Resistance of Gram-Positive Bacteria to Current Antibacterial Agents and Overcoming Approaches




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
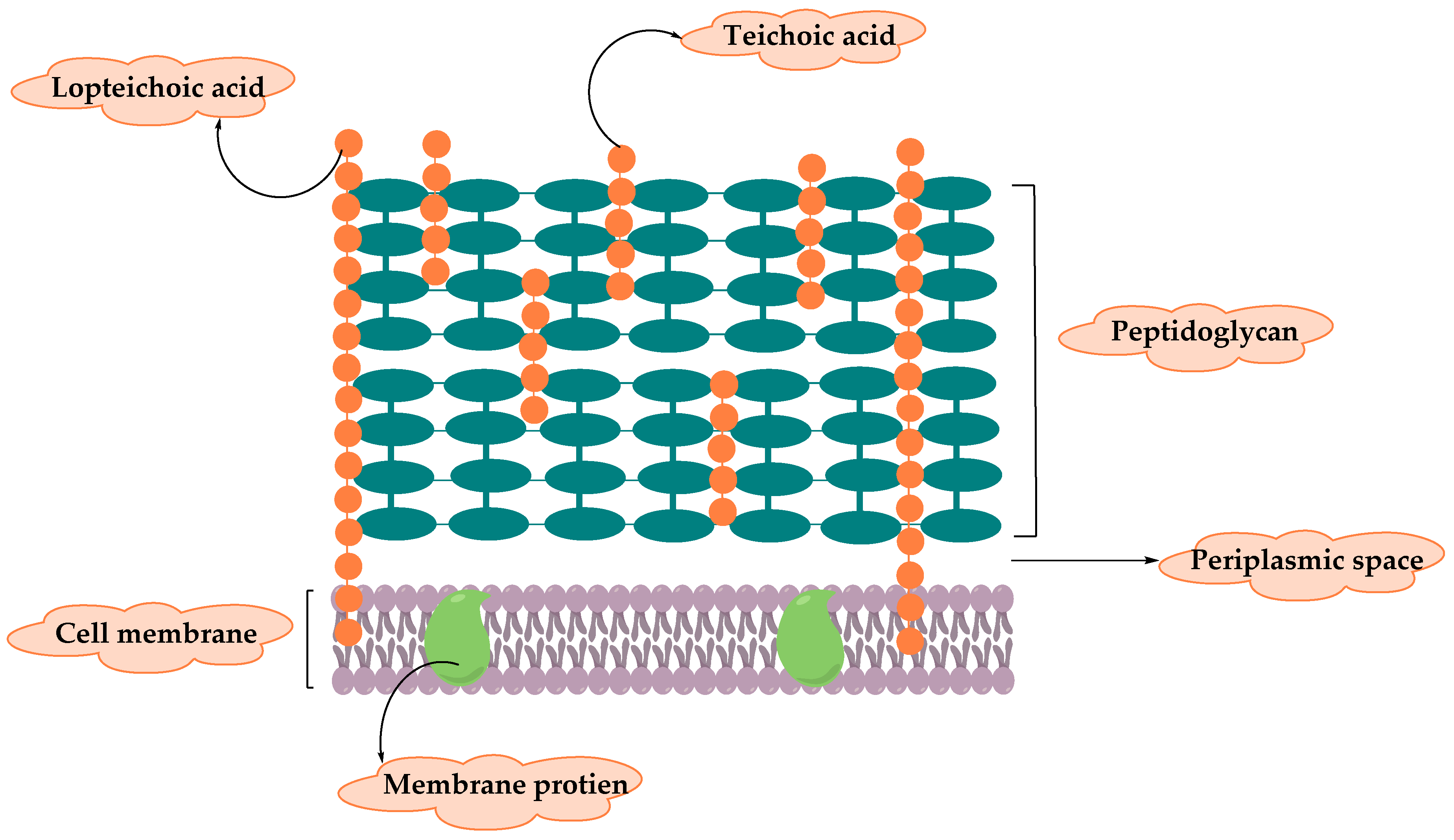
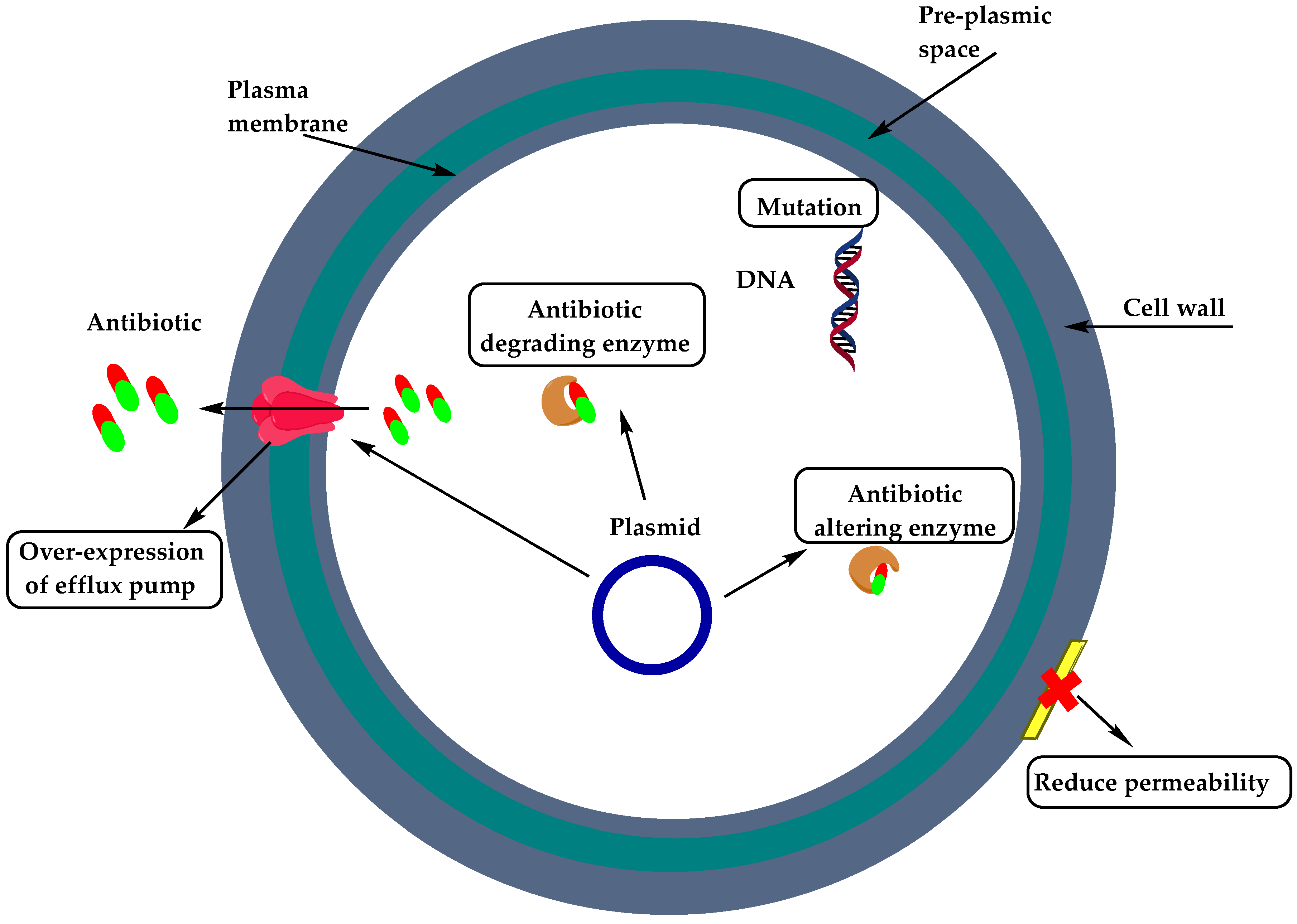
2. Resistance of Gram-Positive Bacteria
2.1. Staphylococcus Aureus
2.1.1. Staphylococcus Aureus—Methicillin-Resistant
2.1.2. Staphylococcus Aureus—Vancomycin Intermediate and Resistant
2.1.3. Staphylococcus Aureus—other Antibiotic Resistance
2.2. Enterococcus Faecium
2.2.1. E. faecium—Ampicillin/Penicillin and Cephalosporins resistance
2.2.2. E. faecium—Vancomycin-Resistant
2.2.3. E. faecium—other Antibiotic Resistance
2.3. Streptococcus Pneumoniae
2.3.1. Streptococcus Pneumoniae- Penicillin-non-Susceptible
2.3.2. S. Pneumoniae-other Antibiotic Resistance
2.4. Other Resistant Gram-Positive Bacteria
- ▪
- Staphylococcus epidermidis uses exopolysaccharide matrix or its ability to form biofilms as a mechanism of resistance to reduce penetration and permeability of antibiotics. The majority of S. epidermidis isolates from different nosocomial infections are methicillin-resistant strains due to the transfer of resistant mecA gene that encode PBP2a, in addition, resistance to quinolones and vancomycin were also reported [47].
- ▪
- Staphylococcus saprophyticus is the most common cause of uncomplicated urinary tract infections (UTIs). Resistance of S. saprophyticus occurred to commonly prescribed UTIs antibiotics such as ampicillin, ceftriaxone, cephalexin, and ciprofloxacin [48].
- ▪
- Streptococcus viridans are an upper respiratory tract commensal bacterial that developed resistance to penicillin and other β-lactam antibiotics due to alteration in the penicillin-binding protein. In addition, other reports demonstrated that Streptococcus viridans can serve as reservoirs for resistance genes such as mef(E) and mel genes which develop resistant to the macrolide-lincosamide-streptogramin B (MLS(B)) antibiotics [49,50].
- ▪
- Streptococcus pyogenesis a human pathogen that colonize in the upper respiratory tract and skin. S. pyogenes are resistant to macrolides, lincosamides, and streptogramins (MLS), in addition to tetracyclines, and very uncommon resistance to aminoglycosides or fluoroquinolones [51].
- ▪
- Streptococcus agalactiae or Group B streptococcus (GBS) is responsible for most neonatal infections in humans that can be transferred from mother to child via the maternal genital tract into the amniotic fluid or at delivery. Resistance to antibiotic such as erythromycin and other macrolides is due to either ribosomal modification encoded by erm genes or through efflux pump mediated by mefA genes. Moreover, ribosomal translocation encoded by linB genes results in clindamycin resistance in GBS [52].
- ▪
- Clostridium difficile and C. Perfringens
- ▪
- Bacillus anthracis and Bacillus cereus are spore-forming bacteria. B. anthracis causes anthrax disease by its virulence factors—capsule and toxin that are encoded on plasmids—while B. cereus is a soil bacterium and human pathogen that causes contamination to dairy industry by producing numerous enzymes and aggressins. B. antracis has a good susceptibility to penicillin in contrast to B. cereus which produce potent ß-lactamases and resistance against penicillin, ampicillin, cephalosporins, and trimethoprim [56,57].
- ▪
- Corynebacterium diphtheria is a human pathogen that causes diphtheria disease, an upper respiratory tract illness mediated by potent A-B exotoxin named diphtheria toxin that inhibits protein biosynthesis and kills susceptible host cells. Horizontal gene transfer of antibiotic resistance genes such as cmx, sul1, and tet(W) result in resistant C. diphtheria to chloramphenicol, sulfonamides, and tetracyclines [58].
- ▪
- Listeria monocytogenes is a foodborne pathogen that causes severe infections in humans that can be treated with early administration of aminopenicillin and gentamicin antibiotics. Resistance of L. monocytogenes to tetracyclines and fluoroquinolones developed due to acquisition of conjugative transposons and active efflux, respectively. A low level of resistant strains to streptomycin, chloramphenicol, macrolide, and trimethoprim has been reported [59].
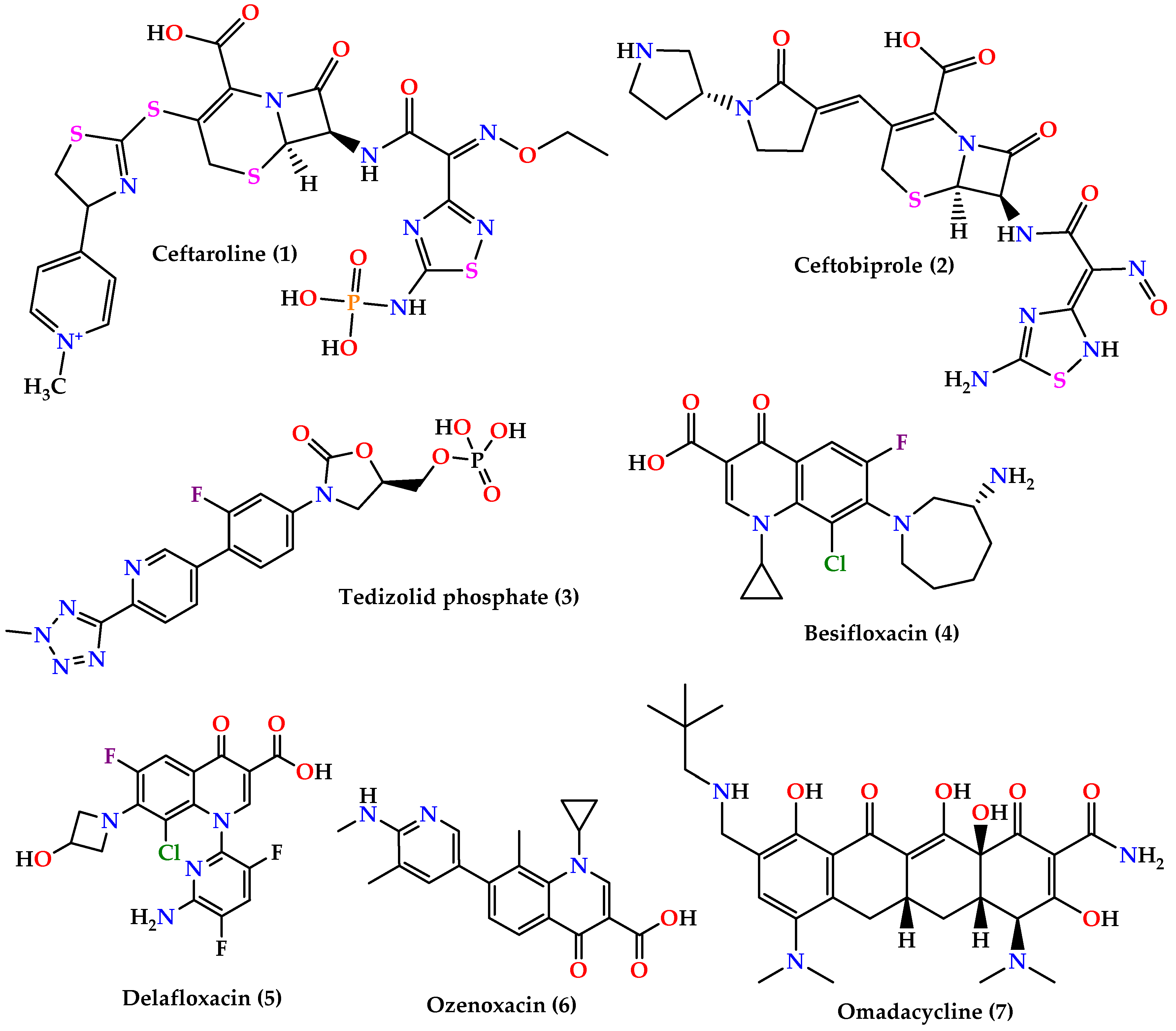
3. New Treatments
- ▪
- Cephalosporins: ceftaroline (1) and ceftobiprole (2) (Figure 3) which are 5th generation cephalosporins that inhibit cell wall synthesis by binding to PBP proteins with higher affinity than other β-lactam drugs. Ceftaroline is active against many Gram-positive organisms like MRSA, VRSA, Streptococcus pyogenes, and others, although resistance increase in MRSA sp. was reported. On the other hand, ceftobiprole is active against Gram-positive and Gram-negative microorganisms [60,61,62].
- ▪
- ▪
- Quinolones: besifloxacin (4), delafloxacin (5), and ozenoxacin (6) (Figure 3) all act by inhibiting DNA synthesis by binding to DNA gyrase and topoisomerase IV. Besifloxacin is active against Gram-positive bacteria, especially S. aureus, Staphylococcus epidermidis, S. pneumoniae, and Haemophilus influenzae, and Gram-negative bacteria, while delafloxacin is active against S. aureus, S. pneumoniae, and fluoroquinolone-resistant strains except for enterococci. On the other hand, ozenoxacin is active against MRSA, MSSA, MRSE, and S. pyogenes and was approved by the FDA to treat impetigo caused by S. aureus and S. pyogenes [62].
- ▪
- Omadacycline (7) (Figure 3) is a tetracycline analog that inhibits protein synthesis by binding on the 30S ribosomal subunit. It is active against a wide spectrum of bacteria such as resistant Gram-positive pathogens (MRSA, VRE, S. pneumoniae, S. pyogenes, and Streptococcus agalactiae), Gram-negative aerobes, anaerobes, and atypical bacteria [64,65].
- ▪
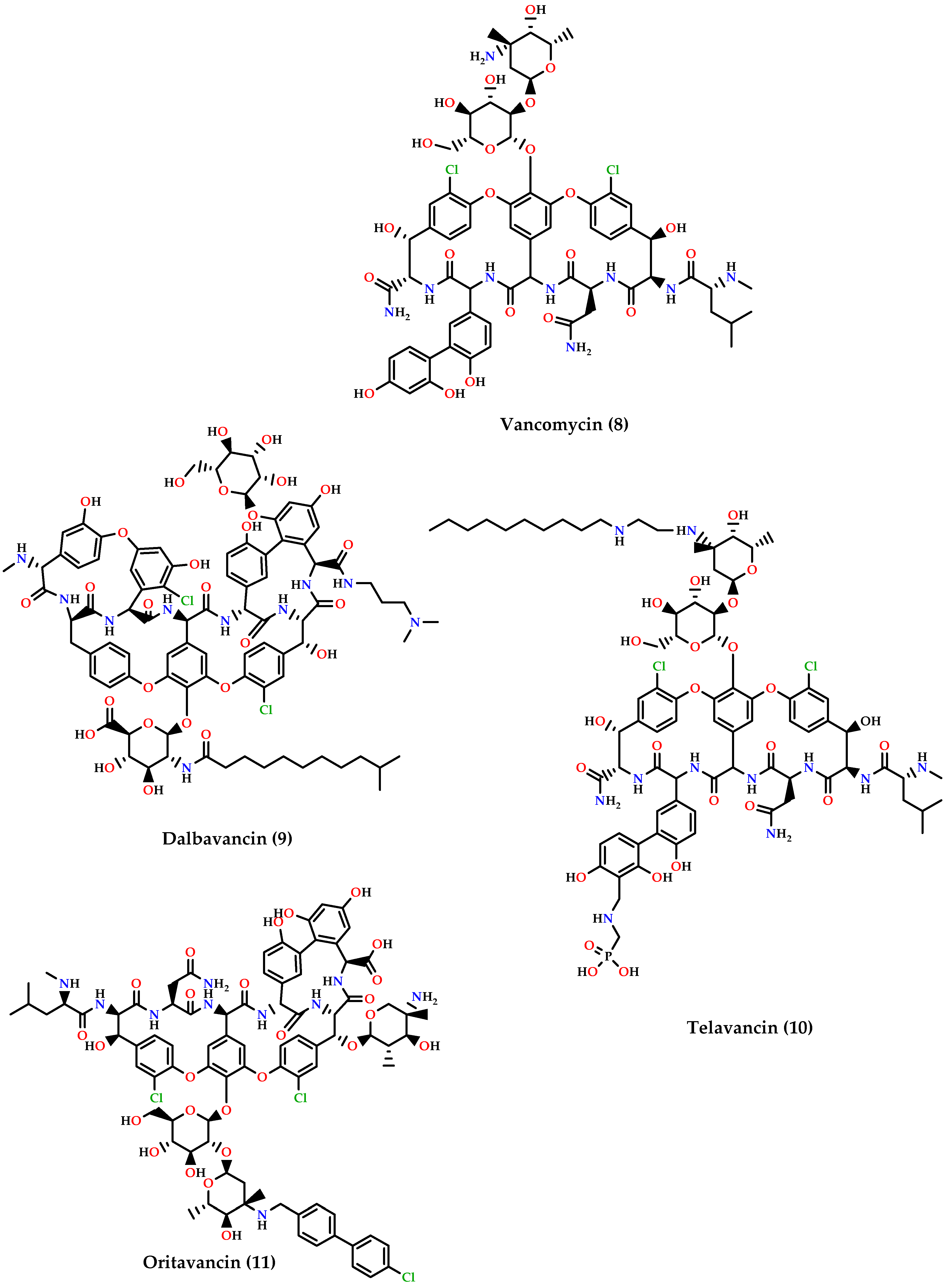
- Glycopeptides: dalbavancin (9), telavancin (10) and oritavancin (11) are vancomycin (8) derivatives and analogs (Figure 4). Dalbavancin inhibits cell wall synthesis and has an additional lipophilic side chain that enhances its activity and potency against wide-spectrum of Gram-positive organisms such as MRSA, S. pyogenes, Streptococcus anginosus, and E. faecalis susceptible to vancomycin. Telavancin inhibits cell wall synthesis and is active against aerobic and anaerobic Gram-positive bacteria. Oritavancin acts by inhibiting cell membranes and also inhibits RNA synthesis. It is active against MSSA, MRSA, VRE, and VISA VRSA [60,62,66].
4. Approaches to Overcome Gram-Positive Resistance
4.1. Novel Antibiotics
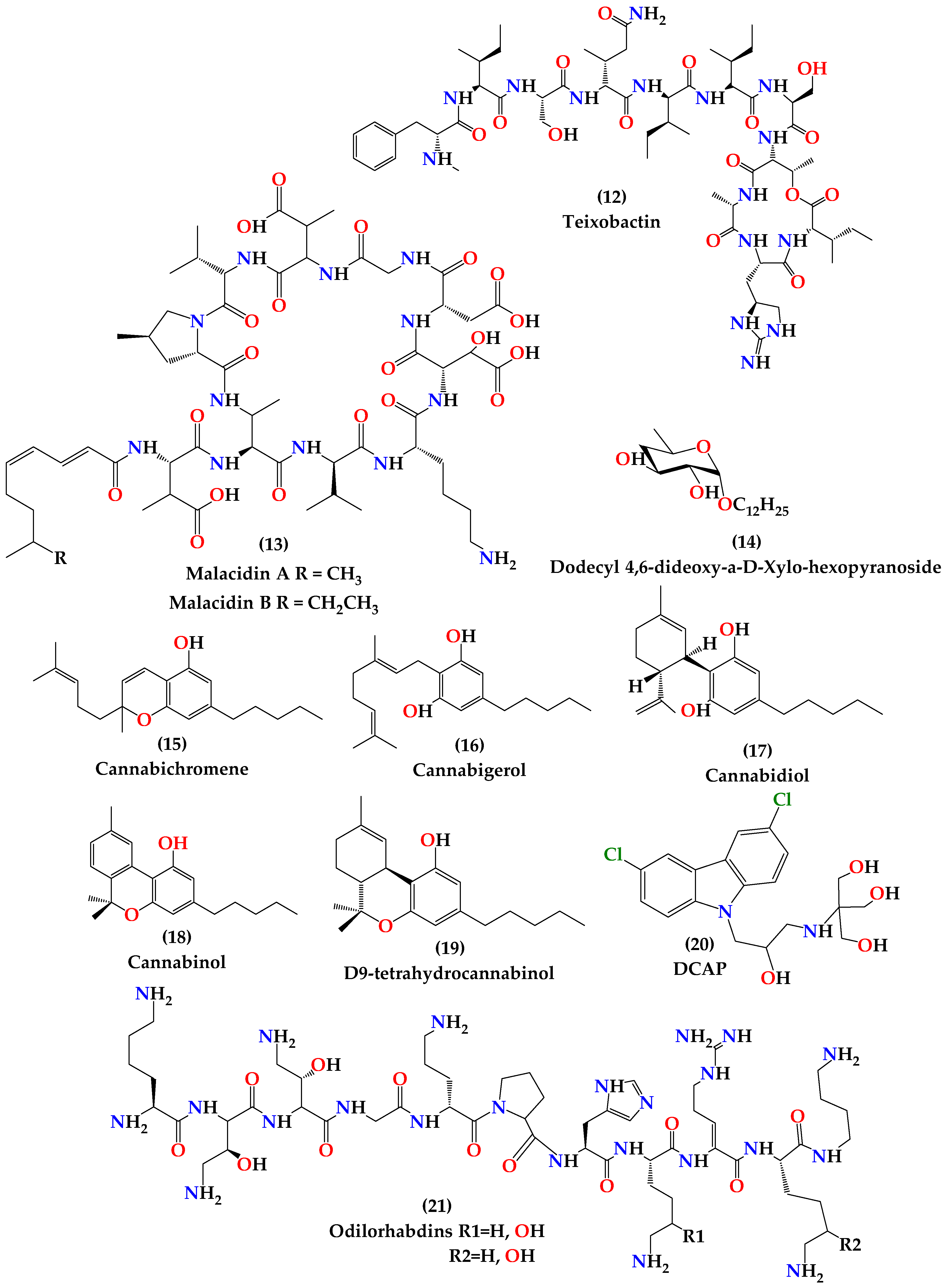
4.1.1. Teixobactin
4.1.2. Malacidins
4.1.3. Antimicrobial Peptides
4.1.4. Dodecyl Deoxy Glycosides (Antimicrobials that Target Membrane Lipid Polymorphism)
4.1.5. Cannabinoids
4.1.6. DCAP
4.1.7. Odilorhabdins
4.2. Bacteriophage Therapy
4.3. The Probiotic Approach to Prevent Antibiotic Resistance
4.4. National and International Approaches and Commitment
4.5. Education of Prudent Antibiotic Use
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Boil. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reygaert, W. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Cornaglia, G. Fighting infections due to multidrug-resistant Gram-positive pathogens. Clin. Microbiol. Infect. 2009, 15, 209–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asokan, G.V.; Ramadhan, T.; Ahmed, E.; Sanad, H. WHO Global Priority Pathogens List: A Bibliometric Analysis of Medline-PubMed for Knowledge Mobilization to Infection Prevention and Control Practices in Bahrain. Oman Med. J. 2019, 34, 184–193. [Google Scholar] [CrossRef]
- Sizar, O.; Unakal, C.G. Gram Positive Bacteria; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Silhavy, T.J.; Kahne, D.; Walker, S. The Bacterial Cell Envelope. Cold Spring Harb. Perspect. Boil. 2010, 2, a000414. [Google Scholar] [CrossRef]
- Yang, D.C.; Blair, K.M.; Salama, N.R. Staying in Shape: The Impact of Cell Shape on Bacterial Survival in Diverse Environments. Microbiol. Mol. Boil. Rev. 2016, 80, 187–203. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, M.; Walker, S.G. Envelope Structures of Gram-Positive Bacteria; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2017; Volume 404, pp. 1–44. [Google Scholar]
- Munita, J.M.; Bayer, A.S.; Arias, C.A. Evolving resistance among Gram-positive pathogens. Clin. Infect. Dis. 2015, 61, S48–S57. [Google Scholar] [CrossRef] [Green Version]
- Berger-Bächi, B. Resistance mechanisms of Gram-positive bacteria. Int. J. Med Microbiol. 2002, 292, 27–35. [Google Scholar] [CrossRef]
- Fisher, J.F.; Mobashery, S. Beta-Lactam Resistance Mechanisms: Gram-Positive Bacteria and Mycobacterium tuberculosis. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, G.; Saigal, S.; Elongavan, A. Action and resistance mechanisms of antibiotics: A guide for clinicians. J. Anaesthesiol. Clin. Pharmacol. 2017, 33, 300–305. [Google Scholar] [CrossRef]
- Sujatha, S.; Praharaj, I. Glycopeptide Resistance in Gram-Positive Cocci: A Review. Interdiscip. Perspect. Infect. Dis. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of Quinolone Action and Resistance. Biochemistry 2014, 53, 1565–1574. [Google Scholar] [CrossRef]
- Hooper, D.C. Fluoroquinolone resistance among Gram-positive cocci. Lancet Infect. Dis. 2002, 2, 530–538. [Google Scholar] [CrossRef]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a027029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pechère, J.-C. Macrolide resistance mechanisms in Gram-positive cocci. Int. J. Antimicrob. Agents 2001, 18, 25–28. [Google Scholar] [CrossRef]
- Dweba, C.; Zishiri, O.T.; El Zowalaty, M.E. Methicillin-resistant Staphylococcus aureus: Livestock-associated, antimicrobial, and heavy metal resistance. Infect. Drug Resist. 2018, 11, 2497–2509. [Google Scholar] [CrossRef] [Green Version]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [Green Version]
- Chambers, H.F.; deLeo, F.R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Genet. 2009, 7, 629–641. [Google Scholar] [CrossRef]
- Crisóstomo, M.I.; Westh, H.; Tomasz, A.; Chung, M.; Oliveira, D.; de Lencastre, H. The evolution of methicillin resistance in Staphylococcus aureus: Similarity of genetic backgrounds in historically early methicillin-susceptible and -resistant isolates and contemporary epidemic clones. Proc. Natl. Acad. Sci. USA 2001, 98, 9865–9870. [Google Scholar] [CrossRef] [Green Version]
- Pantosti, A.; Sanchini, A.; Monaco, M. Mechanisms of antibiotic resistance inStaphylococcus aureus. Futur. Microbiol. 2007, 2, 323–334. [Google Scholar] [CrossRef]
- David, M.Z.; Glikman, D.; Crawford, S.E.; Peng, J.; King, K.J.; Hostetler, M.A.; Boyle-Vavra, S.; Daum, R.S. What Is Community-Associated Methicillin-Resistant Staphylococcus aureus? J. Infect. Dis. 2008, 197, 1235–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieradzki, K.; Tomasz, A. Inhibition of cell wall turnover and autolysis by vancomycin in a highly vancomycin-resistant mutant of Staphylococcus aureus. J. Bacteriol. 1997, 179, 2557–2566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, L.; Iwamoto, A.; Lian, J.-Q.; Neoh, H.-M.; Maruyama, T.; Horikawa, Y.; Hiramatsu, K. Novel Mechanism of Antibiotic Resistance Originating in Vancomycin-Intermediate Staphylococcus aureus. Antimicrob. Agents Chemother. 2006, 50, 428–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Wang, T.; Onodera, Y.; Uchida, Y.; Sato, K. Mechanism of quinolone resistance in Staphylococcus aureus. J. Infect. Chemother. 2000, 6, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Foster, T. Antibiotic resistance in Staphylococcus aureus. Status and prospects. FEMS Microbiol. Rev. 2017, 41, 430–449. [Google Scholar] [CrossRef]
- Bayer, A.S.; Schneider, T.; Sahl, H.-G. Mechanisms of daptomycin resistance in Staphylococcus aureus: Role of the cell membrane and cell wall. Ann. New York Acad. Sci. 2012, 1277, 139–158. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.R.; Bayer, A.S.; Arias, C.A. Mechanism of Action and Resistance to Daptomycin in Staphylococcus aureus and Enterococci. Cold Spring Harb. Perspect. Med. 2016, 6, a026997. [Google Scholar] [CrossRef] [Green Version]
- Babaoglu, K.; Qi, J.; Lee, R.E.; White, S.W. Crystal Structure of 7,8-Dihydropteroate Synthase from Bacillus anthracis. Struct. 2004, 12, 1705–1717. [Google Scholar] [CrossRef]
- Rouch, D.A.; Messerotti, L.J.; Loo, L.S.L.; Jackson, C.A.; Skurray, R.A. Trimethoprim resistance transposon Tn4003 from Staphylococcus aureus encodes genes for a dihydrofolate reductase and thymidylate synthetase flanked by three copies of IS257. Mol. Microbiol. 1989, 3, 161–175. [Google Scholar] [CrossRef]
- Emaneini, M.; Bigverdi, R.; Kalantar, D.; Soroush, S.; Jabalameli, F.; Khoshgnab, B.N.; Asadollahi, P.; Taherikalani, M. Distribution of genes encoding tetracycline resistance and aminoglycoside modifying enzymes in Staphylococcus aureus strains isolated from a burn center. Ann. Burn. Fire Disasters 2013, 26, 76–80. [Google Scholar]
- Prabhu, K.; Rao, S.; Rao, I.V. Inducible Clindamycin Resistance in Staphylococcus aureus Isolated from Clinical Samples. J. Lab. Phys. 2011, 3, 025–027. [Google Scholar] [CrossRef] [PubMed]
- Cetinkaya, Y.; Falk, P.; Mayhall, C.G. Vancomycin-resistant enterococci. Clin. Microbiol. Rev. 2000, 13, 686–707. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Pang, S.; Abraham, S.; Coombs, G.W. Antimicrobial-resistant CC17 Enterococcus faecium: The past, the present and the future. J. Glob. Antimicrob. Resist. 2019, 16, 36–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, W.R.; Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance in enterococci. Expert Rev. Anti-Infect.Ther. 2014, 12, 1221–1236. [Google Scholar] [CrossRef]
- Fontana, R.; Aldegheri, M.; Ligozzi, M.; Lopez, H.; Sucari, A.; Satta, G. Overproduction of a low-affinity penicillin-binding protein and high-level ampicillin resistance in Enterococcus faecium. Antimicrob. Agents Chemother. 1994, 38, 1980–1983. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, E.; Keynan, Y. Vancomycin-resistant enterococci. Crit. Care Clin. 2013, 29, 841–852. [Google Scholar] [CrossRef]
- Cattoir, V.; Giard, J.-C. Antibiotic resistance in Enterococcus faecium clinical isolates. Exp. Rev. Anti-Infect. Ther. 2014, 12, 239–248. [Google Scholar] [CrossRef]
- Zivich, P.N.; Grabenstein, J.D.; Becker-Dreps, S.I.; Weber, D.J. Streptococcus pneumoniae outbreaks and implications for transmission and control: A systematic review. Pneumonia 2018, 10, 11. [Google Scholar] [CrossRef]
- Mamishi, S.; Moradkhani, S.; Mahmoudi, S.; Sadeghi, R.H.; Pourakbari, B. Penicillin-Resistant trend of Streptococcus pneumoniae in Asia: A systematic review. Iran. J. Microbiol. 2014, 6, 198–210. [Google Scholar]
- Hampton, L.M.; Farley, M.M.; Schaffner, W.; Thomas, A.; Reingold, A.; Harrison, L.H.; Lynfield, R.; Bennett, N.M.; Petit, S.; Gershman, K.; et al. Prevention of Antibiotic-Nonsusceptible Streptococcus pneumoniaeWith Conjugate Vaccines. J. Infect. Dis. 2011, 205, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Kim, L.; McGee, L.; Tomczyk, S.; Beall, B. Biological and Epidemiological Features of Antibiotic-Resistant Streptococcus pneumoniae in Pre- and Post-Conjugate Vaccine Eras: A United States Perspective. Clin. Microbiol. Rev. 2016, 29, 525–552. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, M.R. Drug-resistant Streptococcus pneumoniae: Rational antibiotic choices. Am. J. Med. 1999, 106, 19–25. [Google Scholar] [CrossRef]
- Wang, E.E.; Kellner, J.D.; Arnold, S. Antibiotic-resistant Streptococcus pneumoniae. Implications for medical practice. Can. Fam. Phys. Med. Fam. Can. 1998, 44, 1881–1888. [Google Scholar]
- Reinert, R. The antimicrobial resistance profile of Streptococcus pneumoniae. Clin. Microbiol. Infect. 2009, 15, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Namvar, A.E.; Bastarahang, S.; Abbasi, N.; Ghehi, G.S.; Farhadbakhtiarian, S.; Arezi, P.; Hosseini, M.; Baravati, S.Z.; Jokar, Z.; Chermahin, S.G. Clinical characteristics of Staphylococcus epidermidis: A systematic review. GMS Hyg. Infect. Control. 2014, 9, 23. [Google Scholar]
- Lo, D.; Shieh, H.H.; Barreira, E.R.; Ragazzi, S.L.B.; Gilio, A.E. High Frequency of Staphylococcus saprophyticus Urinary Tract Infections Among Female Adolescents. Pediatr. Infect. Dis. J. 2015, 34, 1025. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.; Huh, H.J.; Lee, N.Y. Species-Specific Difference in Antimicrobial Susceptibility Among Viridans Group Streptococci. Ann. Lab. Med. 2015, 35, 205–211. [Google Scholar] [CrossRef]
- Nakajima, T.; Nakanishi, S.; Mason, C.; Montgomery, J.; Leggett, P.; Matsuda, M.; Coulter, W.A.; Millar, B.C.; Goldsmith, C.E.; Moore, J.E. Population structure and characterization of viridans group streptococci (VGS) isolated from the upper respiratory tract of patients in the community. Ulst. Med. J. 2013, 82, 164–168. [Google Scholar]
- Chen, Z.; Itzek, A.; Malke, H.; Ferretti, J.J.; Kreth, J. Multiple Roles of RNase Y in Streptococcus pyogenes mRNA Processing and Degradation. J. Bacteriol. 2013, 195, 2585–2594. [Google Scholar] [CrossRef] [Green Version]
- Bolukaoto, J.Y.; Monyama, C.M.; Chukwu, M.O.; Lekala, L.M.; Nchabeleng, M.; Maloba, M.R.B.; Mavenyengwa, R.T.; Lebelo, S.L.; Monokoane, S.; Tshepuwane, C.; et al. Antibiotic resistance of Streptococcus agalactiae isolated from pregnant women in Garankuwa, South Africa. BMC Res. Notes 2015, 8, 364. [Google Scholar] [CrossRef] [Green Version]
- Spigaglia, P. Recent advances in the understanding of antibiotic resistance inClostridium difficileinfection. Ther. Adv. Infect. Dis. 2015, 3, 23–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chon, J.-W.; Seo, K.-H.; Bae, D.; Park, J.-H.; Khan, S.; Sung, K. Prevalence, toxin gene profile, antibiotic resistance, and molecular characterization of Clostridium perfringens from diarrheic and non-diarrheic dogs in Korea. J. Veter Sci. 2018, 19, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Hamza, D.; Dorgham, S.; Hakim, A. Toxinotyping and Antimicrobial Resistance of Clostridium Perfringens Isolated from Processed Chicken Meat Products. J. Veter. Res. 2017, 61, 53–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helgason, E.; Økstad, O.A.; Caugant, D.A.; Johansen, H.A.; Fouet, A.; Mock, M.; Hegna, I.; Kolstø, A.-B. Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis—One Species on the Basis of Genetic Evidence. Appl. Environ. Microbiol. 2000, 66, 2627–2630. [Google Scholar] [CrossRef] [Green Version]
- Loesche, W.J. Microbiology of dental decay and periodontal disease. In Medical Microbiology, 4th ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Trost, E.; Blom, J.; Soares, S.C.; Huang, I.-H.; Al-Dilaimi, A.; Schröder, J.; Jaenicke, S.; Dorella, F.A.; Rocha, F.; Miyoshi, A.; et al. Pangenomic Study of Corynebacteriumdiphtheriae That Provides Insights into the Genomic Diversity of Pathogenic Isolates from Cases of Classical Diphtheria, Endocarditis, and Pneumonia. J. Bacteriol. 2012, 194, 3199–3215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morvan, A.; Moubareck, C.; Leclercq, A.; Hervé-Bazin, M.; Bremont, S.; Lecuit, M.; Courvalin, P.; le Monnier, A. Antimicrobial Resistance of Listeria monocytogenes Strains Isolated from Humans in France. Antimicrob. Agents Chemother. 2010, 54, 2728–2731. [Google Scholar] [CrossRef] [Green Version]
- Kinney, K.K. Treatment of Infections Caused By Antimicrobial-Resistant Gram-Positive Bacteria. Am. J. Med. Sci. 2010, 340, 209–217. [Google Scholar] [CrossRef]
- Morosini, M.-I.; Díez-Aguilar, M.; Cantón, R. Mechanisms of action and antimicrobial activity of ceftobiprole. Rev. Esp.Quimioter. 2019, 32, 3–10. [Google Scholar]
- Koulenti, D.; Xu, E.; Mok, I.Y.S.; Song, A.; Karageorgopoulos, D.E.; Armaganidis, A.; Lipman, J.; Tsiodras, S. Novel Antibiotics for Multidrug-Resistant Gram-Positive Microorganisms. Microorganisms 2019, 7, 270. [Google Scholar] [CrossRef] [Green Version]
- Rybak, J.M.; Roberts, K. Tedizolid Phosphate: A Next-Generation Oxazolidinone. Infect. Dis. Ther. 2015, 4, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Burgos, R.M.; Rodvold, K.A. Omadacycline: A novel aminomethylcycline. Infect. Drug Resist. 2019, 12, 1895–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, J. Omadacycline: A Modernized Tetracycline. Clin. Infect. Dis. 2019, 69, S1–S5. [Google Scholar] [CrossRef] [PubMed]
- Zhanel, G.G.; Calic, D.; Schweizer, F.; Zelenitsky, S.A.; Adam, H.; Lagacé-Wiens, P.R.S.; Rubinstein, E.; Gin, A.S.; Hoban, D.J.; Karlowsky, J.A.; et al. New Lipoglycopeptides. Drugs 2010, 70, 859–886. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Müller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogen without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef]
- McCarthy, M.W. Teixobactin: A novel anti-infective agent. Expert Rev. Anti-Infect. Ther. 2018, 17, 1–3. [Google Scholar] [CrossRef]
- Hover, B.M.; Kim, S.-H.; Katz, M.; Charlop-Powers, Z.; Owen, J.G.; Ternei, M.A.; Maniko, J.; Estrela, A.B.; Molina, H.; Park, S.; et al. Culture-independent discovery of the malacidins as calcium-dependent antibiotics with activity against multidrug-resistant Gram-positive pathogens. Nat. Microbiol. 2018, 3, 415–422. [Google Scholar] [CrossRef] [Green Version]
- James, G.; Swogger, E.; Wolcott, R.; Pulcini, E.D.; Secor, P.; Sestrich, J.; Costerton, J.W.; Stewart, P.S. Biofilms in chronic wounds. Wound Repair Regen. 2008, 16, 37–44. [Google Scholar] [CrossRef]
- Lai, Y.; Gallo, R.L. AMPed up immunity: How antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009, 30, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Bahar, A.; Ren, D. Antimicrobial peptides. Pharmaceuticals 2013, 6, 1543–1575. [Google Scholar] [CrossRef] [Green Version]
- Chung, P.Y.; Khanum, R. Antimicrobial peptides as potential anti-biofilm agents against multidrug-resistant bacteria. J. Microbiol. Immunol. Infect. 2017, 50, 405–410. [Google Scholar] [CrossRef]
- Zhou, Y.; Peng, Y. Synergistic effect of clinically used antibiotics and peptide antibiotics against Gram-positive and Gram-negative bacteria. Exp. Ther. Med. 2013, 6, 1000–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogs with phospholipid membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Harroun, T.A.; Weiss, T.M.; Ding, L.; Huang, H.W. Barrel-stave model, or toroidal model? A case study on melittin pores. Biophys. J. 2001, 81, 1475–1485. [Google Scholar] [CrossRef] [Green Version]
- Kaconis, Y.; Kowalski, I.; Howe, J.; Brauser, A.; Richter, W.; Razquin-Olazarán, I.; Íñigo, M.; Garidel, P.; Rössle, M.; de Tejada, G.M.; et al. Biophysical Mechanisms of Endotoxin Neutralization by Cationic Amphiphilic Peptides. Biophys. J. 2011, 100, 2652–2661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, K.; Murase, O.; Fujii, N.; Miyajima, K. An Antimicrobial Peptide, Magainin 2, Induced Rapid Flip-Flop of Phospholipids Coupled with Pore Formation and Peptide Translocation. Biochemistry 1996, 35, 11361–11368. [Google Scholar] [CrossRef] [PubMed]
- Samy, R.P.; Stiles, B.G.; Franco, O.L.; Sethi, G.; Lim, L. Animal venoms as antimicrobial agents. Biochem. Pharmacol. 2017, 134, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.A.; Yoon, K.A.; Yoon, K.A. Differential Properties of Venom Peptides and Proteins in Solitary vs. Social Hunting Wasps. Toxins 2016, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Souza, B.M.; Mendes, M.A.; dos Santos, L.D.; Marques, M.R.; César, L.M.; Almeida, R.N.; Pagnocca, F.C.; Konno, K.; Palma, M.S. Structural and functional characterization of two novel peptide toxins isolated from the venom of the social wasp Polybiapaulista. Peptides 2005, 26, 2157–2164. [Google Scholar] [CrossRef]
- Wang, K.; Yan, J.; Chen, R.; Dang, W.; Zhang, B.; Zhang, W.; Song, J.; Wang, R. Membrane-Active Action Mode of Polybia-CP, a Novel Antimicrobial Peptide Isolated from the Venom of Polybiapaulista. Antimicrob. Agents Chemother. 2012, 56, 3318–3323. [Google Scholar] [CrossRef] [Green Version]
- Torres, M.D.T.; Pedron, C.N.; Higashikuni, Y.; Kramer, R.M.; Cardoso, M.H.; Oshiro, K.G.N.; Franco, O.L.; Junior, P.I.S.; Silva, F.D.; Junior, V.X.O.; et al. Structure-function-guided exploration of the antimicrobial peptide polybia-CP identifies activity determinants and generates synthetic therapeutic candidates. Commun. Boil. 2018, 1, 221. [Google Scholar] [CrossRef]
- Das Neves, R.C.; Mortari, M.R.; Schwartz, E.F.; Kipnis, A.; Junqueira-Kipnis, A.P. Antimicrobial and Antibiofilm Effects of Peptides from Venom of Social Wasp and Scorpion on Multidrug-Resistant Acinetobacterbaumannii. Toxins 2019, 11, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, B.; Augusto, M.; Felício, M.R.; Hollmann, A.; Franco, O.L.; Gonçalves, S.; Santos, N.C. Designing improved active peptides for therapeutic approaches against infectious diseases. Biotechnol. Adv. 2018, 36, 415–429. [Google Scholar] [CrossRef]
- Dias, C.; Pais, J.P.; Nunes, R.; Blazquez-Sanchez, M.T.; Marquês, J.T.; Almeida, A.F.; Serra, P.; Xavier, N.M.; Vila-Viçosa, D.; Machuqueiro, M.; et al. Sugar-based bactericides targeting phosphatidylethanolamine-enriched membranes. Nat. Commun. 2018, 9, 4857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, A.; Santos, M.S.C.; Dias, C.; Serra, P.; Cachatra, V.; Pais, J.; Caio, J.; Teixeira, V.; Machuqueiro, M.; Silva, M.S.; et al. Tuning the Bioactivity of TensioactiveDeoxy Glycosides to Structure: Antibacterial Activity Versus Selective Cholinesterase Inhibition Rationalized by Molecular Docking. Eur. J. Org. Chem. 2013, 1448–1459. [Google Scholar] [CrossRef]
- Baulamycins, A.B. Broad-spectrum antibiotics identified as inhibitors of siderophore biosynthesis in Staphylococcus aureus and Bacillus anthracisTripathi. Ashootosh 2006, 49. [Google Scholar]
- Appendino, G.; Gibbons, S.; Giana, A.; Pagani, A.; Grassi, G.; Stavri, M.; Smith, E.; Rahman, M.M. Antibacterial Cannabinoids fromCannabis sativa: A Structure−Activity Study. J. Nat. Prod. 2008, 71, 1427–1430. [Google Scholar] [CrossRef]
- Chakraborty, S.; Afaq, N.; Singh, N.; Majumdar, S. Antimicrobial activity of Cannabis sativa, Thujaorientalis and Psidiumguajava leaf extracts against methicillin-resistant Staphylococcus aureus. J. Integr. Med. 2018, 16, 350–357. [Google Scholar] [CrossRef]
- Thanbichler, M.; Shapiro, L. MipZ, a Spatial Regulator Coordinating Chromosome Segregation with Cell Division in Caulobacter. Cell 2006, 126, 147–162. [Google Scholar] [CrossRef] [Green Version]
- Eun, Y.J.; Foss, M.H.; Kiekebusch, D.; Pauw, D.A.; Westler, W.M.; Thanbichler, M.; Weibel, D.B. DCAP: A Broad-Spectrum Antibiotic That Targets the Cytoplasmic Membrane of Bacteria. J. Am. Chem. Soc. 2012, 134, 11322–11325. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, V.; Hurley, K.; Santos, T.; Weibel, D. DCAP: A broad-spectrum antibiotic that targets the cytoplasmic membrane of bacteria. FASEB J. 2015, 29, 575. [Google Scholar]
- Hurley, K.A.; Heinrich, V.; Hershfield, J.R.; Demons, S.T.; Weibel, D.B. Membrane-Targeting DCAP Analogues with Broad-Spectrum Antibiotic Activity against Pathogenic Bacteria. ACS Med. Chem. Lett. 2015, 6, 466–471. [Google Scholar] [CrossRef] [Green Version]
- Pantel, L.; Florin, T.; Dobosz-Bartoszek, M.; Racine, E.; Sarciaux, M.; Serri, M.; Houard, J.; Campagne, J.-M.; de Figueiredo, R.M.; Midrier, C.; et al. Odilorhabdins, Antibacterial Agents that Cause Miscoding by Binding at a New Ribosomal Site. Mol. Cell 2018, 70, 83–94.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polikanov, Y.S.; Aleksashin, N.A.; Beckert, B.; Wilson, D.N. The Mechanisms of Action of Ribosome-Targeting Peptide Antibiotics. Front. Mol. Biosci. 2018, 5, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- d’Herelle, F. Sur un microbe invisible antagoniste des bacillesdysentériques. CR Acad. Sci. Paris 1917, 165, 373–375. [Google Scholar]
- Schultz, E.W. One hundred patients with Staphylococcus septicemia receiving bacteriophage service. Calif. West. Med. 1929, 31, 5–10. [Google Scholar]
- MacNeal, W.J.; Frisbee, F.C. A review of phage therapy against bacterial pathogens of aquatic and terrestrial organisms. Am. J. Med. Sci. 1936, 191, 179–195. [Google Scholar] [CrossRef]
- Doss, J.; Culbertson, K.; Hahn, D.; Camacho, J.; Barekzi, N. A Review of Phage Therapy against Bacterial Pathogens of Aquatic and Terrestrial Organisms. Viruses 2017, 9, 50. [Google Scholar] [CrossRef] [Green Version]
- Doolittle, M.M.; Cooney, J.J.; Caldwell, D.E. Tracing the interaction of bacteriophage with bacterial biofilms using fluorescent and chromogenic probes. J. Ind. Microbiol. Biotechnol. 1996, 16, 331–341. [Google Scholar] [CrossRef]
- Parasion, S.; Kwiatek, M.; Gryko, R.; Mizak, L.; Malm, A. Bacteriophages as an Alternative Strategy for Fighting Biofilm Development. Pol. J. Microbiol. 2014, 63, 137–145. [Google Scholar] [CrossRef]
- Kortright, K.E.; Chan, B.K.; Koff, J.L.; Turner, P.E. Phage Therapy: A Renewed Approach to Combat Antibiotic-Resistant Bacteria. Cell Host Microbe 2019, 25, 219–232. [Google Scholar] [CrossRef] [Green Version]
- Chanishvili, N.; Aminov, R.I. Bacteriophage therapy: Coping with the growing antibiotic resistance problem. Microbiol. Aust. 2019, 40, 5. [Google Scholar] [CrossRef]
- Kumaran, D.; Taha, M.; Yi, Q.; Ramirez-Arcos, S.; Diallo, J.-S.; Carli, A.; Abdelbary, H. Does Treatment Order Matter? Investigating the Ability of Bacteriophage to Augment Antibiotic Activity against Staphylococcus aureus Biofilms. Front. Microbiol. 2018, 9, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, C.; Guarnef, F.; Gibson, G.R. Expert consensus document: The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef] [Green Version]
- Hotel, A.C.P.; Cordoba, A. Health, and nutritional properties of probiotics in food including powder milk with live lactic acid bacteria. Prevention 2001, 5, 1–10. [Google Scholar]
- Ouwehand, A.C.; Forssten, S.; Hibberd, A.; Lyra, A.; Stahl, B. Probiotic approach to prevent antibiotic resistance. Ann. Med. 2016, 48, 1–10. [Google Scholar] [CrossRef]
- Applegate, J.A.; Walker, C.L.F.; Ambikapathi, R.; Black, R.E. Systematic review of probiotics for the treatment of community-acquired acute diarrhea in children. BMC Public Heal. 2013, 13, S16. [Google Scholar] [CrossRef] [Green Version]
- Newberry, S.J.; Hempel, S.; Maher, A.R.; Wang, Z.; Miles, J.N.V.; Shanman, R.; Johnsen, B.; Shekelle, P.G. Probiotics for the Prevention and Treatment of Antibiotic-Associated Diarrhea. JAMA 2012, 307, 1959. [Google Scholar] [CrossRef] [Green Version]
- King, S.; Glanville, J.; Sanders, M.E.; Fitzgerald, A.; Varley, D. Effectiveness of probiotics on the duration of illness in healthy children and adults who develop common acute respiratory infectious conditions: A systematic review and meta-analysis. Br. J. Nutr. 2014, 112, 41–54. [Google Scholar] [CrossRef]
- Wang, J.; Ji, H.; Wang, S.; Liu, H.; Zhang, W.; Zhang, N.; Wang, Y. Probiotic Lactobacillus plantarum Promotes Intestinal Barrier Function by Strengthening the Epithelium and Modulating Gut Microbiota. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- de Vuyst, L.; Leroy, F. Bacteriocins from Lactic Acid Bacteria: Production, Purification, and Food Applications. J. Mol. Microbiol. Biotechnol. 2007, 13, 194–199. [Google Scholar] [CrossRef]
- Ennahar, S.; Sashihara, T.; Sonomoto, K.; Ishizaki, A. Class IIabacteriocins: Biosynthesis, structure, and activity. FEMS Microbiol. Rev. 2000, 24, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Atassi, F.; Servin, A.L. Individual and co-operative roles of lactic acid and hydrogen peroxide in the killing activity of enteric strain Lactobacillus johnsonii NCC933 and vaginal strain Lactobacillus gasseri KS120.1 against enteric, uropathogenic and vaginosis-associated pathog. FEMS Microbiol. Lett. 2010, 304, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Midolo, P.D.; Lambert, J.R.; Hull, R.; Luo, F.; Grayson, M.L. In vitro inhibition of Helicobacter pylori NCTC 11637 by organic acids and lactic acid bacteria. J. Appl. Bacteriol. 1995, 79, 475–479. [Google Scholar] [CrossRef]
- Lin, X.; Chen, X.; Chen, Y.; Jiang, W.; Chen, H. The effect of five probiotic lactobacilli strains on the growth and biofilm formation of S treptococcusmutans. Oral Dis. 2015, 21, e128–e134. [Google Scholar] [CrossRef] [PubMed]
- Haller, D.; Bode, C.; Hammes, W.P. Cytokine secretion by stimulated monocytes depends on the growth phase and heat treatment of bacteria: A comparative study between lactic acid bacteria and invasive pathogens. Microbiol. Immunol. 1999, 43, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Tejada-Simon, M.V.; Pestka, J.J. Proinflammatory Cytokine and Nitric Oxide Induction in Murine Macrophages by Cell Wall and Cytoplasmic Extracts of Lactic Acid Bacteria. J. Food Prot. 1999, 62, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Hessle, C.; Hanson, L.A.; Wold, A. Lactobacilli from human gastrointestinal mucosa are strong stimulators of IL-12 production. Clin. Exp. Immunol. 1999, 116, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Perdigon, G.; Alvarez, S.; Holgado, A.P.D.R. Immunoadjuvant activity of oralLactobacilluscasei: Influence of dose on the secretory immune response and protective capacity in intestinal infections. J. Dairy Res. 1991, 58, 485–496. [Google Scholar] [CrossRef]
- Stone, S.P.; Fuller, C.; Savage, J.; Cookson, B.; Hayward, A.; Cooper, B.; Duckworth, G.; Michie, S.; Murray, M.; Jeanes, A.; et al. Evaluation of the national Cleanyourhands campaign to reduce Staphylococcus aureus bacteraemia and Clostridium difficile infection in hospitals in England and Wales by improved hand hygiene: Four year, prospective, ecological, interrupted time series study. BMJ 2012, 344, e3005. [Google Scholar] [CrossRef] [Green Version]
- Schwaber, M.J.; Lev, B.; Israeli, A.; Solter, E.; Smollan, G.; Rubinovitch, B.; Shalit, I.; Carmeli, Y. Containment of a Country-wide Outbreak of Carbapenem-Resistant Klebsiellapneumoniae in Israeli Hospitals via a Nationally Implemented Intervention. Clin. Infect. Dis. 2011, 52, 848–855. [Google Scholar] [CrossRef]
- Brinsley, K.; Srinivasan, A.; Sinkowitz-Cochran, R.; Lawton, R.; McIntyre, R. Implementation of the campaign to prevent antimicrobial resistance in healthcare settings: I2 steps to prevent antimicrobial resistance among hospitalized adults-experiences from 3 institutions. Am. J. Infect. Control 2005, 33, 53–54. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.P. Mandatory surveillance of methicillin-resistant Staphylococcus aureus (MRSA) bacteraemia in England: The first 10 years. J. Antimicrob. Chemother. 2012, 67, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.M.; Wertheim, H.F.L.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H.; et al. Antibiotic resistance—The need for global solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef] [Green Version]
- Winters, C.; Gelband, H. Part I. The global antibiotic resistance partnership (GARP). SAMJ South Afr. Med. J. 2011, 101, 556–557. [Google Scholar]
- Sirijatuphat, R.; Sripanidkulchai, K.; Boonyasiri, A.; Rattanaumpawan, P.; Supapueng, O.; Kiratisin, P.; Thamlikitkul, V. Implementation of global antimicrobial resistance surveillance system (GLASS) in patients with bacteremia. PLoS ONE 2018, 13, e0190132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Rasool, M.H.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; et al. Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-R.; Lee, J.H.; Kang, L.-W.; Jeong, B.C.; Lee, S.H. Educational Effectiveness, Target, and Content for Prudent Antibiotic Use. BioMed Res. Int. 2015, 2015, 1–13. [Google Scholar] [CrossRef]
- Price, L.; Gozdzielewska, L.; Young, M.; Smith, F.; Macdonald, J.; McParland, J.; Williams, L.; Langdridge, D.; Davis, M.D.M.; Flowers, P. Effectiveness of interventions to improve the public’s antimicrobial resistance awareness and behaviours associated with prudent use of antimicrobials: A systematic review. J. Antimicrob. Chemother. 2018, 73, 1464–1478. [Google Scholar] [CrossRef] [Green Version]
- de Bont, E.G.P.M.; Alink, M.; Falkenberg, F.C.J.; Dinant, G.-J.; Cals, J. Patient information leaflets to reduce antibiotic use and reconsultation rates in general practice: A systematic review. BMJ Open 2015, 5. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jubeh, B.; Breijyeh, Z.; Karaman, R. Resistance of Gram-Positive Bacteria to Current Antibacterial Agents and Overcoming Approaches. Molecules 2020, 25, 2888. https://doi.org/10.3390/molecules25122888
Jubeh B, Breijyeh Z, Karaman R. Resistance of Gram-Positive Bacteria to Current Antibacterial Agents and Overcoming Approaches. Molecules. 2020; 25(12):2888. https://doi.org/10.3390/molecules25122888
Chicago/Turabian StyleJubeh, Buthaina, Zeinab Breijyeh, and Rafik Karaman. 2020. "Resistance of Gram-Positive Bacteria to Current Antibacterial Agents and Overcoming Approaches" Molecules 25, no. 12: 2888. https://doi.org/10.3390/molecules25122888