
Inequality in the Frequency of the Open States Occurrence Depends on Single 2H/1H Replacement in DNA

 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Mathematical Model
- —is the angular deflection of the i-th nitrogen base of the j-th chain counted counterclockwise at time t;
- —is the rotational inertia of the i-th nitrogen base of the j-th chain;
- —is the distance between the center of inertia of the i-th nitrogen base of the j-th chain to sugar phosphate chain;
- —is the constant characterizing the torsion moment of the i-th segment of the j-th sugar phosphate chain;
- —is the constant characterizing the bond elastic properties of the i-th nitrogen base pairs;
- —external influence on the i-th nitrogen base of the j-th chain at a time ,
- —is the number of nitrogene base pairs in the system.
2.2. The Effect of 2H/1H Exchange on the Probability of OS Formation
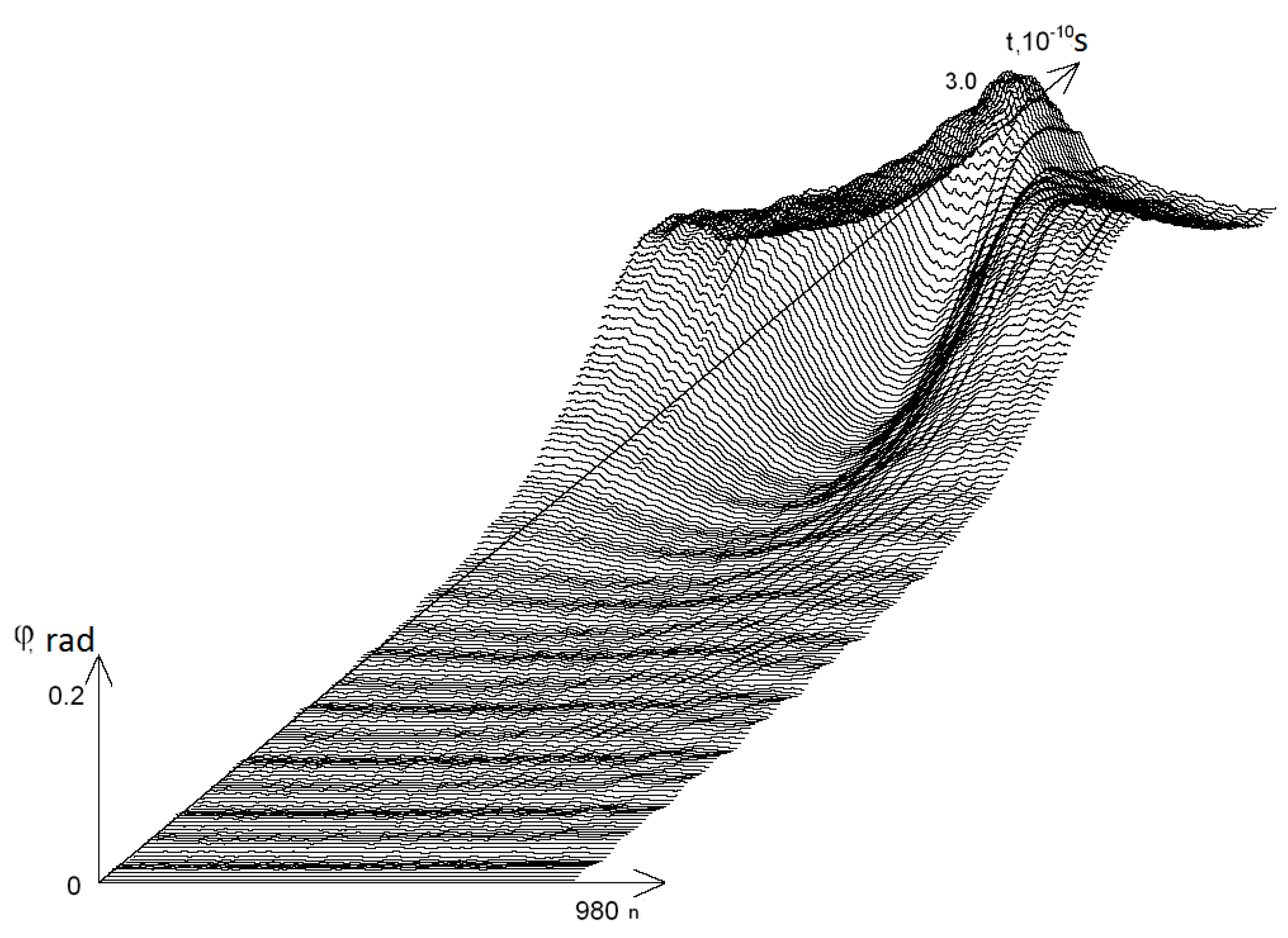
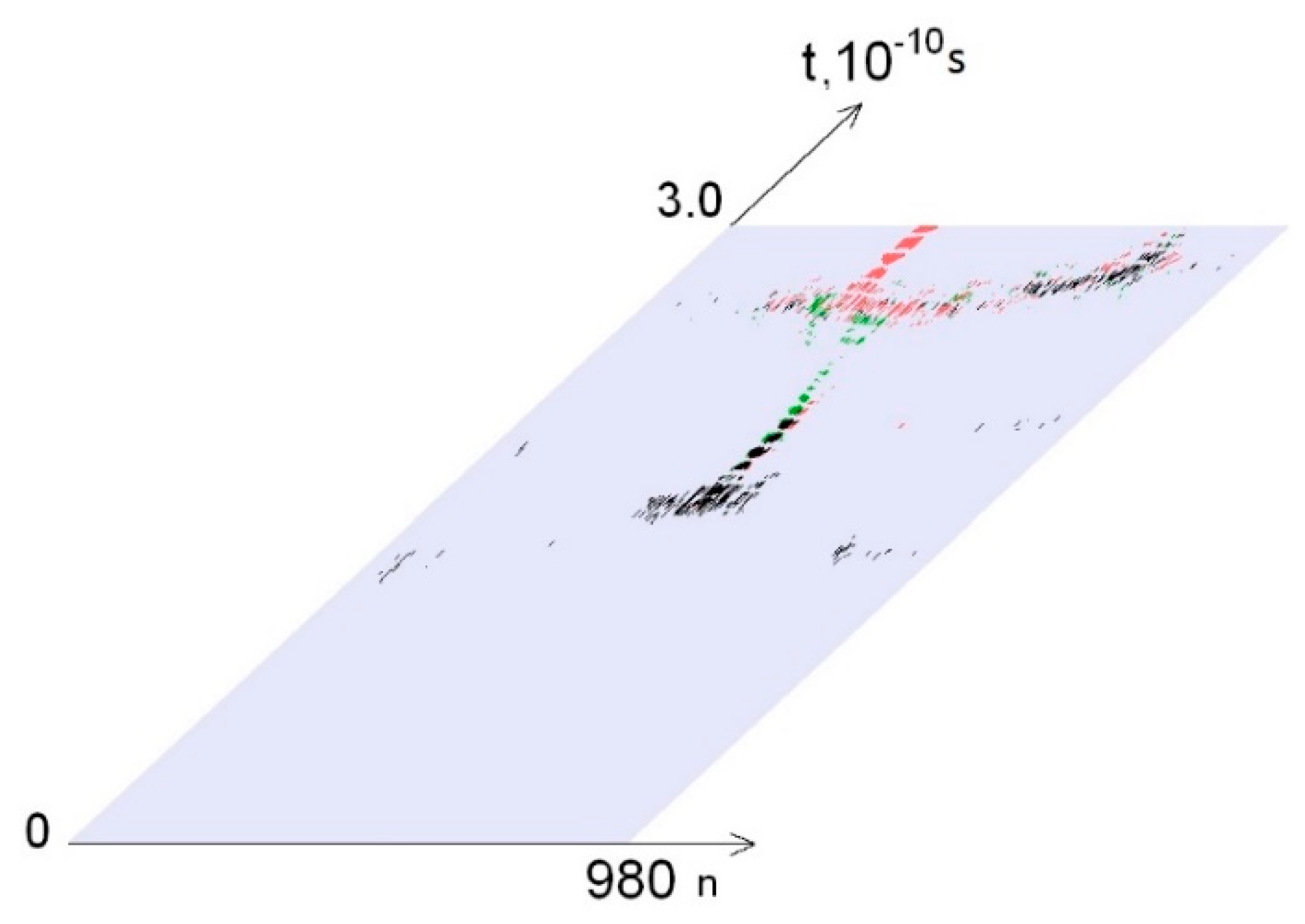
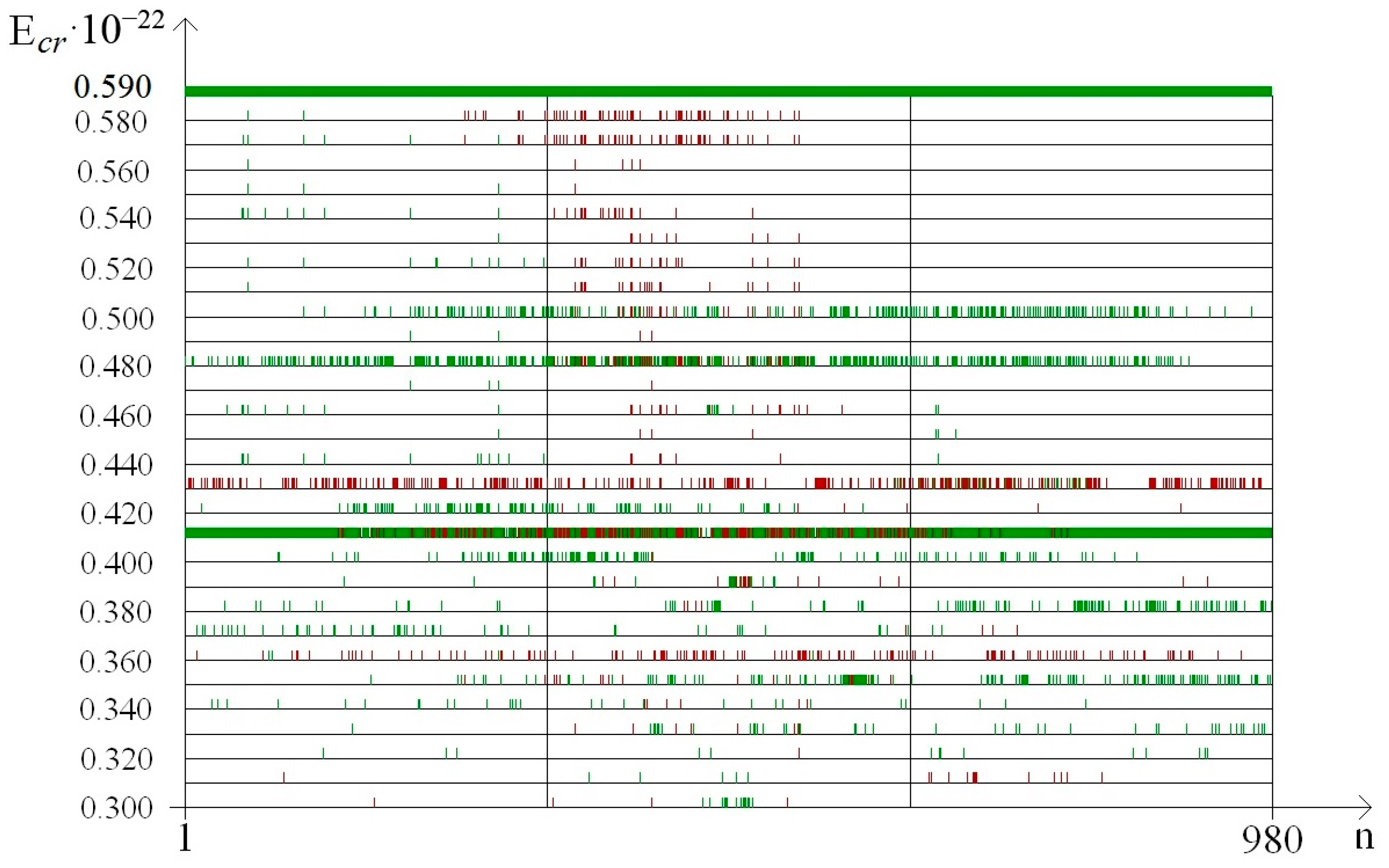
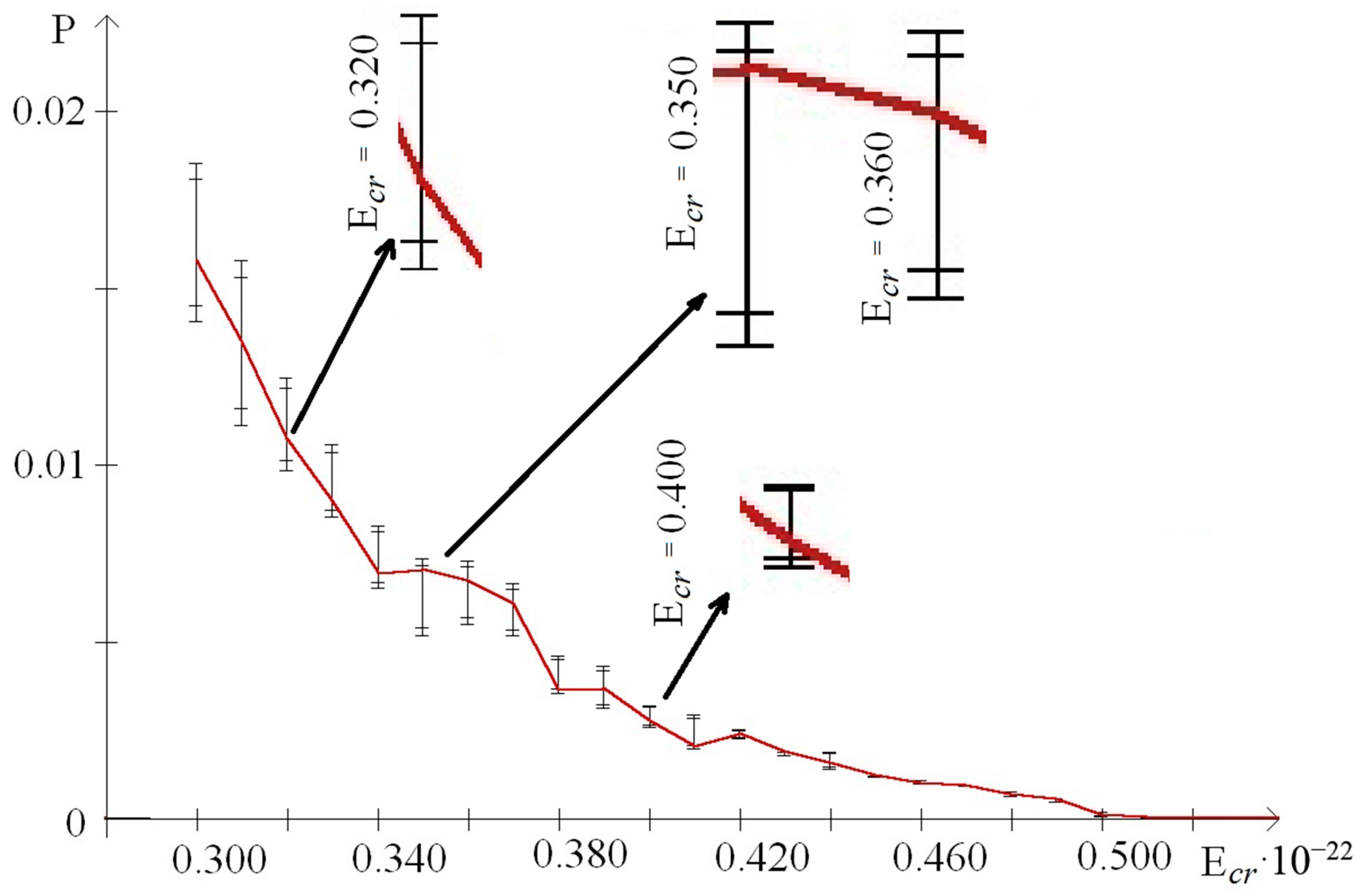
3. Results
- Decile method:
- (1)
- i ϵ range “Maximum” (top 10th decile, Dec10):Pi ≥ Pimax − 1/10·(Pimax − Pimin) ⇒ nmax = ƩnPi;
- (2)
- i ϵ range “Minimum” (bottom 1st decile, Dec1):Pi ≤ Pimin + 1/10·(Pimax − Pimin) ⇒ nmin = ƩnPi.
- Quartile method*:
- (1)
- i ϵ range “Maximum” (1st quartile in range “Maximum”, Q1-max):Pi ≥ Pimax − 1/4·(Pimax − P0) ⇒ nmax = ƩnPi;
- (2)
- i ϵ range “Minimum” (4th quartile in range “Minimum”, Q4-min):Pi ≤ Pimin + 1/4·(P0 − Pimin) ⇒ nmin = ƩnPi.
*Note: mathematical description of other quartiles:- i ϵ range Q2-max: Pimax − 1/4·(Pimax − P0) > Pi ≥ Pimax − 1/2·(Pimax − P0);
- i ϵ range Q3-max: Pimax − 1/2·(Pimax − P0) > Pi ≥ Pimax − 3/4·(Pimax − P0);
- i ϵ range Q4-max: Pimax − 3/4·(Pimax − P0) > Pi > P0;
- i ϵ range Q1-min: Pimin + 3/4·(P0 − Pimin) < Pi < P0;
- i ϵ range Q2-min: Pimin + 1/2·(P0 − Pimin) < Pi ≤ Pimin + 3/4·(P0 − Pimin);
- i ϵ range Q3-min: Pimin + 1/4·(P0 − Pimin) < Pi ≤ Pimin + 1/2·(P0 − Pimin).
- Basov-Jimack algorithm (BJ-algorithm):
- (1)
- i ϵ range “Maximum” (BJ-max):
- if Pimax − 1/10·(Pimax − Pimin) ≥ P0 + 1/2·(Pimax − P0) and Pimax > P0 ≥ Pimin ≥ 0: Pi ≥ Pimax − 1/10·(Pimax − Pimin) ⇒ nmax = ƩnPi; or else:
- if Pimax − 1/10·(Pimax − Pimin) < P0 + 1/2·(Pimax − P0) and Pimax > P0 ≥ Pimin ≥ 0: Pi ≥ Pimax − 1/4·(Pimax − P0) ⇒ nmax = ƩnPi;
- (2)
- i ϵ range “Minimum” (BJ-min):
- if Pimin + 1/10·(Pimax − Pimin) ≤ P0 − 1/2·(P0 − Pimin), Pimax ≠ Pimin, and ƩPi ϵ Q2-Q4-min > 0: Pi ≤ Pimin + 1/10·(Pimax − Pimin) ⇒ nmin = ƩnPi; or else:
- if Pimin + 1/10·(Pimax − Pimin) > P0 − 1/2·(P0 − Pimin), Pimax ≠ Pimin, and ƩPi ϵ Q2-Q4-min > 0: Pi ≤ Pimin + 1/4·(P0 − Pimin) ⇒ nmin = ƩnPi; or else:
- if Pimax = Pimin = ƩPi ϵ Q2-Q4-min = Pi = 0: nmin = 0;
where: nmin is nitrogen base pair numbers (i), which were included in the range “Minimum”; nmax is number of i, which was included in the range “Maximum”; if Pi ≤ Pimin + 3/4·(P0 − Pimin): i ϵ range from Q2-min to Q4-min (Q2-Q4-min).
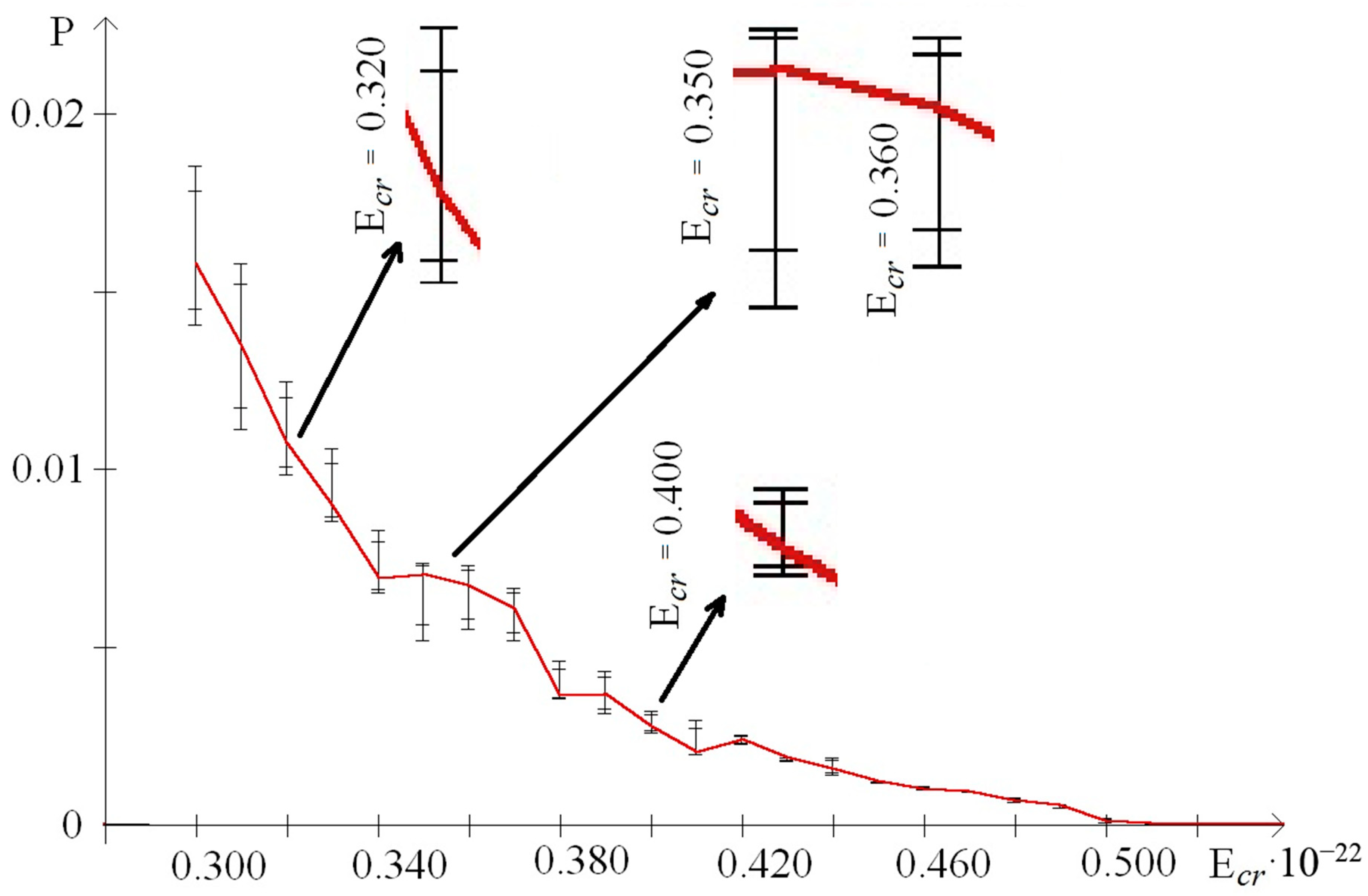
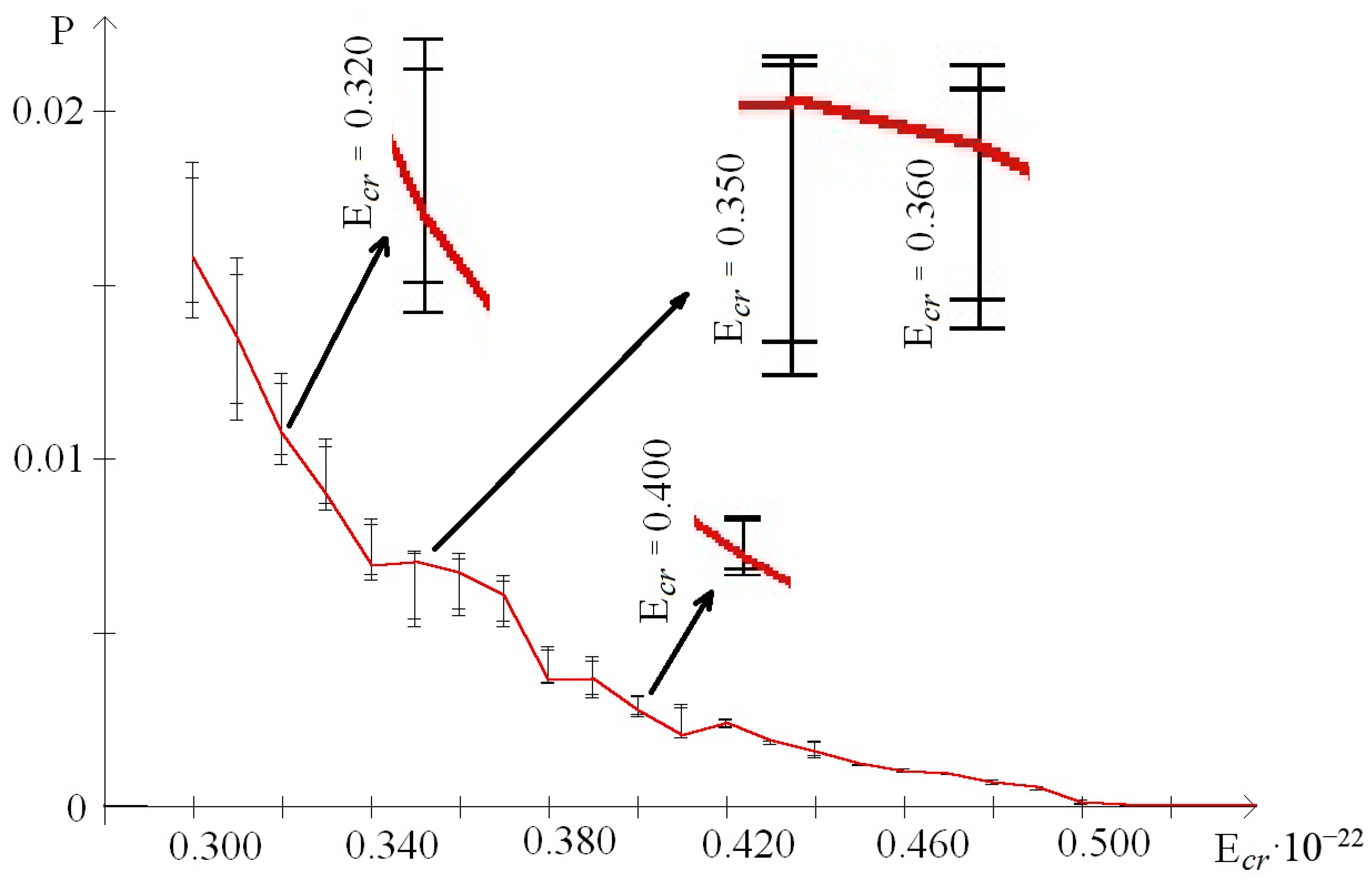
- from 0.44 to 0.59 that were selected because of III part, which in this range had summary nmax equaled 0; its nmax, measured by the each approach: decile method, quartile method, or BJ-algorithm, always equaled 0 (Figure 3).
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Somlyai, G.; Somlyai, I.; Fórizs, I.; Czuppon, G.; Papp, A.; Molnár, M. Effect of systemic subnormal deuterium level on metabolic syndrome related and other blood parameters in humans: A preliminary study. Molecules 2020, 25, 1376. [Google Scholar] [CrossRef] [Green Version]
- Basov, A.A.; Fedulova, L.V.; Baryshev, M.G.; Dzhimak, S.S. Deuterium-depleted water influence on the isotope 2H/1H regulation in body and individual adaptation. Nutrients 2019, 11, 1903. [Google Scholar] [CrossRef] [Green Version]
- Halenova, T.; Zlatskiy, I.; Syroeshkin, A.; Maximova, T.; Pleteneva, T. Deuterium-depleted water as adjuvant therapeutic agent for treatment of diet-induced obesity in rats. Molecules 2020, 25, 23. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-Q.; Zhao, C.-R.; Xiong, A.-B. Effect of deuterium depleted water combined with platelet-rich plasma on wound healing of diabetic ulcer in rats. J. Shanghai Jiaotong Univ. (Med. Sci.) 2020, 40, 326–332. [Google Scholar]
- Volchenko, N.N.; Samkov, A.A.; Malyshko, V.V.; Khudokormov, A.A.; Moiseev, A.V.; Elkina, A.A.; Baryshev, M.G.; Pershin, S.M. Influence of the Environmental Isotope Composition Modification on Growth and Metabolic Activity of Rhodococcus and Saccharomyces. Biol. Bull. 2020, 47, 326–330. [Google Scholar] [CrossRef]
- Zlatskiy, I.A.; Zlatska, A.V.; Antipova, N.V.; Dolenko, S.A.; Gordiienko, I.M.; Gubar, O.S.; Vasyliev, R.G.; Zubov, D.A.; Novikova, S.N.; Syroeshkin, A.V. Comparative analysis of the different dyes’ potential to assess human normal and cancer cell viability in vitro under different d/h ratios in a culture medium. Sci. World J. 2020, 2020, 1. [Google Scholar] [CrossRef] [Green Version]
- Yavari, K.; Kooshesh, L. Deuterium Depleted Water Inhibits the Proliferation of Human MCF7 Breast Cancer Cell Lines by Inducing Cell Cycle Arrest. Nutr. Cancer 2019, 71, 1019–1029. [Google Scholar] [CrossRef]
- Zlatska, A.; Vasyliev, R.G.; Gordiienko, I.M.; Rodnichenko, A.E.; Morozova, M.A.; Vulf, M.A.; Zubov, D.O.; Novikova, S.N.; Litvinova, L.S.; Grebennikova, T.; et al. Effect of the deuterium on efficiency and type of adipogenic differentiation of human adipose-derived stem cells in vitro. Sci. Rep. 2020, 10, 5217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasooli, A.; Fatemi, F.; Hajihosseini, R.; Vaziri, A.; Akbarzadeh, K.; Mohammadi Malayeri, M.R.; Dini, S.; Foroutanrad, M. Synergistic effects of deuterium depleted water and Mentha longifolia L. essential oils on sepsis-induced liver injuries through regulation of cyclooxygenase-2. Pharm. Biol. 2019, 57, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatemi, F.; Golbodagh, A.; Hojihosseini, R.; Dadkhah, A.; Akbarzadeh, K.; Dini, S.; Malayeri, M.R.M. Anti-inflammatory effects of deuterium-depleted water plus rosa damascena mill. Essential oil via cyclooxygenase-2 pathway in rats. Turk. J. Pharm. Sci. 2020, 17, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Avila, D.S.; Somlyai, G.; Somlyai, I.; Aschner, M. Anti-aging effects of deuterium depletion on Mn-induced toxicity in a C. elegans model. Toxicol. Lett. 2012, 211, 319–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzhimak, S.S.; Basov, A.A.; Elkina, A.A.; Fedulova, L.V.; Kotenkova, E.A.; Vasilevskaya, E.R.; Lyasota, O.M.; Baryshev, M.G. Influence of deuterium-depleted water on hepatorenal toxicity. Jundishapur J. Nat. Pharm. Prod. 2018, 13, e69557. [Google Scholar] [CrossRef]
- Rehakova, R.; Klimentova, J.; Cebova, M.; Barta, A.; Matuskova, Z.; Labas, P.; Pechanova, O. Effect of Deuterium-Depleted Water on Selected Cardiometabolic Parameters in Fructose-Treated Rats. Physiol. Res. 2016, 65, S401–S407. [Google Scholar] [CrossRef]
- Gaponov, N.V.; Svistunov, S.V.; Bondarenko, N.N.; Romanenko, I.A. Effect of deuterium water on blood values and digestibility of nutrients of rhesus macaque. Bull. Natl. Acad. Sci. Repub. Kazakhstan 2020, 2, 22–28. [Google Scholar] [CrossRef]
- Kozin, S.V.; Kravtsov, A.A.; Elkina, A.A.; Zlishcheva, E.I.; Barysheva, E.V.; Shurygina, L.V.; Moiseev, A.V.; Baryshev, M.G. Isotope exchange of deuterium for protium in rat brain tissues changes brain tolerance to hypoxia. Biophysics 2019, 64, 272–278. [Google Scholar] [CrossRef]
- Wu, Y.; Qin, D.; Yang, H.; Wang, W.; Xiao, J.; Zhou, L.; Fu, H. Neuroprotective Effects of Deuterium-Depleted Water (DDW) Against H2O2-Induced Oxidative Stress in Differentiated PC12 Cells Through the PI3K/Akt Signaling Pathway. Neurochem. Res. 2020, 45, 1034–1044. [Google Scholar] [CrossRef]
- Basov, A.A.; Kozin, S.V.; Bikov, I.M.; Popov, K.A.; Moiseev, A.V.; Elkina, A.A.; Dzhimak, S.S. Changes in prooxidant-antioxidant system indices in the blood and brain of rats with modelled acute hypoxia which consumed a deuterium-depleted drinking diet. Biol. Bull. 2019, 46, 531–535. [Google Scholar] [CrossRef]
- Basov, A.A.; Fedulova, L.V.; Vasilevskaya, E.R.; Dzhimak, S.S. Possible mechanisms of biological effects observed in living systems during 2H/1H isotope fractionation and deuterium interactions with other biogenic isotopes. Molecules 2019, 24, 4101. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Gaetani, M.; Chernobrovkin, A.; Zubarev, R.A. Anticancer effect of deuterium depleted water—Redox disbalance leads to oxidative stress. Mol. Cell. Proteom. 2019, 18, 2373–2387. [Google Scholar] [CrossRef] [Green Version]
- Pomytkin, I.A.; Kolesova, O.E. Relationship between natural concentration of heavy water isotopologs and rate of H2O2 generation by mitochondria. Bull. Exp. Biol. Med. 2006, 142, 570–572. [Google Scholar] [CrossRef]
- Basov, A.A.; Elkina, A.A.; Samkov, A.A.; Volchenko, N.N.; Moiseev, A.V.; Fedulova, L.V.; Baryshev, M.G.; Dzhimak, S.S. Influence of deuterium depleted water on the isotope D/H composition of liver tissue and morphological development of rats at different periods of ontogenesis. Iran. Biomed. J. 2019, 23, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Boros, L.G.; D’Agostino, D.P.; Katz, H.E.; Roth, J.P.; Meuillet, E.J.; Somlyai, G. Submolecular regulation of cell transformation by deuterium depleting water exchange reactions in the tricarboxylic acid substrate cycle. Med. Hypotheses 2016, 87, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Stanga, S.; Caretto, A.; Boido, M.; Vercelli, A. Mitochondrial Dysfunctions: A Red Thread across Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 3719. [Google Scholar] [CrossRef]
- Pedersen, L.G.; Bartolotti, L.; Li, L. Deuterium and its role in the machinery of evolution. J. Theor. Biol. 2006, 238, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Reaser, P.B.; Burch, G.E. Determination of deuterium oxide in water by measurement of freezing point. Science 1958, 128, 415–416. [Google Scholar] [CrossRef]
- Parmon, V.N. On the possibility of observing kinetic isotopic effects in the life cycles of living organisms at ultralow concentrations of deuterium. Her. Russ. Acad. Sci. 2015, 85, 170–172. [Google Scholar] [CrossRef]
- Shigaev, A.S.; Ponomarev, O.A.; Lakhno, V.D. Theoretical and experimental investigations of DNA open states. Math. Biol. Bioinform. 2013, 8, 553–664. [Google Scholar] [CrossRef] [Green Version]
- Manghi, M.; Destainville, N. Physics of base-pairing dynamics in DNA. Phys. Rep. 2016, 631, 1–41. [Google Scholar] [CrossRef] [Green Version]
- Vologodskii, A.; Frank-Kamenetskii, M.D. DNA melting and energetics of the double helix. Phys. Life Rev. 2018, 25, 1–21. [Google Scholar] [CrossRef]
- Yakushevich, L.V.; Krasnobaeva, L.A. Forced Oscillations of DNA Bases. Biophysics 2016, 61, 241–250. [Google Scholar] [CrossRef]
- Grinevich, A.A.; Yakushevich, L.V. The influence of the DNA torque on the dynamics of transcription bubbles in plasmid PTTQ18. J. Theor. Biol. 2018, 453, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Drobotenko, M.I.; Dzhimak, S.S.; Svidlov, A.A.; Basov, A.A.; Lyasota, O.M.; Baryshev, M.G. A Mathematical Model for Basepair Opening in a DNA Double Helix. Biophysics 2018, 63, 177–182. [Google Scholar] [CrossRef]
- Grinevich, A.A.; Yakushevich, L.V. On the modeling of the motion of a transcription bubble under constant torque. Biophysics 2016, 61, 539–546. [Google Scholar] [CrossRef]
- Dzhimak, S.S.; Drobotenko, M.I.; Basov, A.A.; Svidlov, A.A.; Barishev, M.G. Mathematical modeling of open states in a double-stranded DNA molecule depending on 2H/1H ratio. Math. Biol. Bioinform. 2019, 14, 612–624. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, B.L. Basic Statistics; New Age International: New Delhi, India, 2009; p. 832. [Google Scholar]
- Coman, D.; Russu, I.M. Probing hydrogen bonding in a DNA triple helix using protium-deuterium fractionation factors. J. Am. Chem. Soc. 2003, 125, 6626–6627. [Google Scholar] [CrossRef]
- Yakushevich, L.V.; Krasnobaeva, L.A. Trajectories of the DNA kinks in the sequences containing CDS regions. Math. Biol. Bioinf. 2017, 12, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Frank-Kamenetskiia, M.D.; Prakash, S. Fluctuations in the DNA double helix: A critical review. Phys. Life Rev. 2014, 11, 153–170. [Google Scholar] [CrossRef]
- Krueger, A.; Protozanova, E.; Frank-Kamenetskii, M.D. Sequence-dependent basepair opening in DNA double helix. Biophys. J. 2006, 90, 3091–3099. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Bushnell, D.A.; Kornberg, R.D. Lock and key to transcription: Sigma-DNA interaction. Cell 2011, 147, 1218–1219. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, V.; Villa, A.; Hess, B. Sequence dependency of canonical base pair opening in the DNA double helix. PLoS Comput. Biol. 2017, 13, e1005463. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Yang, W.; Karplus, M.; Verdine, G.L. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damages DNA. Nature 2005, 434, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Lariviere, L.; Morera, S. A base-flipping mechanism for the T4 phage beta-glucosyltransferase and identification of a transition-state analog. J. Mol. Biol. 2002, 324, 483–490. [Google Scholar] [CrossRef]
- Cao, C.; Jiang, Y.L.; Stivers, J.T.; Song, F. Dynamic opening of DNA during the enzymatic search for a damaged base. Nat. Struct. Mol. Biol. 2004, 11, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Roberts, R.J. AdoMet-dependent methylation, DNA methyltransferases and base flipping. Nucleic Acids Res. 2001, 29, 3784–3795. [Google Scholar] [CrossRef] [PubMed]
- Grinevich, A.A.; Ryasik, A.A.; Yakushevich, L.V. Motion of DNA open states influenced by random force. Comput. Res. Modeling 2015, 7, 1295–1307. [Google Scholar] [CrossRef]
- Kantorovitz, M.R.; Rapti, Z.; Gelev, V.; Usheva, A. Computing DNA duplex instability profiles efficiently with a two-state model: Trends of promoters and binding sites. BMC Bioinform. 2010, 11, 604. [Google Scholar] [CrossRef] [Green Version]
- Metzler, R.; Ambjornsson, T.; Hanke, A.; Fogedby, H.C. Single DNA denaturation and bubble dynamics. J. Phys. Condens. Matter 2009, 21, 14. [Google Scholar] [CrossRef]
- Van Erp, T.S.; Cuesta-López, S.; Peyrard, M. Bubbles and denaturation in DNA. Eur. Phys. J. E 2006, 20, 421–434. [Google Scholar] [CrossRef]
- Yakushevich, L.V.; Krasnobaeva, L.A. Ensemble of DNA Kinks. EPJ Web Conf. 2019, 224, 03005. [Google Scholar] [CrossRef] [Green Version]
- Sontz, P.A.; Mui, T.P.; Fuss, J.O.; Tainer, J.A.; Barton, J.K. DNA charge transport as a first step in coordinating the detection of lesions by repair proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 1856–1861. [Google Scholar] [CrossRef] [Green Version]
- Wagenknecht, H.-A. Electron transfer processes in DNA: Mechanisms, biological relevance and applications in DNA analytics. Nat. Prod. Rep. 2006, 23, 973–1006. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Tainer, J.A. DNA repair without flipping out. Nature 2015, 527, 168–169. [Google Scholar] [CrossRef] [PubMed]
- Lonsdorf, B. Professor Frisby’s Mostly Adequate Guide to Functional Programming. Available online: https://drboolean.gitbooks.io/mostly-adequate-guide/content/ (accessed on 20 April 2020).
- Sicard, F.; Destainville, N.; Manghi, M. DNA denaturation bubbles: Free-energy landscape and nucleation/closure rates. J. Chem. Phys. 2015, 142, 034903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priyakumar, U.D.; MacKerell, A.D. Computational approaches for investigating base flipping in oligonucleotides. Chem. Rev. 2006, 106, 489–505. [Google Scholar] [CrossRef] [PubMed]
- Dow, B.J.; Malik, S.S.; Drohat, A.C. Defining the role of nucleotide flipping in enzyme specificity using 19F NMR. J. Am. Chem. Soc. 2019, 141, 4952–4962. [Google Scholar] [CrossRef]
- Hong, S.; Cheng, X. DNA base flipping: A general mechanism for writing, reading, and erasing DNA modifications. Adv. Exp. Med. Biol. 2016, 945, 321–341. [Google Scholar] [CrossRef] [Green Version]
- Grosjean, H. DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution; CRC Press: Boca Raton, FL, USA, 2018; p. 682. [Google Scholar] [CrossRef]
- Poptsova, M.S.; Il’Icheva, I.A.; Nechipurenko, D.Y.; Panchenko, L.A.; Khodikov, M.V.; Oparina, N.Y.; Polozov, R.V.; Nechipurenko, Y.D.; Grokhovsky, S.L. Non-random DNA fragmentation in next-generation sequencing. Sci. Rep. 2014, 4, 4532. [Google Scholar] [CrossRef] [Green Version]
- Dubois, A.; Francois, C.; Descamps, V.; Fournier, C.; Wychowski, C.; Dubuisson, J.; Castelain, S.; Duverlie, G. Enhanced anti-HCV activity of interferon alpha 17 subtype. Virology 2009, 6, 70. [Google Scholar] [CrossRef] [Green Version]
- GenBank: Homo Sapiens Interferon α 17 (IFNA17), mRNA. Available online: http://www.ncbi.nlm.nih.gov/nuccore/NM_021268.2 (accessed on 29 March 2020).
- Hwa, T.; Marinari, E.; Sneppen, K.; Tang, L. Localization of denaturation bubbles in random DNA sequences. Proc. Natl. Acad. Sci. USA 2003, 100, 4411–4416. [Google Scholar] [CrossRef] [Green Version]
- Brovarets, O.O.; Tsiupa, K.S.; Hovorun, D.M. Surprising Conformers of the Biologically Important A·T DNA Base Pairs: QM/QTAIM Proofs. Front. Chem. 2018. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds “ECM program “Calculation of nonlinear excitations in a DNA molecule taking into account D/H exchange”” are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Base | ||||
|---|---|---|---|---|
| 7.61 | 4.86 | 8.22 | 4.11 | |
| R, Å | 5.80 | 4.80 | 5.70 | 4.70 |
| , | 2.35 | 1.61 | 2.27 | 1.54 |
| 6.20 | 6.20 | 9.60 | 9.60 | |
| 4.25 | 2.91 | 4.10 | 2.79 |
| m | P0 | imin, or nCS | imax | ||
|---|---|---|---|---|---|
| 0.30 | 0.0158002 | 498 | 544 | 0.0140623 | 0.0185293 |
| 0.31 | 0.0135005 | 365 | 711 | 0.0111363 | 0.0157753 |
| 0.32 | 0.0107263 | 475 | 555 | 0.0098598 | 0.0124513 |
| 0.33 | 0.0089972 | 858 | 405 | 0.0085322 | 0.0105687 |
| 0.34 | 0.0069408 | 299 | 435 | 0.0065120 | 0.0082811 |
| 0.35 | 0.0070181 | 512 | 383 | 0,0051732 | 0.0073687 |
| 0.36 | 0.0067221 | 80 | 459 | 0.0054978 | 0.0072946 |
| 0.37 | 0.0060742 | 102 | 651 | 0.0051911 | 0.0066520 |
| 0.38 | 0.0036320 | 191 | 451 | 0.0035676 | 0.0046222 |
| 0.39 | 0.0036706 | 553 | 506 | 0.0031209 | 0.0043160 |
| 0.40 | 0.0027459 | 688 | 422 | 0.0026089 | 0.0032148 |
| 0.41 | 0.0020461 | 284 | 487 | 0.0019833 | 0.0029466 |
| 0.42 | 0.0024118 | 526 | 341 | 0.0022690 | 0.0025324 |
| 0.43 | 0.0018781 | 676 | 509 | 0.0018024 | 0.0018882 |
| 0.44 | 0.0015812 | 284 | 429 | 0.0014226 | 0.0018898 |
| 0.45 | 0.0012137 | 678 | 422 | 0.0011902 | 0.0012356 |
| 0.46 | 0.0010036 | 284 | 422 | 0.0009866 | 0.0010921 |
| 0.47 | 0.0009419 | 284 | 422 | 0.0009282 | 0.0009590 |
| 0.48 | 0.0006775 | 284 | 422 | 0.0006355 | 0.0007785 |
| 0.49 | 0.0005409 | 284 | 422 | 0.0004716 | 0.0005861 |
| 0.50 | 0.0001030 | 284 | 422 | 0.0000739 | 0.0002028 |
| 0.51 | 0.0000386 | 58 | 422 | 0.0000297 | 0.0000598 |
| 0.52 | 0.0000279 | 284 | 395, 403, 404, 411,422, 513 | 0.0000273 | 0.0000284 |
| 0.53 | 0.0000252 | 284 | 411, 422 | 0.0000243 | 0.0000261 |
| 0.54 | 0.0000151 | 52, 53, 58, 73, 93,108, 127, 204, 284 | 334, 345, 353, 358, 359, 361, 362, 376, 378, 383, 388, 389, 392, 395, 403,404, 411, 444, 513 | 0.0000148 | 0.0000153 |
| 0.55 | 0.0000142 | 58, 108, 284 | 353 | 0.0000138 | 0.0000145 |
| 0.56 | 0.0000132 | 58 | 353, 395, 404, 411 | 0.0000127 | 0.0000135 |
| 0.57 | 0.0000120 | 58 | 353, 395, 403, 404, 411 | 0.0000114 | 0.0000124 |
| 0.58 | 0.0000104 | 58 | 353 | 0.0000090 | 0.0000111 |
| 0.59 | 0.0000000 | nCS = 980 | - | 0.0000000 | - |
| S | F | ||
|---|---|---|---|
| A | a | b | NA |
| B | c | d | NB |
| NS | NF | N |
m, n | Part of IFNA17 | Approach | ||
|---|---|---|---|---|
| Decile Method | Quartile Method | BJ-Algorithm | ||
| 0.30–0.59 nmax | I | 212 | 110 | 109 |
| II | 586 # | 589 # | 517 # | |
| III | 203 ¤ | 88 ¤ | 87 ¤ | |
| 0.30–0.59 nmin | I | 998 | 1124 | 386 |
| II | 1014 | 1029 * | 472 # | |
| III | 1080 | 757 #, ¤ | 363 ¤ | |
| 0.30–0.43 nmax | I | 195 | 92 | 92 |
| II | 321 # | 285 # | 252 # | |
| III | 203 ¤ | 88 ¤ | 87 ¤ | |
| 0.30–0.43 nmin | I | 439 | 556 | 162 |
| II | 444 | 543 | 235 # | |
| III | 550 #, ¤ | 310 #, ¤ | 162 ¤ | |
| 0.44–0.59 nmax | I | 17 | 18 | 17 |
| II | 265 # | 304 # | 265 # | |
| III | 0 #, ¤ | 0 #, ¤ | 0 #, ¤ | |
| 0.44–0.59 nmin | I | 559 | 568 | 224 |
| II | 570 | 486 # | 237 | |
| III | 530 | 447 # | 201 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basov, A.; Drobotenko, M.; Svidlov, A.; Gerasimenko, E.; Malyshko, V.; Elkina, A.; Baryshev, M.; Dzhimak, S. Inequality in the Frequency of the Open States Occurrence Depends on Single 2H/1H Replacement in DNA. Molecules 2020, 25, 3753. https://doi.org/10.3390/molecules25163753
Basov A, Drobotenko M, Svidlov A, Gerasimenko E, Malyshko V, Elkina A, Baryshev M, Dzhimak S. Inequality in the Frequency of the Open States Occurrence Depends on Single 2H/1H Replacement in DNA. Molecules. 2020; 25(16):3753. https://doi.org/10.3390/molecules25163753
Chicago/Turabian StyleBasov, Alexander, Mikhail Drobotenko, Alexandr Svidlov, Eugeny Gerasimenko, Vadim Malyshko, Anna Elkina, Mikhail Baryshev, and Stepan Dzhimak. 2020. "Inequality in the Frequency of the Open States Occurrence Depends on Single 2H/1H Replacement in DNA" Molecules 25, no. 16: 3753. https://doi.org/10.3390/molecules25163753