Cytotoxic and Anti-Plasmodial Activities of Stephania dielsiana Y.C. Wu Extracts and the Isolated Compounds

 , , ,
, , ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Bioassay-Guided Fractionation and Isolation of S. dielsiana Y.C. Wu
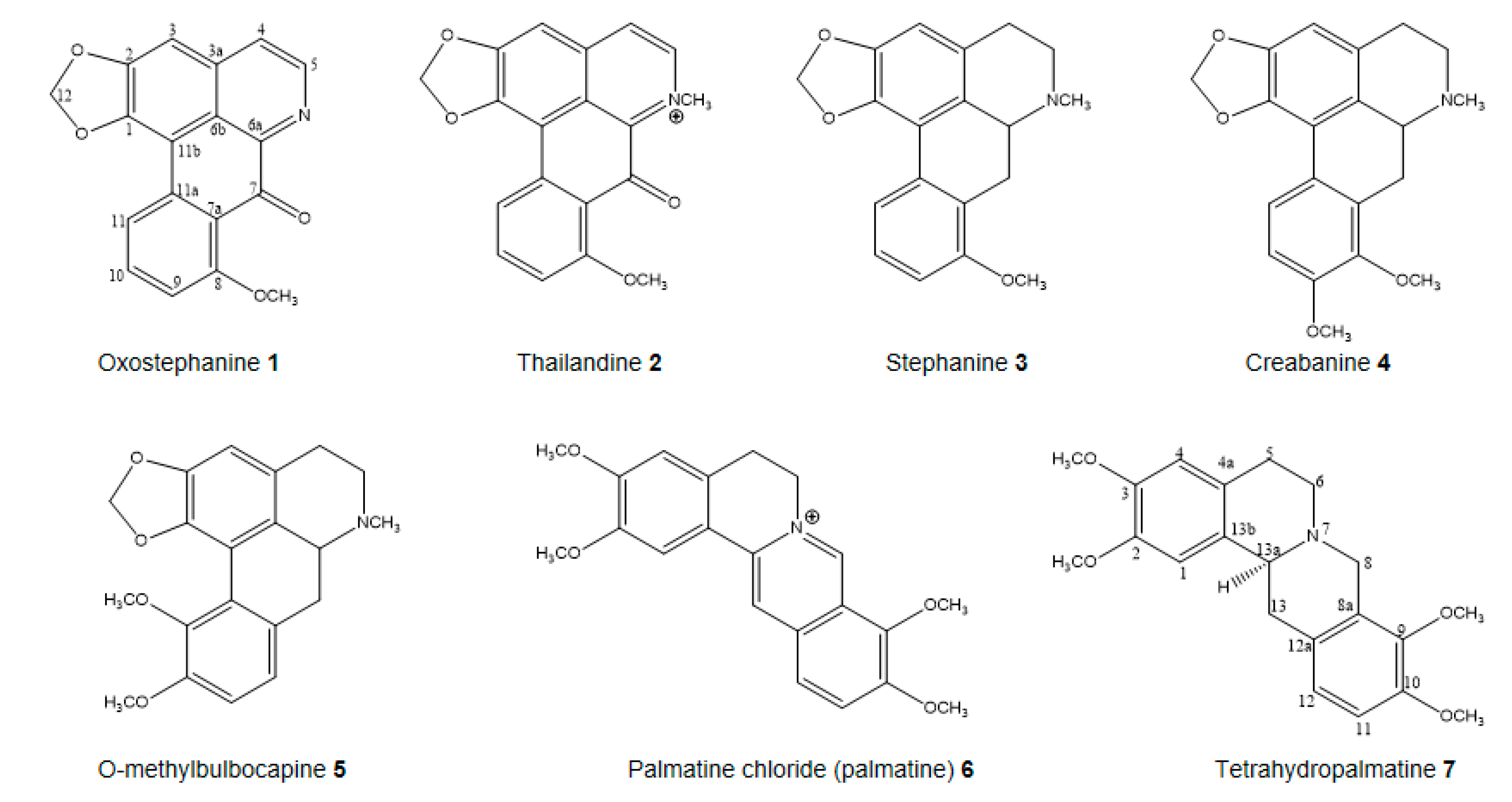
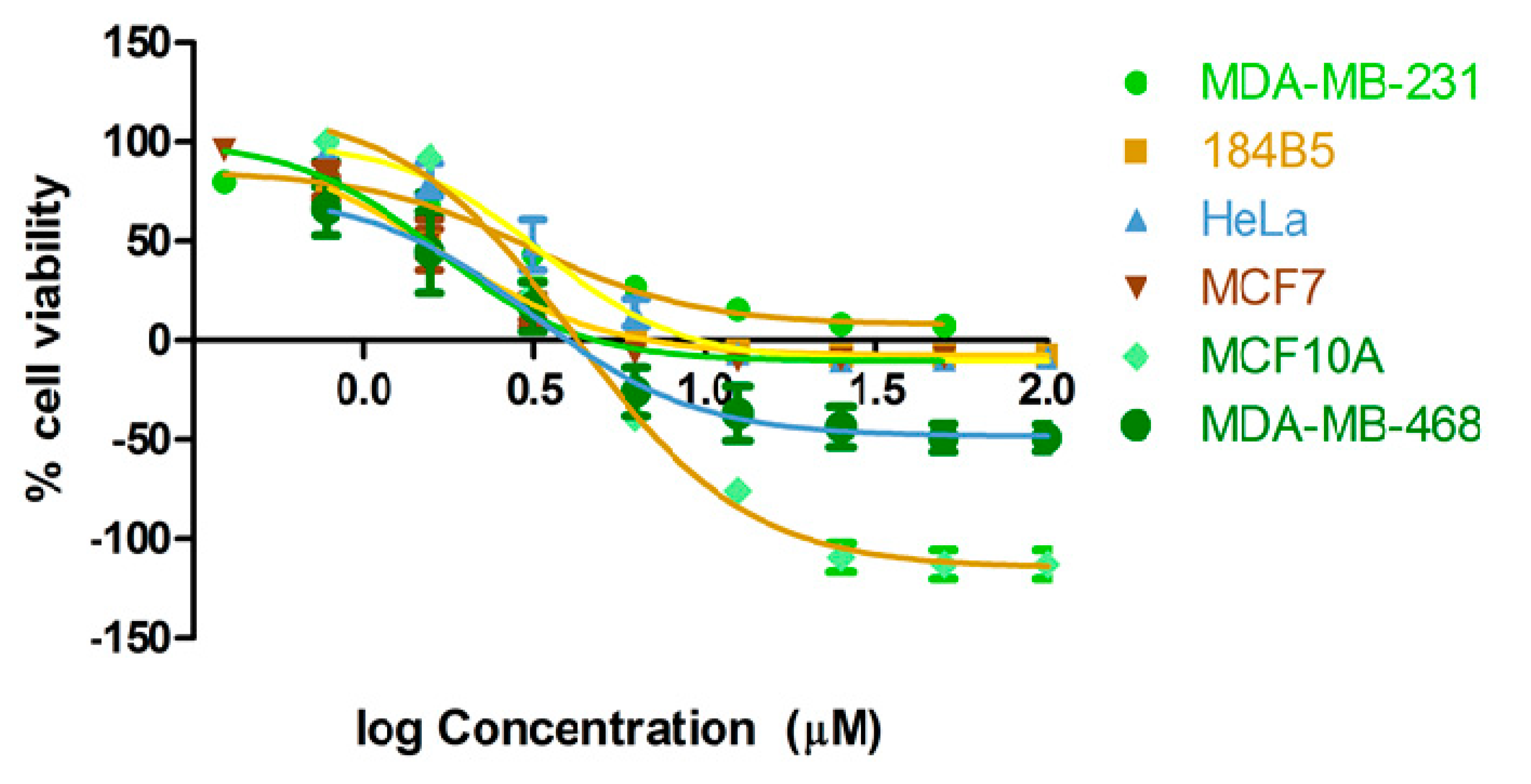
2.2. In Vitro Antiproliferative and Structure-Activity Relationship (SAR) of Isolated Compounds
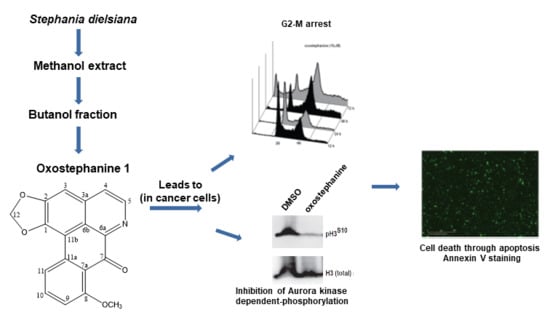
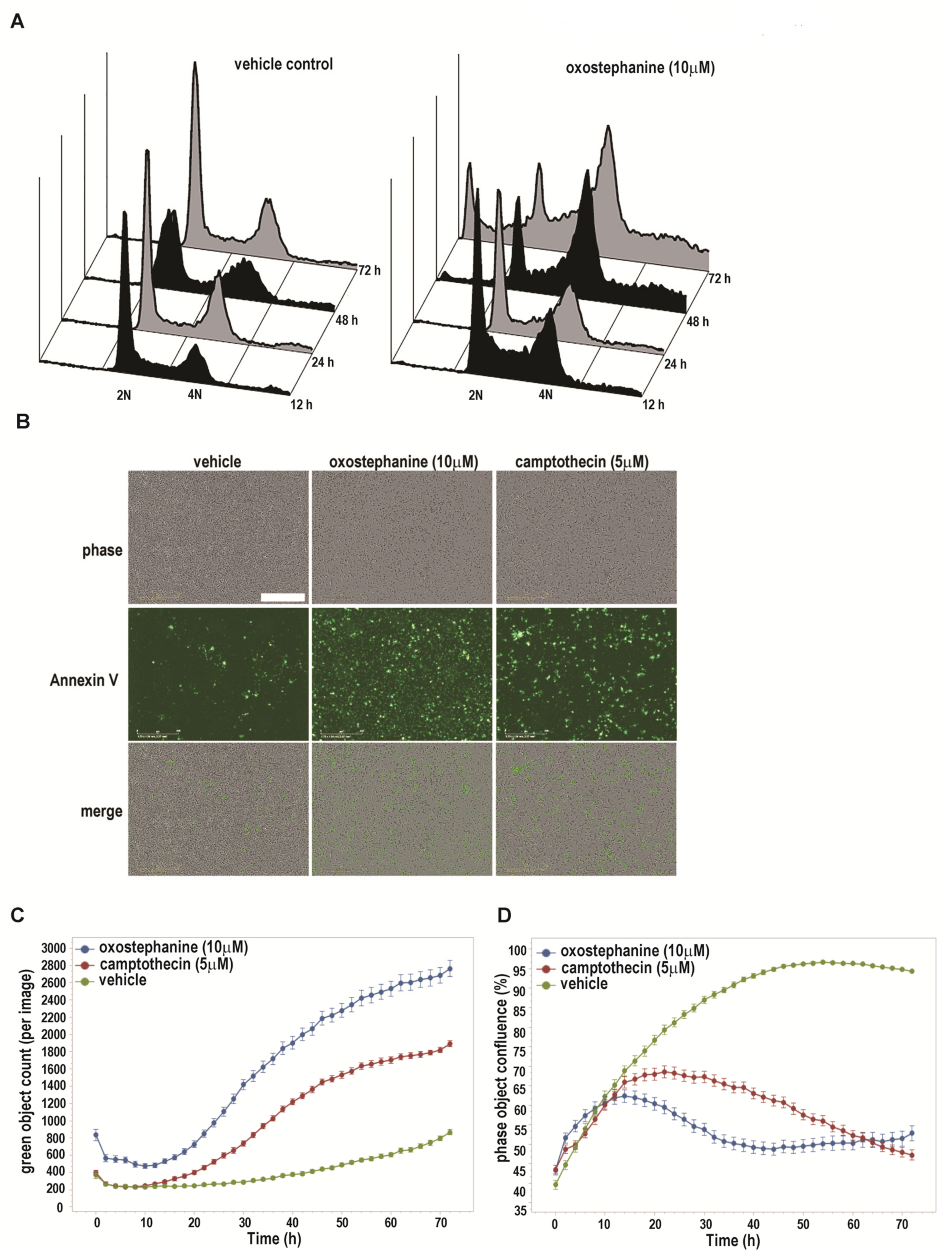
2.3. Oxostephanine Leads to A Transient G2/M Arrest and Apoptotic Cell Death
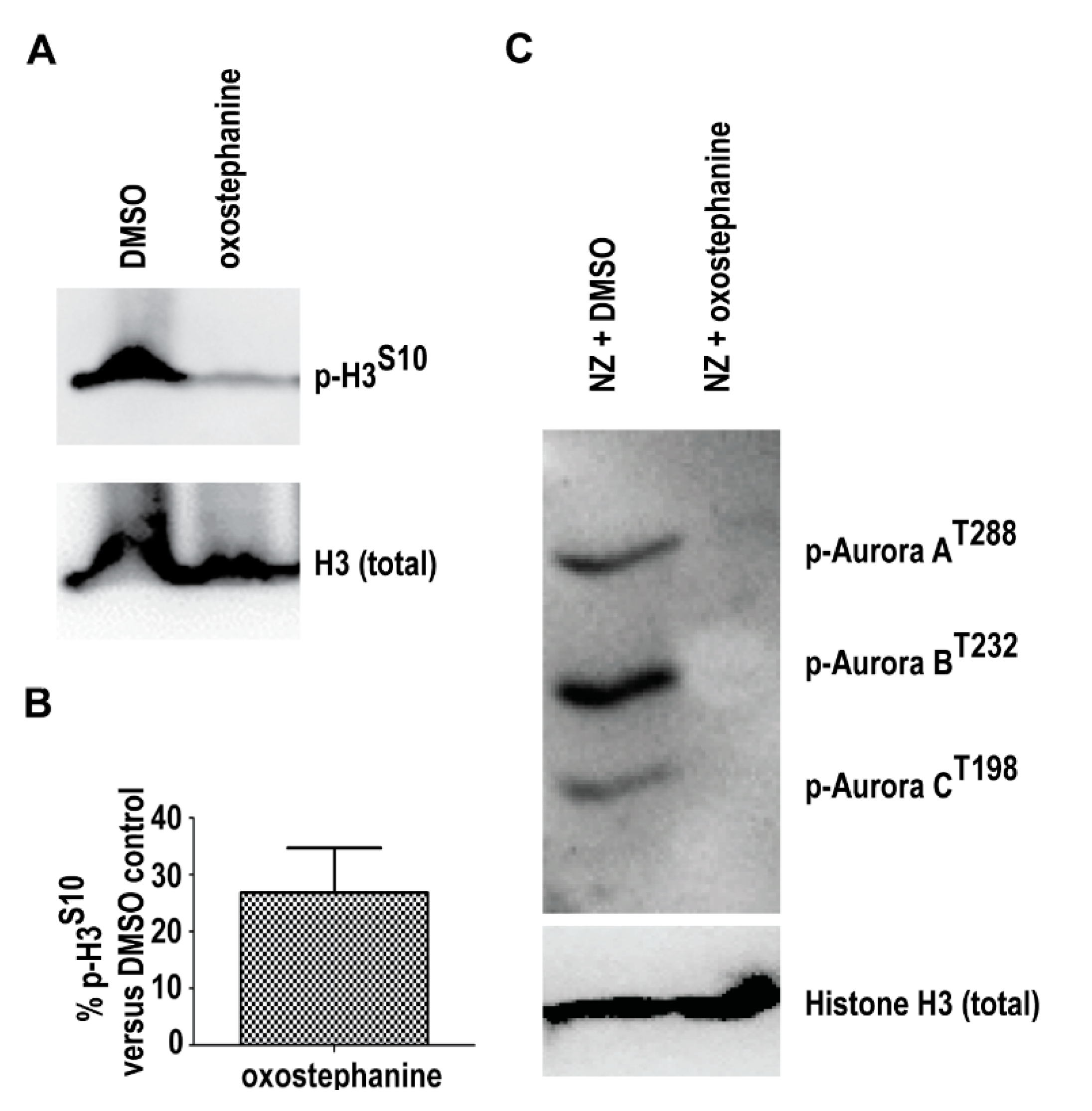
2.4. Oxostephanine Inhibits Aurora Kinase Activity
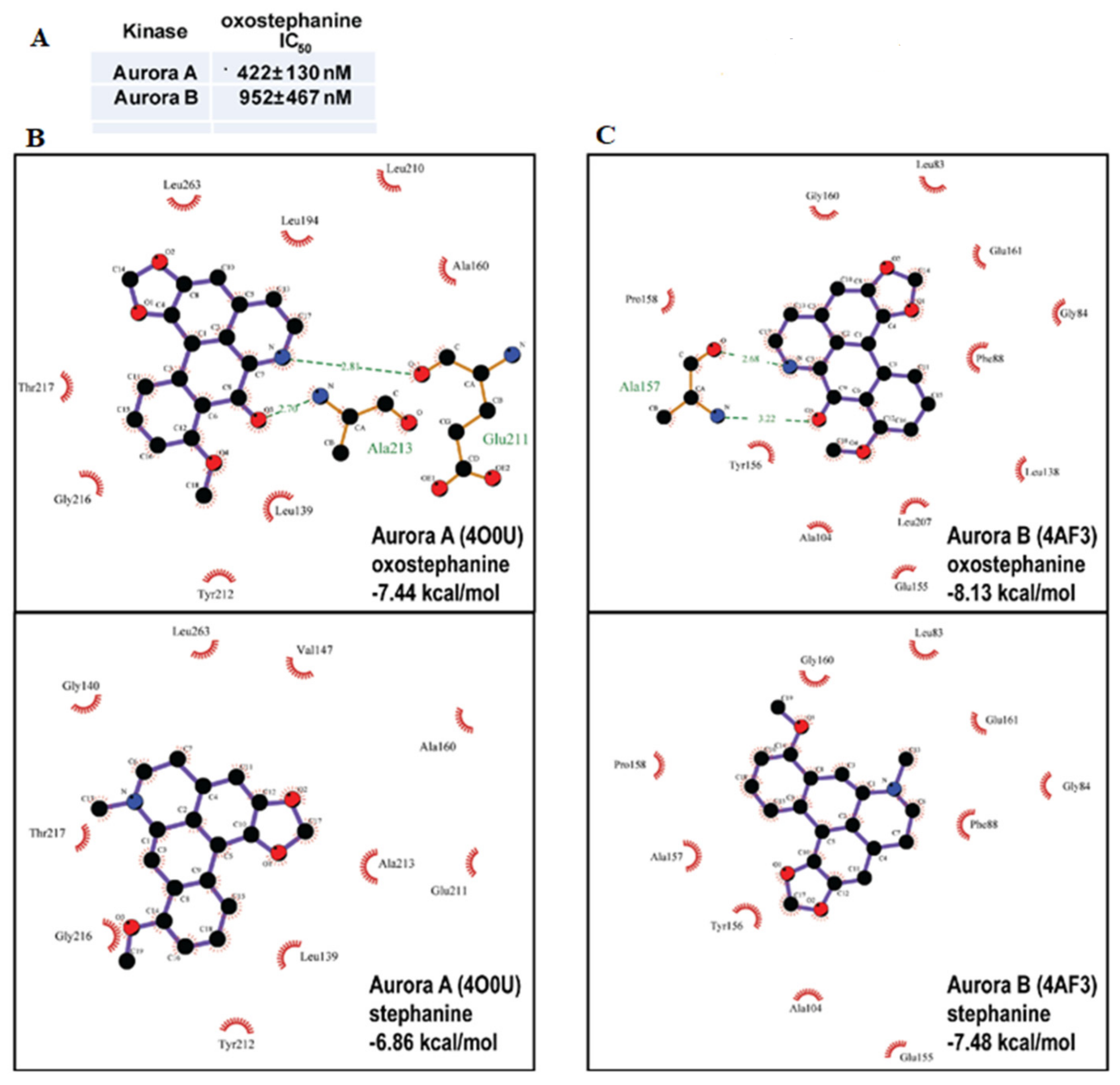
2.5. Oxostephanine Inhibits Aurora Kinase Activity In Vitro and Is Predicted to Bind to the ATP Binding Pocket of Both Aurora A and B
2.6. In Vitro Anti-Plasmodial Activity of Isolated Compounds from S. dielsiana Y.C. Wu
2.7. Thailandine as A Potential Anticancer Agent
3. Material and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Cell Lines and Cell Culture
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Borisa, A.C.; Bhatt, H.G. A comprehensive review on Aurora kinase: Small molecule inhibitors and clinical trial studies. Eur. J. Med. Chem. 2017, 140, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Damodaran, A.P.; Vaufrey, L.; Gavard, O.; Prigent, C. Aurora a kinase is a priority pharmaceutical target for the treatment of cancers. Trends Pharmacol. Sci. 2017, 38, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Lumapat, P.N.; Rogister, B. The functional diversity of Aurora kinases: A comprehensive review. Cell Div. 2018, 13, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quartuccio, S.M.; Schindler, K. Functions of Aurora kinase C in meiosis and cancer. Front. Cell Dev. Biol. 2015, 3, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reininger, L.; Wilkes, J.M.; Bourgade, H.; Miranda-Saavedra, D.; Doerig, C. An essential Aurora-related kinase transiently associates with spindle pole bodies during Plasmodiul falciparum erythrocytic schizogony. Mol. Microbiol. 2011, 79, 205–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, T.G.; Doerig, C.; Reininger, L. Nima- and Aurora-related kinases of malaria parasites. Biochim. Biophys. Acta 2013, 1834, 1336–1345. [Google Scholar] [CrossRef]
- Berry, L.; Chen, C.T.; Reininger, L.; Carvalho, T.G.; El Hajj, H.; Morlon-Guyot, J.; Bordat, Y.; Lebrun, M.; Gubbels, M.J.; Doerig, C.; et al. The conserved apicomplexan Aurora kinase TgArk3 is involved in endodyogeny, duplication rate and parasite virulence. Cell Microbiol. 2016, 18, 1106–1120. [Google Scholar] [CrossRef] [Green Version]
- Patel, G.; Roncal, N.E.; Lee, P.J.; Leed, S.E.; Erath, J.; Rodriguez, A.; Sciotti, R.J.; Pollastri, M.P. Repurposing human Aurora kinase inhibitors as leads for anti-protozoan drug discovery. MedChemComm 2014, 5, 655–658. [Google Scholar] [CrossRef] [Green Version]
- WHO. World Malaria Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Amaratunga, C.; Lim, P.; Suon, S.; Sreng, S.; Mao, S.; Sopha, C.; Sam, B.; Dek, D.; Try, V.; Amato, R.; et al. Dihydroartemisinin–piperaquine resistance in Plasmodium falciparum malaria in Cambodia: A multisite prospective cohort study. Lancet Infect. Dis. 2016, 16, 357–365. [Google Scholar] [CrossRef]
- Phuc, B.Q.; Rasmussen, C.; Duong, T.T.; Dong, L.T.; Loi, M.A.; Ménard, D.; Tarning, J.; Bustos, D.; Ringwald, P.; Galappaththy, G.L.; et al. Treatment failure of dihydroartemisinin/piperaquine for Plasmodium falciparum malaria, Vietnam. Emerg. Infect. Dis. 2017, 23, 715–717. [Google Scholar] [CrossRef] [Green Version]
- Mohammdi, S.; Jafari, B.; Asgharian, P.; Martorell, M.; Sharifi-Rad, J. Medicinal plants used in the treatment of malaria. A key emphasis to Artemisia, Cinchona, Cryptolepis, and Tabebuia genera. Phytother. Res. 2020, 34, 1556–1569. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y. A gift from traditional Chinese medicine to the world (Nobel lecture). Angew. Chem. Int. Ed. Engl. 2016, 55, 10210–10226. [Google Scholar] [CrossRef] [PubMed]
- Tajuddeen, N.; van Heerden, F.R. Antiplasmodial natural products: An update. Malar. J. 2019, 18, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omara, T.; Kiprop, A.K.; Ramkat, R.C.; Cherotoi, J.; Kagoya, S.; Moraa Nyangena, D.; Azeze Tebo, T.; Nteziyaremye, P.; Nyambura Karanja, L.; Jepchirchir, A.; et al. Medicinal plants used in traditional management of cancer in Uganda: A review of ethnobotanical surveys, phytochemistry, and anticancer studies. Evid. Based Complelment. Alternat. Med. 2020, 2020, 3529081. [Google Scholar] [CrossRef] [Green Version]
- McColm, A.A.; Hommel, M.; Trigg, P.I. Inhibition of malaria parasite invasion into erythrocytes pretreated with membrane-active drugs. Mol. Biochem. Parasitol. 1980, 1, 119–127. [Google Scholar] [CrossRef]
- Zhang, D.; Kanakkanthara, A. Beyond the paclitaxel and Vinca alkaloids: Next generation of plant-derived microtubule-targeting agents with potential anticancer activity. Cancers 2020, 12, 1721. [Google Scholar] [CrossRef]
- Pouvelle, B.; Farley, P.J.; Long, C.A.; Taraschi, T.F. Taxol arrests the development of blood-stages Plasmodium falciparum in vitro and Plasmodium chabaudi adami in malaria-infected mice. J. Clin. Investig. 1994, 94, 413–417. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Rawat, P.S.; Cooke, B.M.; Coppel, R.L.; Patankar, S. Cellular effects of curcumin on Plasmodium falciparum include disruption of microtubules. PLoS ONE 2013, 8, e57302. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Ma, Q.; Sun, R.; Cai, J.; Liao, H.; Xu, L.; Xia, J.; Huang, G.; Yao, Y.; Cai, Y.; et al. Dihydroartemisinin inhibits HepG2.2.15 proliferation by inducing cellular senescence and autophagy. BMB Rep. 2019, 52, 520–525. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lu, J.; Chen, Q.; Han, S.; Shao, H.; Chen, P.; Jin, Q.; Yang, M.; Shangguan, F.; Fei, M.; et al. Artemisinin supresses hepatocellular carcinoma cell growth, migration and invasion by targeting cellular bioenetgetics and Hippo-YAP signaling. Arch Toxicol. 2019, 93, 3367–3383. [Google Scholar] [CrossRef]
- Fontinha, D.; Sousa, S.A.; Morais, T.S.; Prundencio, M.; Leitao, J.H.; Le Gal, Y.; Lorcy, D.; Silva, R.A.L.; Velho, M.F.G.; Belo, D.; et al. Gold(III) bis(dithilene) complexes: From molecular conductors to prospective anticancer, antimicrobial and antiplasmodial agents. Metallomics 2020, 12, 974–987. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Lumb, I.; Mehra, V.; Kumar, V. Ferrocene-appended pharmacophores: An exciting approach for moduling biological potential of organic scaffolds. Dalton Trans. 2019, 48, 2840–2860. [Google Scholar] [CrossRef] [PubMed]
- Peter, S.; Aderibigbe, B.A. Ferrocene-based compounds with antimalaria/anticancer activity. Molecules 2019, 24, 3604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradines, B.; Fusai, T.; Daries, W.; Laloge, V.; Rogier, C.; Millet, P.; Panconi, E.; Kombila, M.; Parzy, D. Ferrocene-chloroquine analogues as antimalarial agents: In vitro activity of ferrochloroquine against 103 Gabonese isolates of Plasmodium falciparum. J. Antimicrob. Chemother. 2001, 48, 179–184. [Google Scholar] [CrossRef]
- Henry, M.; Briolant, S.; Fontaine, A.; Mosnier, J.; Baret, E.; Amalvict, R.; Fusai, T.; Fraisse, L.; Rogier, C.; Pradines, B. In vitro activity of ferroquine is independent of polymorphisms in transport protein genes implicated in quinoline resistance in Plasmodium falciparum. Antimicrob. Agents Chemother. 2008, 52, 2755–2759. [Google Scholar] [CrossRef] [Green Version]
- Dubar, F.; Egan, T.J.; Pradines, B.; Kuter, D.; Ncokazi, K.K.; Forge, D.; Paul, J.F.; Pierrot, C.; Kalamou, H.; Khalife, J.; et al. The antimalarial ferroquine: Role of the metal and intramolecular hydrogen bond in activity and resistance. ACS Chem. Biol. 2011, 6, 275–287. [Google Scholar] [CrossRef]
- Kondratskyi, A.; Kondratska, K.; Vanden Abeele, F.; Gordienko, D.; Dubois, C.; Toillon, R.A.; Slomianny, C.; Lemière, S.; Delcourt, P.; Dewailly, E.; et al. Ferroquine, the next generation antimalarial drug, has antitumor activity. Sci. Rep. 2017, 7, 15896. [Google Scholar] [CrossRef] [Green Version]
- Semwal, D.K.; Badoni, R.; Semwal, R.; Kothiyal, S.K.; Singh, G.J.P.; Rawat, U. The genus Stephania (Menispermaceae): Chemical and pharmacological perspectives. J. Ethnopharmacol. 2010, 132, 369–383. [Google Scholar] [CrossRef]
- Blanchfield, J.T.; Sands, D.P.A.; Kennard, C.H.L.; Byriel, K.A.; Kitching, W. Characterisation of alkaloids from some Australian Stephania (Menispermaceae) species. Phytochemistry 2003, 63, 711–720. [Google Scholar] [CrossRef]
- Makarasen, A.; Sirithana, W.; Mogkhuntod, S.; Khunnawutmanotham, N.; Chimnoi, N.; Techasakul, S. Cytotoxic and antimicrobial activities of aporphine alkaloids isolated from Stephania venosa (Blume) Spreng. Planta Med. 2011, 77, 1519–1524. [Google Scholar] [CrossRef] [Green Version]
- Baghdikian, B.; Mahiou-Leddet, V.; Bory, S.; Bun, S.S.; Dumetre, A.; Mabrouki, F.; Hutter, S.; Azas, N.; Ollivier, E. New antiplasmodial alkaloids from Stephania rotunda. J. Ethnopharmacol. 2013, 145, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Chimmoi, N.; Khunnawutmanotham, N.; Feungfuloy, P.; Chatrewongwan, K.; Techasakul, S. Facine isolation and purification of Thailandine, a biologically active oxoaprophine alkaloids, from Stephania venosa leaves using ion-pair liquid-liquid extraction. Res. J. Med. Plant 2013, 7, 68–76. [Google Scholar]
- Le, P.M.; Srivastava, V.; Nguyen, T.T.; Pradines, B.; Madamet, M.; Mosnier, J.; Trinh, T.T.; Lee, H. Stephanine from Stephania venosa (Blume) Spreng Showed Effective Antiplasmodial and Anticancer Activities, the Latter by Inducing Apoptosis through the Reverse of Mitotic Exit. Phytother. Res. 2017, 31, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.A.; Bello, A.; Rubio, L.L.; Rodriguez, C.; Galan, L.; Caudales, E.; Alvarez, J.L. Calcium antagonist properties of the bisbenzylisoquinoline alkaloid cycleanine. Fundarn. Clin. Pharmmol. 1998, 12, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Likhitwitayawuid, K.; Angergofer, C.K.; Chai, H.; Pezzuto, H.M.; Cordell, G.A. Cytotoxic and antimalarial alkaloids from the tubers of Stephania pierrei. J. Nat. Prod. 1993, 56, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.X.; Liang, Y.; Liu, X.B.; Zheng, N.; Xu, W.F.; Li, J.; Yang, R.Y. Aporphine alkaloids from Stephania dielsiana. Chem. Nat. Compd. 2018, 54, 1202–1204. [Google Scholar] [CrossRef]
- Dao, D.T.; Trinh, T.T.; Nguyen, Q.H.; Hoang, V.T.; Le, T.T.D.; Nguyen, T.T. Cytotoxic alkaloids from Stephania dielsiana. Chem. Nat. Compd. 2018, 54, 613–616. [Google Scholar]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)-chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 2017, 11, 781. [Google Scholar] [CrossRef] [Green Version]
- Boyd, M.R. The NCI Human Tumor Cell Line (60-Cell) Screen; Anticancer Drug Development Guide; Humana Press: Totowa, NJ, USA, 2004. [Google Scholar]
- Rodrıguez, M.; Hasegawa, M.; Mendez, J.; Pereira, G.; Arvelob, F. Bioactive oxoaporphine alkaloids from Guatteria Calva. Fitoterapia 1999, 70, 74–76. [Google Scholar]
- Kaestner, P.; Stolz, A.; Bastians, H. Determinants for the efficiency of anticancer drugs targeting either Aurora-A or Aurora-B kinases in human colon carcinoma cells. Mol. Cancer Ther. 2009, 8, 2046–2056. [Google Scholar] [CrossRef] [Green Version]
- Crosio, C.; Fimia, G.M.; Loury, R.; Kimura, M.; Okano, Y.; Zhou, H.; Sen, S.; David Allis, C.; Sassone-Corsi, P. Mitotic phosphorylation of histone H3: Spatio-temporal regulation by mammalian Aurora kinases. Mol. Cell. Biol. 2002, 22, 874–885. [Google Scholar] [CrossRef] [Green Version]
- Donnella, H.J.; Webber, J.T.; Levin, R.S.; Camarda, R.; Momcilovic, O.; Bayani, N.; Shah, K.N.; Korkola, J.E.; Shokat, K.M.; Goga, A.; et al. Kinome rewiring reveals AURKA limits PI3K-pathway inhibitor efficacy in breast cancer. Nat. Chem. Biol. 2018, 14, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Likhitwitayawuid, K.; Dej-adesai, S.; Jongbunprasert, V.; Krungkrai, J. Antimalarials from Stephania venosa, Prismatomeris sessiliflora, Diospyros montana and Murraya siamensis. Planta Med. 1999, 65, 754–756. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, H.C.; Van Schalkwyk, D.A.; Wiehart, U.I.M.; Meredith, A.A.; Egan, J.; Weber, B.W. Antimalarial quinolines and artemisinin inhibits endocytosis in Plasmodium falciparum. Antimicrob. Agents Chemother. 2004, 48, 2370–2378. [Google Scholar] [CrossRef] [Green Version]
- Pisonero-Vaquero, S.; Medina, D.L. Lysosomotropic drugs: Pharmacological tools to study lysosomal function. Curr. Drug Metab. 2017, 18, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, V.; Lee, H. Synthesis and bio-evaluation of novel quinolino-stilbene derivatives as potential anticancer agents. Bioorg. Med. Chem. 2015, 23, 7629–7640. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Lambros, C.; Vanderberg, J.P. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 1979, 65, 418–420. [Google Scholar] [CrossRef]
- Bogreau, H.; Renaud, F.; Bouchiba, H.; Durand, P.; Assi, S.B.; Henry, M.C.; Garnotel, E.; Pradines, B.; Fusao, T.; Wade, B.; et al. Genetic diversity and structure of African Plasmodium falciparum populations in urban and rural areas. Am. J. Trop. Med. Hyg. 2006, 74, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.; Diallo, I.; Bordes, J.; Ka, S.; Pradines, B.; Diatta, B.; M’Baye, P.S.; Sane, M.; Thiam, M.; Gueye, P.M.; et al. Urban malaria in Dakar, Senegal: Chemosusceptibility and genetic diversity of Plasmodium falciparum isolates. Am. J. Trop. Med. Hyg. 2006, 75, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Fall, B.; Madamet, M.; Camara, C.; Amalvict, R.; Fall, M.; Nakoulima, A.; Diatta, B.; Diémé, Y.; Wade, B.; Pradines, B. Plasmodium falciparum in vitro resistance to monodesethylamodiaquine, Dakar, Senegal, 2014. Emerg. Infect. Dis. 2016, 22, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hout, S.; Chea, A.; Bun, S.S.; Elias, R.; Gasquet, M.; Timon-David, P.; Balansard, G.; Azas, N. Screening of selected indigenous plants of Cambodia for antiplasmodial activity. J. Ethnopharmacol. 2006, 107, 12–18. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extracts | IC50 (μg/mL) | |||||||
|---|---|---|---|---|---|---|---|---|
| HeLa | MDA-MB231 | MDA-MB-468 | MCF7 | 184B5 | MCF 10A | 3D7 | W2 | |
| MB2L | 6.1 ± 1.2 | 7.8 ± 1.3 | 3.8 ± 0.5 | 5.9 ± 0.4 | 4.3 ± 1.8 | 8.9 ± 0.9 | NT | NT |
| MB2L-H | >50 | >50 | >50 | >50 | >50 | >50 | NT | NT |
| MB2L-CH | 1.1 ± 0.3 | 1.1 ± 0.6 | 0.6 ± 0.3 | 1.9 ± 0.6 | 2.2 ± 1.2 | 0.8 ± 0.6 | 4.5 ± 0.9 | 5.8 ± 0.4 |
| MB2L-B | 7.5 ± 0.6 | 9.4 ± 1.2 | 1.8 ± 1.0 | 14.4 ± 5.9 | 5.8 ± 0.6 | 12.5 ± 2.4 | 7.9 ± 1.3 | 7.1 ± 0.9 |
| Compounds | IC50 (μM ± SD) | |||||
|---|---|---|---|---|---|---|
| HeLa | MDA-MB-231 | MDA-MB-468 | MCF7 | 184B5 | MCF 10A | |
| Oxostephanine 1 | 1.76 ± 0.20 | 2.67 ± 0.29 | 2.26 ± 0.54 | 4.35 ± 1.20 | 1.66 ± 0.56 | 2.49 ± 0.11 |
| Thailandine 2 | 4.10 ± 0.40 | 7.11 ± 0.07 | 0.78 ± 0.12 | 1.99 ± 1.36 | 3.02 ± 0.10 | 5.01 ± 0.15 |
| Stephanine 3 | 3.33 ± 0.23 | 5.66 ± 0.16 | 7.14 ± 2.11 | 6.49 ± 0.43 | 6.25 ± 0.14 | 7.19 ± 0.33 |
| Crebanine 4 | 48.13 ± 2.38 | 38.94 ± 7.10 | 17.82 ± 4.63 | 30.50 ± 4.89 | 47.44 ± 2.83 | 47.16 ± 4.37 |
| O-methylbulbocapine 5 | 70.37 ± 11.40 | 56.59 ± 9.08 | 48.13 ± 1.69 | 39.36 ± 6.20 | 73.26 ± 0.47 | 59.47 ± 1.90 |
| Palmatine chloride 6 | >100 | >50 | >50 | >50 | >50 | >50 |
| Tetrahydropalmatine 7 | >50 | >50 | >50 | >50 | >50 | >50 |
| Chloroquine | 29.80 ± 0.70 | 28.90 ± 0.40 | 33.20 ± 0.40 | >50 | >50 | 40.50 ± 9.80 |
| Paclitaxel a | 3.81 ± 0.42 | 1.58 ± 0.75 | 3.96 ± 0.32 | 2.71 ± 1.11 | 2.65 ± 1.91 | 1.67 ± 0.21 |
| 184B5 | MCF10A | |||||||
|---|---|---|---|---|---|---|---|---|
| Compounds | Hela | MDA-MB-231 | MDA-MB-468 | MCF7 | HeLa | MDA-MB-231 | MDA-MB-468 | MCF7 |
| Oxostephanine 1 | 0.94 | 0.62 | 0.73 | 0.38 | 1.40 | 0.93 | 1.10 | 0.57 |
| Thailandine 2 | 0.73 | 0.42 | 3.90 | 1.50 | 1.20 | 0.70 | 6.40 | 2.50 |
| Stephanine 3 | 1.88 | 1.10 | 0.88 | 0.96 | 2.16 | 1.27 | 1.01 | 1.11 |
| Crebanine 4 | 0.99 | 1.22 | 2.66 | 1.56 | 0.98 | 1.21 | 2.65 | 1.55 |
| O-Methylbulbocapine 5 | 1.04 | 1.29 | 1.52 | 1.86 | 0.85 | 1.05 | 1.24 | 1.51 |
| Chloroquine | <0.59 | <0.57 | <0.66 | >1 | 1.40 | 1.40 | 1.20 | <0.91 |
| Paclitaxel | 0.7 | 1.67 | 0.67 | 0.98 | 0.44 | 1.10 | 0.42 | 0.62 |
| Compounds | IC50 (μM ± SD) | SI | ||||||
|---|---|---|---|---|---|---|---|---|
| 3D7 | W2 | 184B5 | MCF 10A | 3D7 | W2 | |||
| 184B5 | MCF10A | 184B5 | MCF10A | |||||
| Oxostephanine 1 | 63.91 ± 18.38 | 215.54 ± 31.49 | 1.66 ± 0.56 | 2.49 ± 0.11 | 0.026 | 0.039 | 0.007 | 0.016 |
| Thailandine 2 | 0.24 ± 0.04 | 0.22 ± 0.02 | 3.02 ± 0.10 | 5.01 ± 0.15 | 12.58 | 20.86 | 13.73 | 22.77 |
| Stephanine 3 | 0.69 ± 0.15 | 1.32 ± 0.38 | 6.25 ± 0.14 | 7.19 ± 0.33 | 9.06 | 10.42 | 4.73 | 5.45 |
| Crebanine 4 | 1.56 ± 0.22 | 2.16 ± 0.38 | 47.44 ± 2.83 | 47.16 ± 4.37 | 30.41 | 30.23 | 21.96 | 21.83 |
| O-methylbulbocapine 5 | 2.81 ± 0.40 | 5.71 ± 0.62 | 73.26 ± 0.47 | 59.47 ± 1.90 | 26.07 | 21.16 | 12.83 | 10.42 |
| Palmatine chloride 6 | 1.25 ± 0.28 | 3.19 ± 0.64 | >50 | >50 | >416.6 | >416.6 | >15.67 | >15.67 |
| Tetrahydropalmatine 7 | 275.73 ± 14.43 | 226.09 ± 19.84 | >50 | >50 | >0.18 | >0.18 | >0.22 | >0.22 |
| Chloroquine a | 0.021 ± 0.005 | 0.38 ± 0.03 | >50 | 40.50 ± 9.80 | >2380 | 1928 | >131.6 | 106.50 |
| Mefloquine a | 0.052 ± 0.009 | 0.022 ± 0.006 | 10.80 ± 0.75 | NT | 207.6 | NA | 490.9 | NA |
| Dihydroartemisinin a | 0.002 ± 0.001 | 0.002 ± 0.001 | NT | NT | NA | NA | NA | NA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knockleby, J.; Pradines, B.; Gendrot, M.; Mosnier, J.; Nguyen, T.T.; Trinh, T.T.; Lee, H.; Le, P.M. Cytotoxic and Anti-Plasmodial Activities of Stephania dielsiana Y.C. Wu Extracts and the Isolated Compounds. Molecules 2020, 25, 3755. https://doi.org/10.3390/molecules25163755
Knockleby J, Pradines B, Gendrot M, Mosnier J, Nguyen TT, Trinh TT, Lee H, Le PM. Cytotoxic and Anti-Plasmodial Activities of Stephania dielsiana Y.C. Wu Extracts and the Isolated Compounds. Molecules. 2020; 25(16):3755. https://doi.org/10.3390/molecules25163755
Chicago/Turabian StyleKnockleby, James, Bruno Pradines, Mathieu Gendrot, Joel Mosnier, Thanh Tam Nguyen, Thi Thuy Trinh, Hoyun Lee, and Phuong Mai Le. 2020. "Cytotoxic and Anti-Plasmodial Activities of Stephania dielsiana Y.C. Wu Extracts and the Isolated Compounds" Molecules 25, no. 16: 3755. https://doi.org/10.3390/molecules25163755