Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes–7

 ,
,  , , , , ,
, , , , ,  ,
,  , , , ,
, , , ,  , , ,
, , ,  , , , , , , , ,
, , , , , , , ,  , ,
, ,  ,
,  , , , ,
, , , ,  , , ,
, , ,  ,
,  , ,
, ,  ,
,  , ,
, ,  , , ,
, , ,  , ,
, ,  and add
Show full author list
and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Targeting MAO Isoforms by cis/trans Isomers
3. A New Lead in the Combat Against the Multi-Drug Resistant Mycobacterium Abscessus
4. New InhA Inhibitors Provided by Fragment-Based Drug Design
5. Combined Modulation of Farnesoid X Receptor and Target Genes: A Promising Therapeutic Strategy in Non-Alcoholic Steatohepatitis
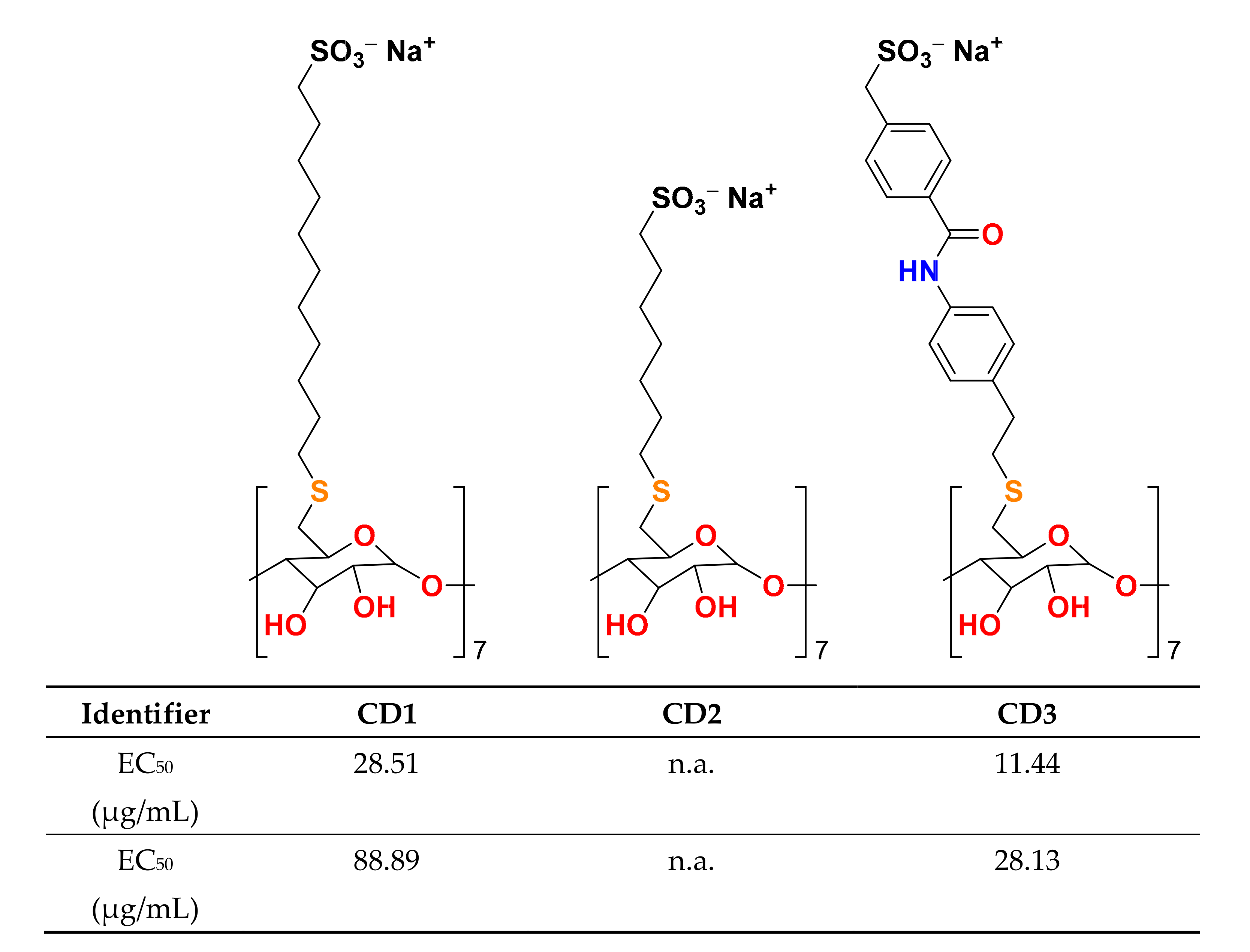
6. Cyclodextrins as a Potent Tool for Fighting Multiple Viral Infections
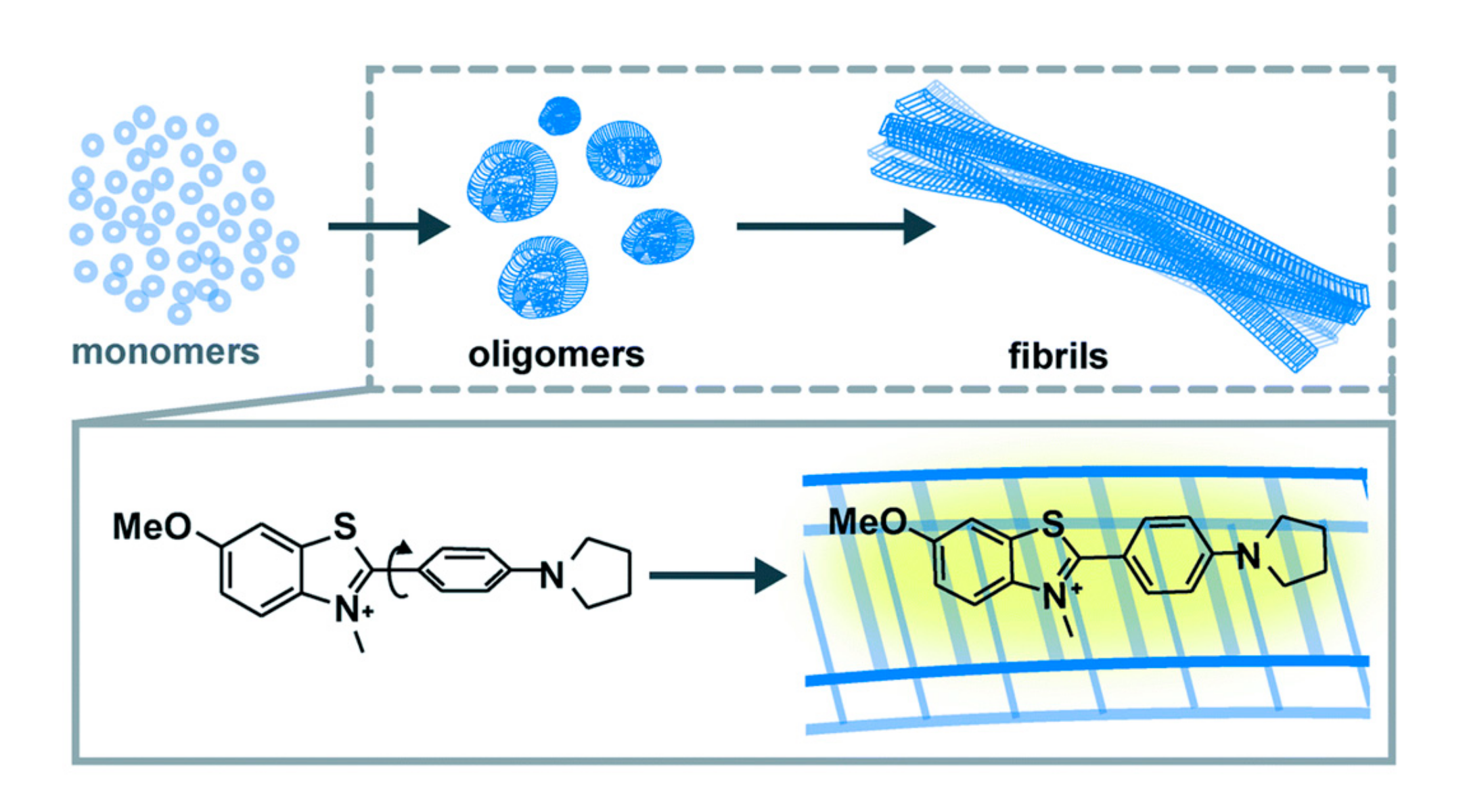
7. Probing Amyloid Oligomers is Key to Solving the Neurodegeneration Puzzle
8. Tyrosyl-DNA Phosphodiesterase 2 as a Novel Target to Enhance the Efficacy of Topoisomerase II Poisons
9. Proteostasis Network: A New Paradigm for Drug Discovery?
10. The Fascinating Perspective of Material Science: Precise Embolization in Tumor Vessels Through Peptide-Based Nanoparticles
11. Promising Anti-Leishmania Compounds in the Telluride Sulfonamide Series
12. New Insight in Fak Inhibitors
13. Tackling Coronas with EIDD-2801 Pills?
14. Developing Efficient Mutant GTPase KRASG12C Inhibitors: Are We Ready for Mutant KRAS Yet?
15. A Senolytic PROTAC Exploits the Restricted Expression Profile of CRBN to Lose Toxicity Associated with Its Parent Inhibitor
16. Possibility to Use Interleukin Itself for Treatment—IL-37 as an Example
17. RNA-Based Nanoconstructs for Targeted Delivery of Paclitaxel to Breast Cancer: The Road to Drug Efficacy and Safety
18. New Insights into Mechanisms for Allosterically Modulating the Kappa Opioid Receptor with Nanobodies
19. Is the Role of LMW Heparin in COVID-19 Infection Really Only Antithrombotic or Not?
20. Identification of Small Molecules Targeting Zika Virus NS2B-NS3 Protease via Fragment-Based Drug Discovery (FBDD) Approach
21. Synthesis and Chemical Profile of New HIV-1 Capsid Targeting Peptidomimetic Analogous of PF74
22. Let Cancer Cells Trip Themselves Up
23. Studies on SARSCoV-2 Spike Glycoprotein Open Doors for Covid-19 Drug Discovery
24. With or Without ssDNA Genome, Pf4 Filamentous Phage Promotes the Stability of P. Aeruginosa Biofilm Formation and Consequently Increases Protection Against Antibiotics
25. An NMR Drug Screening Approach in Human Cells as a New Tool for Drug Potency Early Assessment
26. Histone Deacetylase and Proteasome Dual Inhibitors for Overcoming Bortezomib Resistance
27. BRAF-V600E Degraders: A New Weapon Against Melanoma
28. Target-Guided Synthesis of Noncanonical DNA-Binding Transcriptional Modulators
29. The Crystal Structure of the SARS-CoV-2 Main Protease: New Hopes to Fight a Threatening Disease
30. Stalobacin I: The Newest Member of the Remarkable Family of Peptide-Based Antibiotics
31. Are We Now One Step Closer to the Discovery of new Antibiotics or Overcoming Their Resistance with Artificial Intelligence?
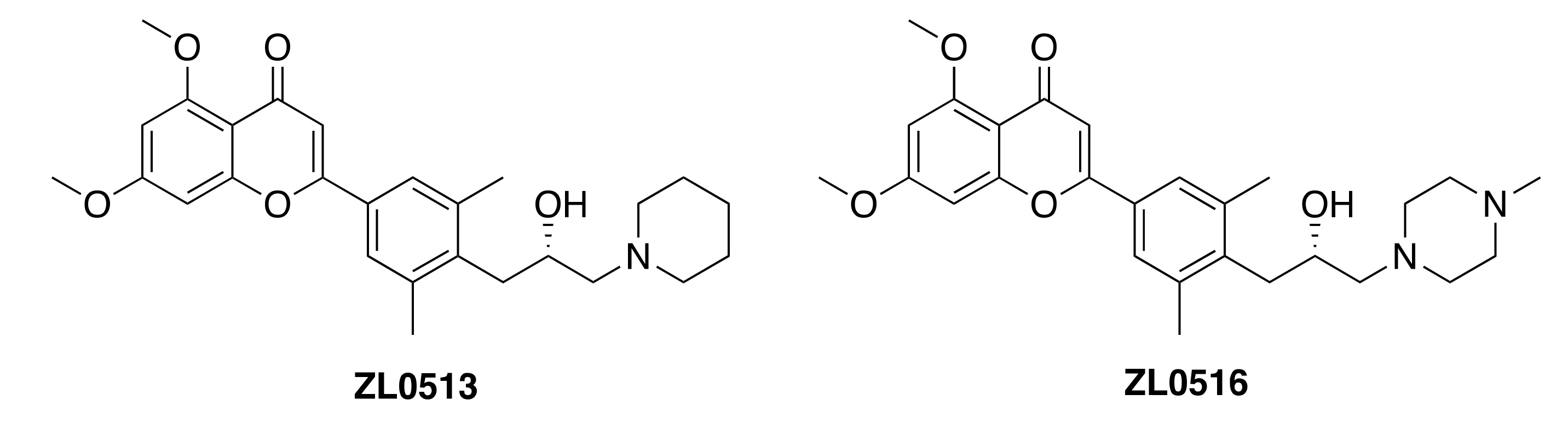
32. Design and Synthesis of Oral Bioavailable Chromone Derivatives as Novel Selective BRD4 Inhibitors
33. Aptamers as a Therapeutic Tool Against Prion Diseases
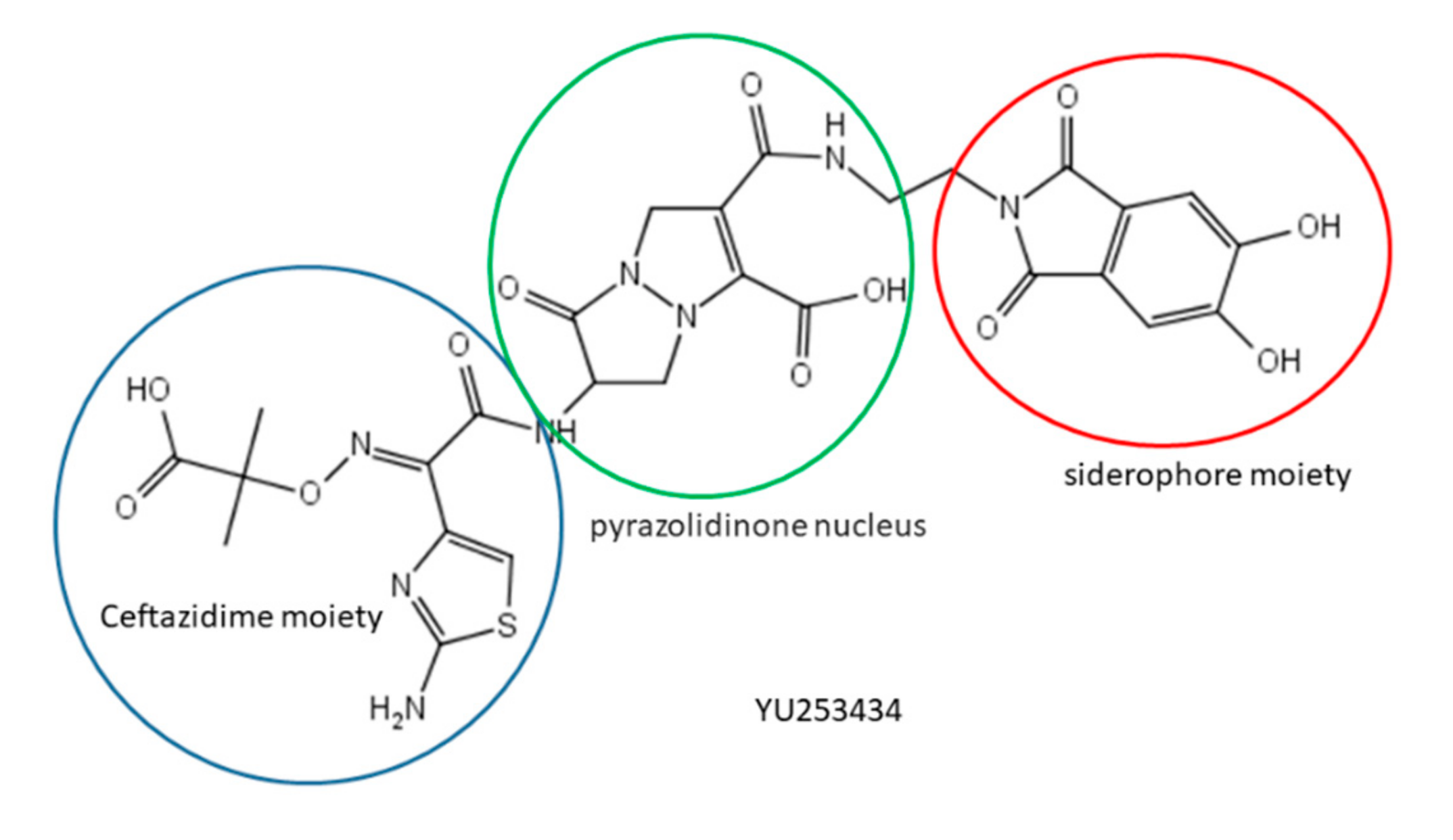
34. A Newly Designed γ–Lactam-Siderophore Conjugate Shows Significant Antibiotic Activity Against Multidrug-Resistant Gram-Negative Bacteria
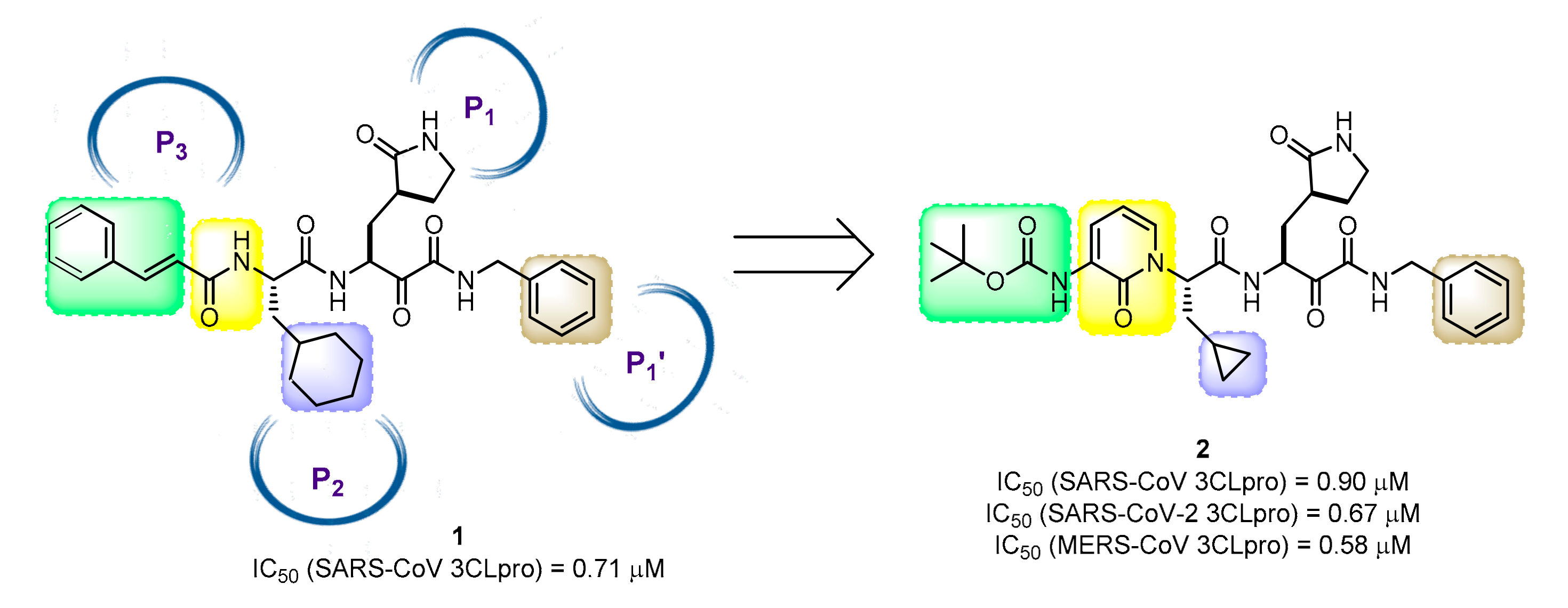
35. Tackling SARS-CoV-2: Key Hints for Structure-Based Design of Broad-Spectrum Inhibitors of 3CL Protease
36. Drug Discovery and Central Nervous System Diseases: Neuroinflammation as a Step Forward
37. Evaluation of Nonvaccination Preventive Mechanisms to Fight Antibiotic Resistence: Evidence from Enteropathogenic E. coli Infection
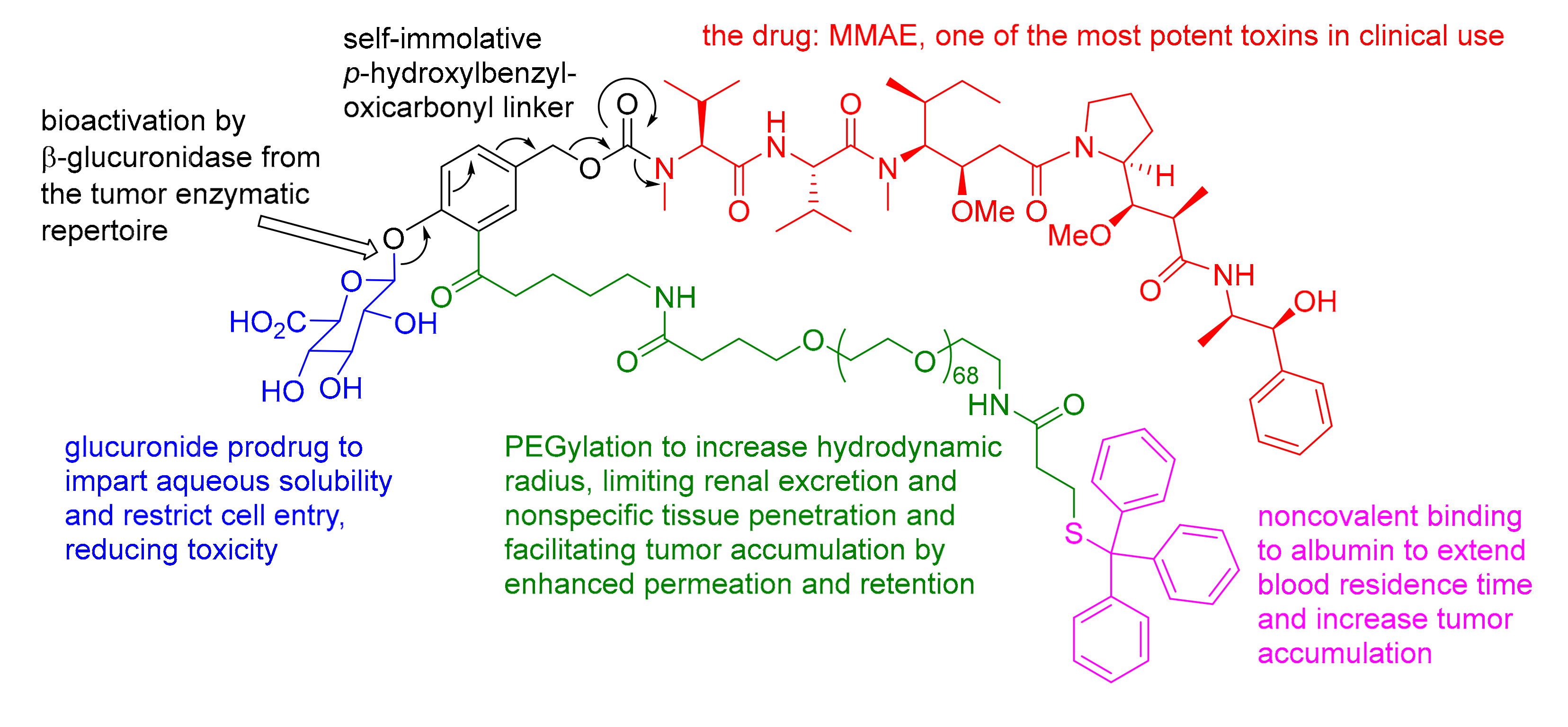
38. Glucuronide Prodrug, PEGylation, Albumin Binder, Self-Immolative Linker, and a Potent Toxin: An Ideal Ensemble for Efficient Tumor Suppression Upon Highly Localized Bioactivation
Funding
Conflicts of Interest
References
- Knez, D.; Colettis, N.; Iacovino, L.G.; Sova, M.; Pišlar, A.; Konc, J.; Lešnik, S.; Higgs, J.; Kamecki, F.; Mangialavori, I.; et al. Stereoselective activity of 1-propargyl-4-styrylpiperidine-like analogues that can discriminate between monoamine oxidase isoforms A and B. J. Med. Chem. 2020, 63, 1361–1387. [Google Scholar] [CrossRef] [PubMed]
- De Ruyck, J.; Dupont, C.; Lamy, E.; Le Moigne, V.; Biot, C.; Guérardel, Y.; Herrmann, J.-L.; Blaise, M.; Grassin-Delyle, S.; Kremer, L.; et al. Structure-based design and synthesis of piperidinol-containing molecules as new Mycobacterium abscessus inhibitors. ChemistryOpen 2020, 9, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Jacquemard, C.; Kellenberger, E. A Bright future for fragment-based drug discovery: What does it hold? Expert Opin. Drug Discov. 2019, 14, 413–416. [Google Scholar] [CrossRef] [Green Version]
- Erlanson, D.A.; Davis, B.J.; Jahnke, W. Fragment-based drug discovery: Advancing fragments in the absence of crystal structures. Cell Chem. Biol. 2019, 26, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbah, M.; Mendes, V.; Vistal, R.G.; Dias, D.M.G.; Zahorszka, M.; Mikusova, K.; Kordulakova, J.; Coyne, A.G.; Blundell, T.L.; Abell, C. Fragment-based design of Mycobacterium tuberculosis InhA inhibitors. J. Med. Chem. 2020, 63, 4749–4761. [Google Scholar] [CrossRef] [Green Version]
- Sayiner, M.; Koenig, A.; Henry, L.; Younossi, Z.M. Epidemiology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in the United States and the rest of the world. Clin. Liver Dis. 2016, 20, 205–214. [Google Scholar] [CrossRef]
- Chianelli, D.; Rucker, P.V.; Roland, J.; Tully, D.C.; Nelson, J.; Liu, X.; Bursulaya, B.; Hernandez, E.D.; Wu, J.; Prashad, M.; et al. Nidufexor (LMB763), a novel FXR modulator for the treatment of nonalcoholic steatohepatitis. J. Med. Chem. 2020, 63, 3868–3880. [Google Scholar] [CrossRef]
- Jones, S.T.; Cagno, V.; Janeček, M.; Ortiz, D.; Gasilova, N.; Piret, J.; Gasbarri, M.; Constant, D.A.; Han, Y.; Vuković, L.; et al. Modified cyclodextrins as broad-spectrum antivirals. Sci. Adv. 2020, 6, eaax9318. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Needham, L.-M.; Weber, J.; Varela, J.A.; Fyfe, J.W.B.; Do, D.T.; Xu, C.K.; Tutton, L.; Cliffe, R.; Keenlyside, B.; Klenerman, D. ThX—A next-generation probe for the early detection of amyloid aggregates. Chem. Sci. 2020, 11, 4578–4583. [Google Scholar] [CrossRef] [Green Version]
- Kiselev, E.; Ravji, A.; Kankanala, J.; Xie, J.; Wang, Z.; Pommier, Y. Novel deazaflavin tyrosyl-DNA phosphodiesterase 2 (TDP2) inhibitors. DNA Repair 2020, 85, 102747. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Georgousaki, K.; Tsafantakis, N.; Gumeni, S.; Lambrinidis, G.; González-Menéndez, V.; Tormo, J.R.; Genilloud, O.; Trougakos, I.P.; Fokialakis, N. Biological evaluation and in silico study of benzoic acid derivatives from Bjerkandera adusta targeting proteostasis network modules. Molecules 2020, 25, 666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Yang, P.-P.; He, P.-P.; Wen, S.-F.; Zou, X.-R.; Fan, Y.; Chen, Z.-M.; Cao, H.; Yang, Z.; Yue, K.; et al. Peptide-based nanoparticles mimic fibrillogenesis of laminin in tumor vessels for precise embolization. ACS Nano 2020. [Google Scholar] [CrossRef]
- D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Are carbonic anhydrases suitable targets to fight protozoan parasitic diseases? Curr. Med. Chem. 2018, 25, 5266–5278. [Google Scholar] [CrossRef]
- Angeli, A.; Etxebeste-Mitxeltorena, M.; Sanmartín, C.; Espuelas, S.; Moreno, E.; Azqueta, A.; Parkkila, S.; Carta, F.; Supuran, C.T. Tellurides bearing sulfonamides as novel inhibitors of leishmanial carbonic anhydrase with potent antileishmanial activity. J. Med. Chem. 2020, 63, 4306–4314. [Google Scholar] [CrossRef]
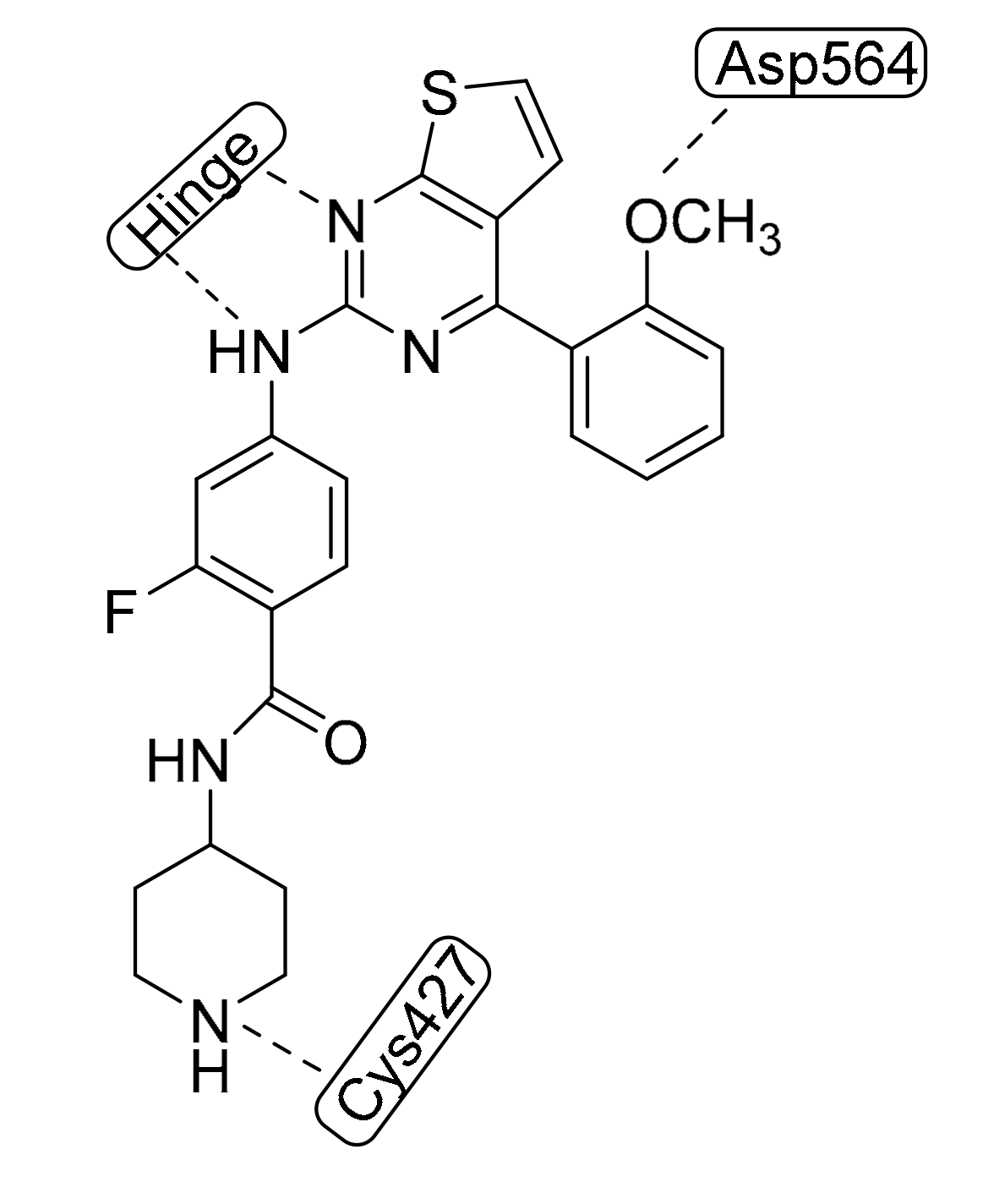
- Wang, R.; Yu, S.; Zhao, X.; Chen, Y.; Yang, B.; Wu, T.; Hao, C.; Zhao, D.; Cheng, M. Design, synthesis, biological evaluation and molecular docking study of novel thieno[3,2-d]pyrimidine derivatives as potent FAK inhibitors. Eur. J. Med. Chem. 2020, 188, 112024. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Zhou, S.; Graham, R.L.; Pruijssers, A.J.; Agostini, M.L.; Leist, S.R.; Schäfer, A.; Dinnon, K.H., 3rd; Stevens, L.J.; et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Trans. Med. 2020, 12, eabb5883. [Google Scholar] [CrossRef] [Green Version]
- Kettle, J.G.; Bagal, S.K.; Bickerton, S.; Bodnarchuk, M.S.; Breed, J.; Carbajo, R.J.; Cassar, D.J.; Chakraborty, A.; Cosulich, S.; Cumming, I.; et al. Structure-based design and pharmacokinetic optimization of covalent allosteric inhibitors of the mutant GTPase KRAS(G12C). J. Med. Chem. 2020, 63, 4468–4483. [Google Scholar] [CrossRef]
- Xue, J.Y.; Zhao, Y.; Aronowitz, J.; Mai, T.T.; Vides, A.; Qeriqi, B.; Kim, D.; Li, C.; de Stanchina, E.; Mazutis, L.; et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 2020, 577, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Luh, L.M.; Scheib, U.; Jünemann, K.; Wortmann, L.; Brands, M.; Cromm, P.M. Prey for the proteasome: Targeted protein degradation—A medicinal chemist´s perspective. Angew. Chem. Int. Ed. 2020. [Google Scholar] [CrossRef]
- He, Y.; Zhang, X.; Chang, J.; Kim, H.-N.; Zhang, P.; Wang, Y.; Khan, S.; Liu, X.; Zhang, X.; Lv, D.; et al. Using proteolysis-targeting chimera technology to reduce navitoclax platelet toxicity and improve its senolytic activity. Nat. Commun. 2020, 11, 1996. [Google Scholar] [CrossRef] [PubMed]
- Ballak, D.B.; Brunt, V.E.; Sapinsley, Z.J.; Ziemba, B.P.; Richey, J.J.; Zigler, M.C.; Johnson, L.C.; Gioscia-Ryan, R.A.; Culp-Hill, R.; Eisenmesser, E.Z.; et al. Short-term interleukin-37 treatment improves vascular endothelial function, endurance exercise capacity, and whole-body glucose metabolism in old mice. Aging Cell 2020, 19, e13074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotfy, H.; Moaaz, M.; Moaaz, M. The novel role of IL-37 to enhance the anti-inflammatory response of regulatory T cells in patients with peripheral atherosclerosis. Vascular 2020. [Google Scholar] [CrossRef]
- Guo, S.; Vieweger, M.; Zhang, K.; Yin, H.; Wang, H.; Li, X.; Li, S.; Hu, S.; Sparreboom, A.; Evers, B.M.; et al. Ultra-thermostable RNA nanoparticles for solubilizing and high-yield loading of paclitaxel for breast cancer therapy. Nat. Commun. 2020, 11, 972. [Google Scholar] [CrossRef] [Green Version]
- Roth, B.L.; Irwin, J.J.; Shoichet, B.K. Discovery of new GPCR ligands to illuminate new biology. Nat. Chem. Biol. 2017, 13, 1143–1151. [Google Scholar] [CrossRef] [Green Version]
- Che, T.; English, J.; Krumm, B.E.; Kim, K.; Pardon, E.; Olsen, R.H.J.; Wang, S.; Zhang, S.; Diberto, J.F.; Sciaky, N.; et al. Nanobody-enabled monitoring of kappa opioid receptor states. Nat. Commun. 2020, 11, 1145. [Google Scholar] [CrossRef]
- Mycroft-West, C.; Su, D.; Elli, S.; Guimond, S.; Miller, G.; Turnbull, J.; Yates, E.; Guerrini, M.; Fernig, D.; Skidmore, M.L.M. The 2019 coronavirus (SARS-CoV-2) surface protein (Spike) S1 receptor binding domain undergoes conformational change upon heparin binding. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Quek, J.P.; Liu, S.; Li, Z.Z.Y.; Ngb, E.Y.; Loh, Y.R.; Hung, A.W.; Luoa, D.; Kang, C.B. Identification and structural characterization of small molecule fragments targeting Zika virus NS2B-NS3 protease. Antivir. Res. 2020, 175, 104707. [Google Scholar] [CrossRef]
- Wang, L.; Casey, M.C.; Vernekar, S.K.V.; Do, H.T.; Sahani, R.L.; Kirby, K.A.; Du, H.; Hachiya, A.; Zhang, H.; Tedbury, P.R.; et al. Chemical profiling of HIV-1 capsid-targeting antiviral PF74. Eur. J. Med. Chem. 2020, 200, 112427–112448. [Google Scholar] [CrossRef] [PubMed]
- Schulze, V.K.; Klar, U.; Kosemund, D.; Wengner, A.M.; Siemeister, G.; Stöckigt, D.; Neuhaus, R.; Lienau, P.; Bader, B.; Prechtl, S.; et al. Treating cancer by spindle assembly checkpoint abrogation: Discovery of two clinical candidates, BAY 1161909 and BAY 1217389, targeting MPS1 kinase. J. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARSCoV-2 spike glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Secor, P.R.; Michaels, L.A.; Smigiel, K.S.; Rohani, M.G.; Jennings, L.K.; Hisert, K.B.; Arrigoni, A.; Braun, K.R.; Birkland, T.P.; Lai, Y.; et al. Filamentous bacteriophage produced by Pseudomonas aeruginosa alters the inflammatory response and promotes noninvasive infection in vivo. Infect. Immun. 2016, 85, e00648-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.W. Bacteria suit up with virus armor. Proc. Natl. Acad. Sci. USA 2020, 117, 6297–6299. [Google Scholar] [CrossRef]
- Tarafder, A.K.; von Kügelgen, A.; Mellul, A.J.; Schulze, U.; Aarts, D.G.A.L.; Bharat, T.A.M. Phage liquid crystalline droplets form occlusive sheaths that encapsulate and protect infectious rod-shaped bacteria. Proc. Natl. Acad. Sci. USA 2020, 117, 4724–4731. [Google Scholar] [CrossRef] [Green Version]
- Luchinat, E.; Barbieri, L.; Cremonini, M.; Nocentini, A.; Supuran, C.T.; Banci, L. Drug screening in human cells by NMR spectroscopy allows the early assessment of drug potency. Angew. Chem. Int. Ed. Engl. 2020, 59, 6535–6539. [Google Scholar] [CrossRef] [Green Version]
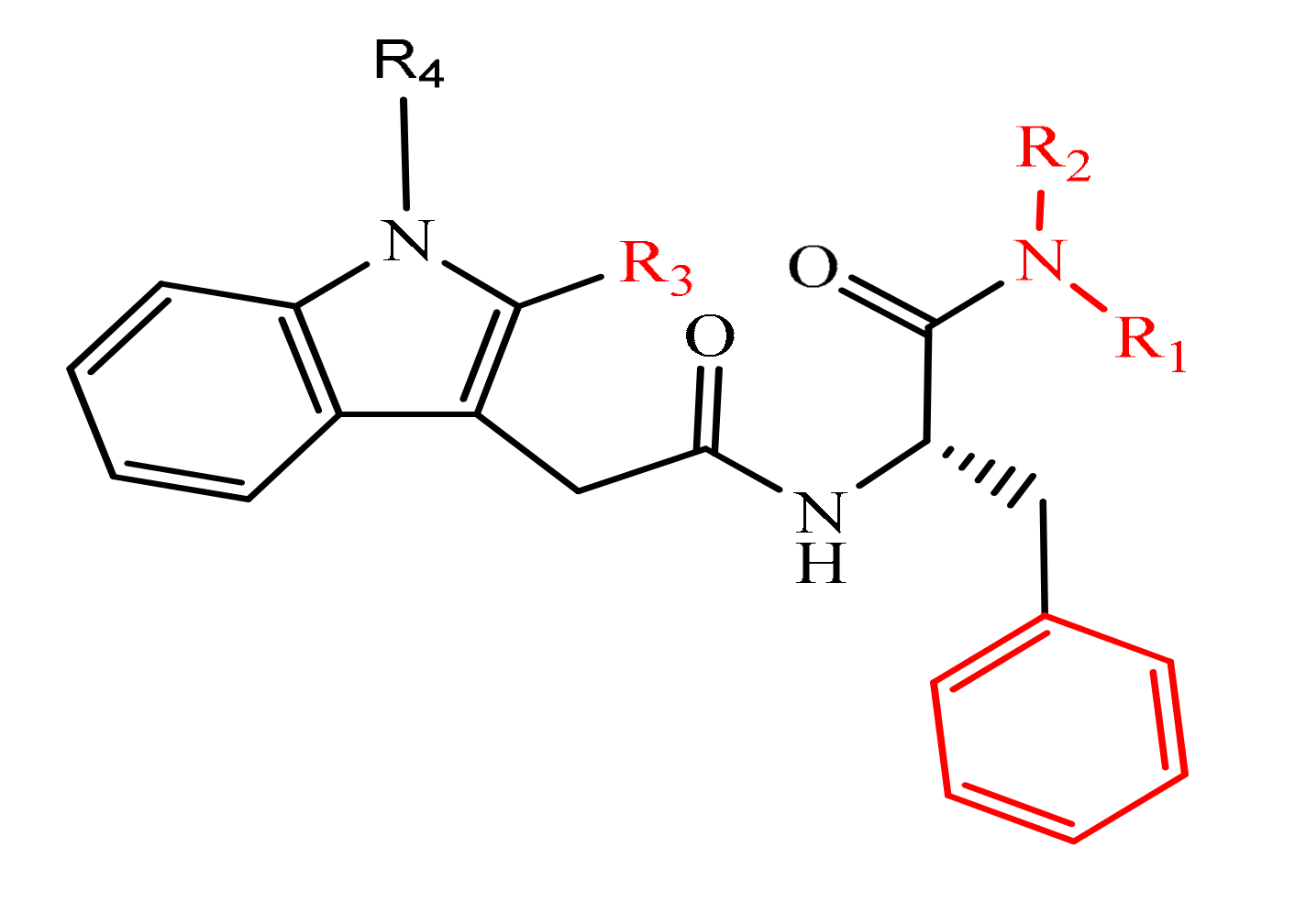
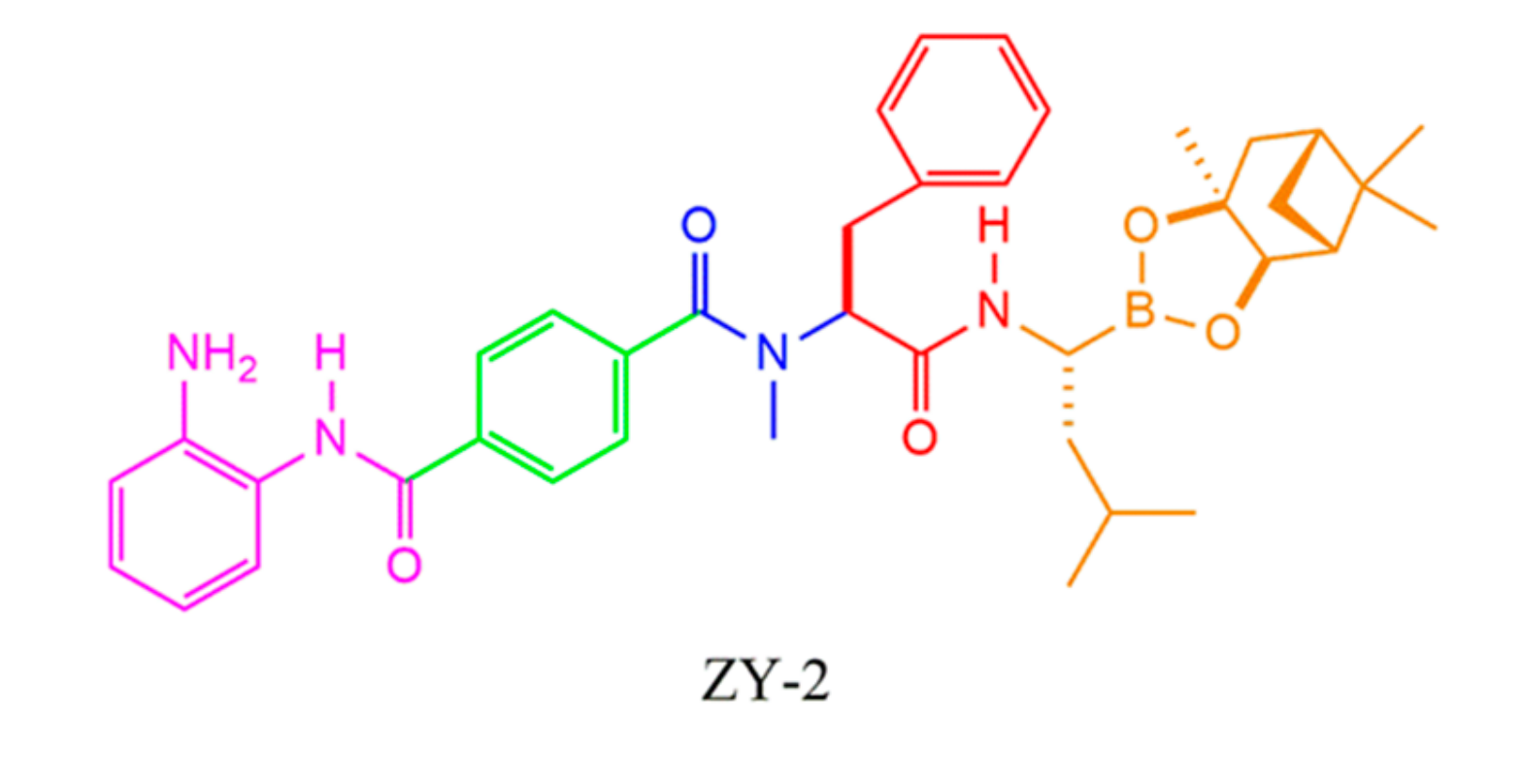
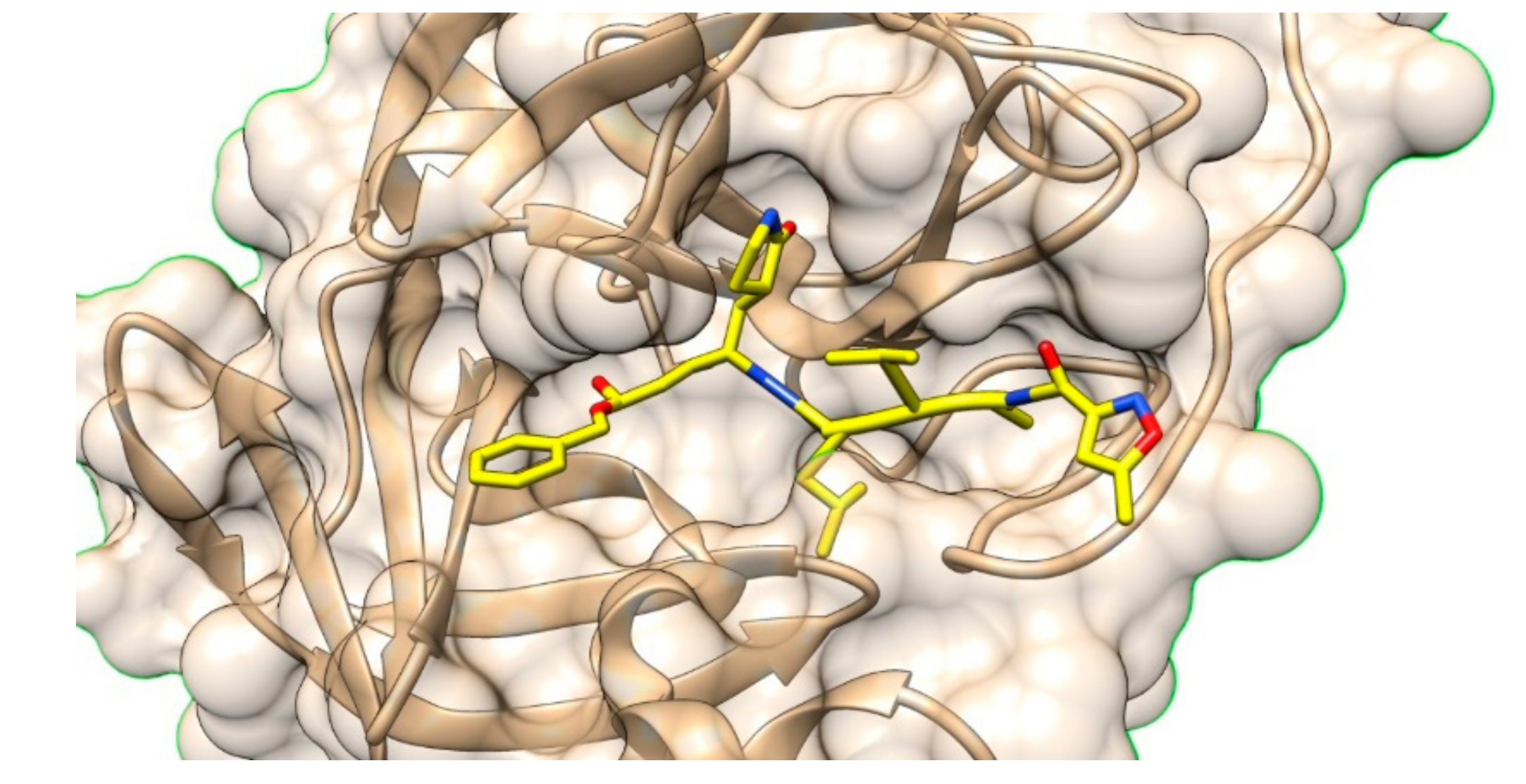
- Zhou, Y.; Liu, X.; Xue, J.; Liu, L.; Liang, T.; Li, W.; Yang, X.; Hou, X.; Fang, H. Discovery of peptide boronate derivatives as histone deacetylase and proteasome dual inhibitors for overcoming bortezomib resistance of multiple myeloma. J. Med. Chem. 2020, 63, 4701–4715. [Google Scholar] [CrossRef]
- Zaman, A.; Wu, W.; Bivona, T.G. Targeting oncogenic BRAF: Past, present, and future. Cancers 2019, 11, 1197. [Google Scholar] [CrossRef] [Green Version]
- Han, X.-R.; Chen, L.; Wei, Y.; Yu, W.; Chen, Y.; Zhang, C.; Jiao, B.; Shi, T.; Sun, L.; Zhang, C.; et al. Discovery of selective small molecule degraders of BRAF-V600E. J. Med. Chem. 2020, 63, 4069–4080. [Google Scholar] [CrossRef]
- Saha, P.; Panda, D.; Müller, D.; Maity, A.; Schwalbe, H.; Dash, J. In situ formation of transcriptional modulators using non-canonical DNA i-motifs. Chem. Sci. 2020, 11, 2058–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.-X.; Wang, C.-Y.; Li, Y.-Y.; Li, J.; Wan, Q.-Q.; Chen, J.-H.; Tay, F.R.; Niu, L.-N. Considerations and caveats in combating ESKAPE pathogens against nosocomial infections. Adv. Sci. 2020, 7, 1901872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef]
- Zipperer, A.; Konnerth, M.C.; Laux, C.; Berscheid, A.; Janek, D.; Weidenmaier, C.; Burian, M.; Schilling, N.A.; Slavetinsky, C.; Marschal, M.; et al. Human commensals producing a novel antibiotic impair pathogen colonization. Nature 2016, 535, 511–516. [Google Scholar] [CrossRef]
- Matsui, K.; Kan, Y.; Kikuchi, J.; Matsushima, K.; Takemura, M.; Maki, H.; Kozono, I.; Ueda, T.; Minagawa, K. Stalobacin: Discovery of novel lipopeptide antibiotics with potent antibacterial activity against multidrug-resistant bacteria. J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackermann, Z.; et al. A deep learning approach to antibiotic discovery. Cell 2020, 180, 688–702.e13. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Chen, H.; Wang, P.; Li, Y.; Wold, E.A.; Leonard, P.G.; Joseph, S.; Brasier, A.L.; Tian, B.; Zhou, J. Discovery of orally bioavailable chromone derivatives as potent and selective BRD4 inhibitors: Scaffold hopping, optimization, and pharmacological evaluation. J. Med. Chem. 2020, 63, 5242–5256. [Google Scholar] [CrossRef]
- Mashima, T.; Lee, J.; Kamatari, Y.O.; Hayashi, T.; Nagata, T.; Nishikawa, F.; Nishikawa, S.; Kinoshita, M.; Kuwata, K.; Katahira, M. Development and structural determination of an anti-PrPC aptamer that blocks pathological conformational conversion of prion protein. Sci. Rep. 2020, 10, 4934. [Google Scholar] [CrossRef] [Green Version]
- Schalk, I.J.; Mislin, G.L.A. Bacterial iron uptake pathways: Gates for the import of bactericide compounds. J. Med. Chem. 2017, 60, 4573–4576. [Google Scholar] [CrossRef]
- Goldberg, J.A.; Nguyen, H.; Kumar, V.; Spencer, E.J.; Hoyer, D.; Marshall, E.K.; Cmolik, A.; O’Shea, M.; Marshall, S.H.; Hujer, A.M.; et al. A γ-lactam siderophore antibiotic effective against multidrug-resistant Gram-negative bacilli. J. Med. Chem. 2020, 63, 5990–6002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J.; et al. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: Structure-based design, synthesis, and activity assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaradhi, R.; Banik, A.; Mohammed, S.; Patro, V.; Rojas, A.; Wang, W.; Motati, D.R.; Dingledine, R.; Ganesh, T. Potent, selective, water soluble, brain-permeable EP2 receptor antagonist for use in central nervous system disease models. J. Med. Chem. 2020, 63, 1032–1050. [Google Scholar] [CrossRef]
- Qiu, J.; Nie, Y.; Zhao, Y.; Zhang, Y.; Li, L.; Wang, R.; Wang, M.; Chen, S.; Wang, J.; Li, Y.Q.; et al. Safeguarding intestine cells against enteropathogenic Escherichia coli by intracellular protein reaction, a preventive antibacterial mechanism. Proc. Natl. Acad. Sci. USA 2020, 117, 5260–5268. [Google Scholar] [CrossRef]
- Jarlstad Olesen, M.T.; Walther, R.; Poier, P.P.; Dagnæs-Hansen, F.; Zelikin, A.N. Molecular, macromolecular, and supramolecular glucuronide prodrugs: Lead identified for anticancer prodrug monotherapy. Angew. Chem. Int. Ed. 2020, 59, 7390–7396. [Google Scholar] [CrossRef]












© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gütschow, M.; Vanden Eynde, J.J.; Jampilek, J.; Kang, C.; Mangoni, A.A.; Fossa, P.; Karaman, R.; Trabocchi, A.; Scott, P.J.H.; Reynisson, J.; et al. Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes–7. Molecules 2020, 25, 2968. https://doi.org/10.3390/molecules25132968
Gütschow M, Vanden Eynde JJ, Jampilek J, Kang C, Mangoni AA, Fossa P, Karaman R, Trabocchi A, Scott PJH, Reynisson J, et al. Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes–7. Molecules. 2020; 25(13):2968. https://doi.org/10.3390/molecules25132968
Chicago/Turabian StyleGütschow, Michael, Jean Jacques Vanden Eynde, Josef Jampilek, CongBao Kang, Arduino A. Mangoni, Paola Fossa, Rafik Karaman, Andrea Trabocchi, Peter J. H. Scott, Jóhannes Reynisson, and et al. 2020. "Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes–7" Molecules 25, no. 13: 2968. https://doi.org/10.3390/molecules25132968