, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ruiz, V.; Pascua, I.; Fernández-Marcelo, T.; Ribelles, P.; Bianchini, G.; Sridharan, V.; Iniesta, P.; Ramos, M.T.; Olives, A.I.; Martín, M.A.; et al. B-Ring-Aryl Substituted Luotonin A Analogues with a New Binding Mode to the Topoisomerase 1-DNA Complex Show Enhanced Cytotoxic Activity. PLoS ONE 2014, 9, e95998. [Google Scholar] [CrossRef]
- Nitiss, J.L.; Soans, E.; Rogojina, A.; Seth, A.; Mishina, M. Topoisomerase Assays. Curr. Protoc. Pharmacol. 2012, 57, 3.3.1–3.3.27. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds only available after repetition of isolation from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mixture M Compound 1 | Mixture M Compound 4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Atom Position | 1H (ppm) | JH.H (Hz) | 13C (ppm) | Atom Position | 1H (ppm) | JH.H (Hz) | 13C (ppm) | ||
| 1 | C | - | - | 165.13 | 1 | C | - | - | 164.96 |
| 2 | CH | 6.120 | s | 93.99 | 2 | CH | 6.118 | s | 93.98 |
| 3 | C | - | - | 167.27 | 3 | C | - | - | 166.94 |
| 4 | C | - | - | 114.12 | 4 | C | - | - | 114.13 |
| 4a | C | - | - | 154.44 | 4a | C | - | - | 154.30 |
| 4b | C | - | - | 148.90 | 4b | C | - | - | e 1 143.66 |
| 5 | C | - | - | 140.89 | 5 | C | - | - | 134.94 |
| 6 | C OCH3 | - 3.966 | s | 143.47 61.19 | 6 | C | - | s | e 143.69 |
| 7 | C | - | - | 141.83 | 7 | C OCH3 | - 3.948 | s | 147.27 56.58 |
| 8 | CH | 7.083 | s | 100.46 | 8 | CH | 7.170 | s | 96.47 |
| 8a | C | - | - | 117.60 | 8a | C | - | 112.93 | |
| 9 | C | - | - | 181.70 | 9 | C | - | - | 181.50 |
| 9a | C | - | - | 104.28 | 9a | C | - | - | 104.08 |
| 11 | C | - | - | 44.95 | 11 | C | - | - | 44.98 |
| 12 | CH3 | 1.604 | s | 25.92 | 12 | CH3 | 1.611 | s | 25.95 |
| 13 | CH3 | 1.316 | s | 21.39 | 13 | CH3 | 1.320 | s | 21.42 |
| 14 | CH | 4.530 | q 6.6 | 92.28 | 14 | CH | 4.525 | q 6.6 | 92.21 |
| 15 | CH3 | 1.398 | q 6.6 | 14.56 | 15 | CH3 | 1.399 | q 6.6 | 14.56 |
| Compound 1 | Compound 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Atom Position | 1H (ppm) | JH.H (Hz) | 13C (ppm) | Atom Position | 1H (ppm) | JH.H (Hz) | 13C (ppm) | ||
| 1 | C | - | - | 165.16 | 1 | C | - | - | 164.98 |
| 2 | CH | 6.139 | s | 93.99 | 2 | CH | 6.145 | s | 94.20 |
| 3 | C | - | - | 167.30 | 3 | C | - | - | 166.92 |
| 4 | C | - | - | 114.14 | 4 | C | - | - | 114.10 |
| 4a | C | - | - | 154.47 | 4a | C | - | - | 154.08 |
| 4b | C | - | - | 148.94 | 4b | C | - | - | 147.98 |
| 5 | C | - | - | 141.00 | 5 | C OCH3 | - 3.969 | - s | 136.75 61.76 |
| 6 | C OCH3 | - 3.967 | - s | 143.52 61.18 | 6 | C | - | - | 151.97 |
| 7 | C | - | - | 141.89 | 7 | C OCH3 | - 3.944 | - s | 148.76 56.59 |
| 8 | CH | 7.090 | s | 100.39 | 8 | CH | 7.359 | s | 100.67 |
| 8a | C | - | - | 117.62 | 8a | C | - | - | 111.67 |
| 9 | C | - | - | 181.75 | 9 | C | - | - | 180.93 |
| 9a | C | - | - | 104.30 | 9a | C | - | - | 104.07 |
| 11 | C | - | - | 44.97 | 11 | C | - | - | 44.97 |
| 12 | CH3 | 1.613 | s | 25.95 | 12 | CH3 | 1.626 | s | 26.12 |
| 13 | CH3 | 1.327 | s | 21.39 | 13 | CH3 | 1.338 | s | 21.72 |
| 14 | CH | 4.547 | q 6.6 | 92.31 | 14 | CH | 4.555 | q 6.6 | 92.13 |
| 15 | CH3 | 1.407 | d 6.6 | 14.58 | 15 | CH3 | 1.417 | d 6.6 | 14.53 |
| Compound 3 | ||||
|---|---|---|---|---|
| Atom Position | 1H (ppm) | JH.H (Hz) | 13C (ppm) | |
| 1 | C | - | - | 164.95 |
| 2 | CH | 6.117 | s | 93.99 |
| 3 | C | - | - | 166.74 |
| 4 | C | - | - | 113.93 |
| 4a | C | - | - | 154.11 |
| 4b | C | - | - | 148.34 |
| 5 | C OCH3 | - 3.954 | - s | 136.60 61.78 |
| 6 | C | - | - | u 1 139.09 |
| 7 | C | - | - | u 141.84 |
| 8 | CH | 7.224 | s, br | 102.75 |
| 8a | C | - | - | u 114.79 |
| 9 | C | - | - | 181.23 |
| 9a | C | - | - | 104.05 |
| 11 | C | - | - | 44.96 |
| 12 | CH3 | 1.624 | s | 26.14 |
| 13 | CH3 | 1.332 | s | 21.72 |
| 14 | CH | 4.536 | q 6.6 | 92.05 |
| 15 | CH3 | 1.410 | d 6.6 | 14.52 |
| % Cytotoxic Activity (12 µM) | SD | |
|---|---|---|
| M | 46.04 | ±3.34 |
| 1 | 51.04 | ±6.00 |
| 2 | 15.79 | ±13.45 |
| OH-XB | 85.32 | ±4.53 |
| XB | 83.99 | ±5.44 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mittermair, E.; Kählig, H.; Tahir, A.; Rindler, S.; Hudec, X.; Schueffl, H.; Heffeter, P.; Marian, B.; Krenn, L. Methylated Xanthones from the Rootlets of Metaxya rostrata Display Cytotoxic Activity in Colorectal Cancer Cells. Molecules 2020, 25, 4449. https://doi.org/10.3390/molecules25194449
Mittermair E, Kählig H, Tahir A, Rindler S, Hudec X, Schueffl H, Heffeter P, Marian B, Krenn L. Methylated Xanthones from the Rootlets of Metaxya rostrata Display Cytotoxic Activity in Colorectal Cancer Cells. Molecules. 2020; 25(19):4449. https://doi.org/10.3390/molecules25194449
Chicago/Turabian StyleMittermair, Eva, Hanspeter Kählig, Ammar Tahir, Stefanie Rindler, Xenia Hudec, Hemma Schueffl, Petra Heffeter, Brigitte Marian, and Liselotte Krenn. 2020. "Methylated Xanthones from the Rootlets of Metaxya rostrata Display Cytotoxic Activity in Colorectal Cancer Cells" Molecules 25, no. 19: 4449. https://doi.org/10.3390/molecules25194449