
How Flexible Is the Concept of Local Thermodynamic Equilibrium?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Basic Approach Leading to the Generalized Phenomenological Irreversible Thermodynamic Theory


3. Transforming GPITT to All Macroscopic Variables
3.1. Gibbs Relation of GPITT with Internal Variables as Fast Variables
3.1.1. Gibbs Relation with Internal Variables as Additional Variables: Fast Domain of Time
3.1.2. Gibbs Relation with Internal Variables as Additional Variables: Slow Domain of Time
3.2. Gibbs Relation of GPITT with Physical Fluxes as Additional Variables
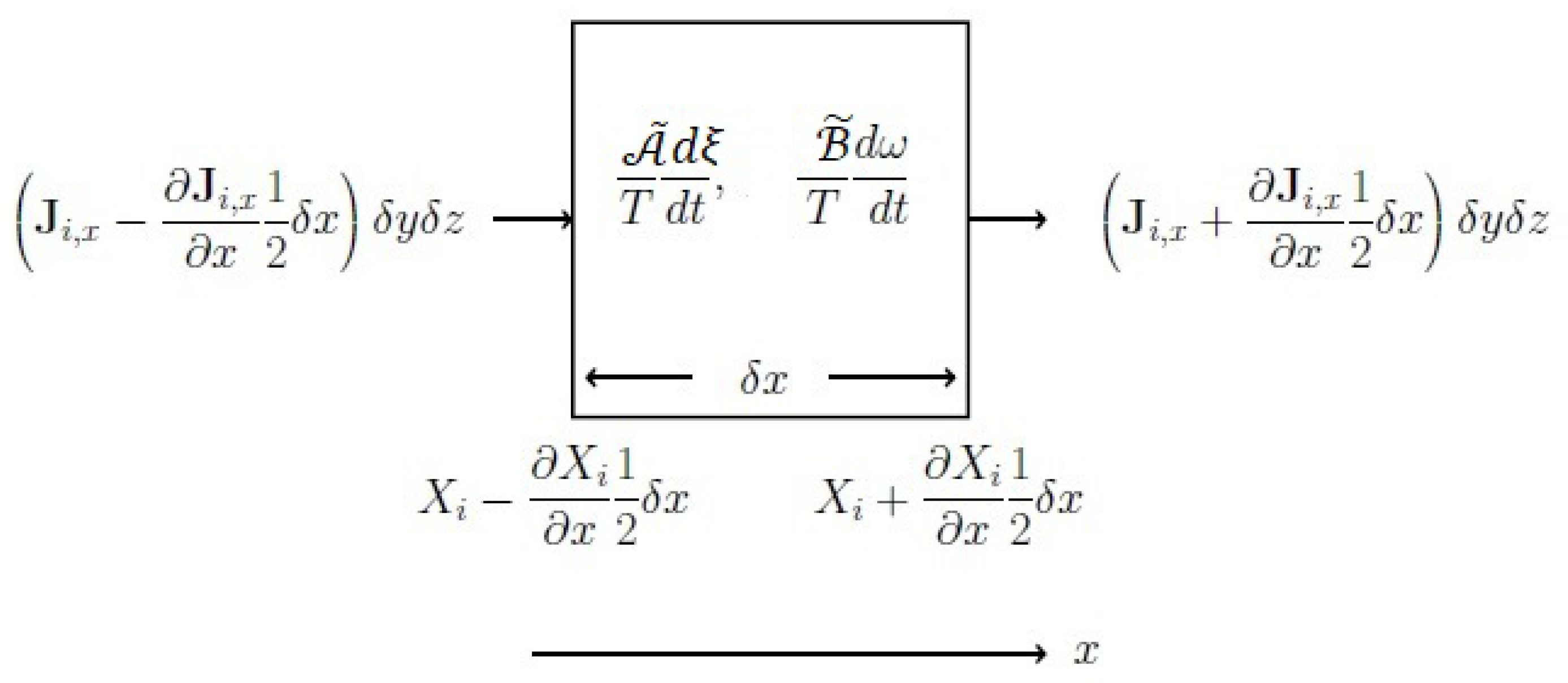
3.2.1. Constitutive Equations of Physical Fluxes for a Spatially Non-Uniform System
3.2.2. Gibbs Relation with Physical Fluxes as Additional Variables. Implications of Fast and Slow Variables
3.2.3. Rate of Change of Entropy in the Fast Domain of Time Using Constitutive Equations for the Physical Fluxes
- In the case of Maxwell–Cattaneo-type constitutive equations given in Equations (87) to (89), the entropy source strength for the fast domain of time reads as,It is easy to realize that the last three terms on the r.h.s. of Equation (92) are individually negative, provided that the -coefficients are positive quantities. However, in this event, the second law of thermodynamics guarantees that the magnitude of the sum of the squared terms preceding them remains greater than the sum of the gradients involved terms. Additionally, with the variation of time, the last three terms on the r.h.s. of Equation (92) contribute only via the variation of respective fluxes.
3.2.4. Rate of Change of Entropy in the Slow Domain of Time Using Constitutive Equations for the Physical Fluxes
- (I).
- Since Equation (96) is the description within the segment belonging to the slow domain of time of the evolution, in general, we do have , and all . This also implies , and all .
- (II).
- In the case of Guyer–Krumhansl’s relation, Equation (68), it reads in the slow domain of time as
- (III).
- (i).
- (ii).
- In the case of rigid body heat conduction on using the expression of Equation (100), the entropy source strength reads as,
- (iii).
3.2.5. The Rate of Change of Entropy Using Physical Fluxes as Additional Variables: Fast and Slow Domains of Time Taken Together
- We use Jeffreys’ type constitutive equations of Equations (69), (73) and (75), and assume the absence of body forces. Since no interaction with radiations is assumed, this exercise generates the following expression of entropy source strength:and is still given by Equation (108). Notice that Equation (109) has three additional terms (the last three terms on its r.h.s.) compared to that of Equation (107).
3.2.6. Fast Versus Slow Domains of Time with Physical Fluxes as Additional Variables
4. The Nature of Temperature in GPITT
“Three tiny volume elements are instantly and simultaneously isolated from their respective nonequilibrium systems and in the same instant they are brought into diathermal contacts as closed rigid systems, 1 with 2 and 2 with 3. If within the short time interval of sensing of the thermal interactions it is found that 1 is in momentary thermal equilibrium with 2 and 2 is with 3 then 3 is also in momentary thermal equilibrium with 1. The momentary thermal equilibrium means that if the volume elements possessed the heat fluxes then they remain unaffected during the minimum short period of thermal interactions and if no heat flux existed, both such nonequilibrium and the equilibrium states included, no heat flux gets generated during the said diathermal contact. The making of a diathermal contact between the tiny volume elements one of which having a heat flux and the other one without it is not forbidden”.
5. Concluding Remarks
and“The universe of the operation of thermodynamics is determined by the instrumental operations of the laboratory. Thus all the measurements that we are now capable of performing the thermodynamic measurements form a sub-group.".
“…in general the analysis of such systems will be furthered by the recognition of a new type of large scale thermodynamic parameters of state, namely the parameters of the state which can be measured but not controlled. Examples are the order-disorder rearrangements in mixed crystals, measurable by X-rays, and dislocations in a solid, measurable by the attenuation of supersonic vibrations. These parameters are measurable, but they are not controllable, which means that they are coupled to no external force variable which might provide the means of control. And not being coupled to a force variable, they cannot take part in mechanical work. Such a parameter of state, which enters into no term in the mechanical work, can be shown by simple analysis to be one which can take part only in irreversible changes."
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CIT | Classical Irreversible Thermodynamics |
| LTE | Local Thermodynamic Equilibrium |
| EIT | Extended Irreversible Thermodynamics |
| GPITT | Generalized Phenomenological Irreversible Thermodynamic Theory |
| TIV | Thermodynamics with internal variables |
| CT | Computational thermodynamics |
| IR | Infrared |
| UV-VIS | Ultra Violet-Visible |
| NMR | Nuclear Magnetic Resonance |
| ESR | Electron Spin Resonance |
| DSMC | Direct Simulation Monte Carlo |
Symbols and Notations
| Symbols/Notations | |
| p | is the local pressure |
| T | is the local temperature |
| s | is the per unit mass local entropy |
| u | is the per unit mass local internal energy |
| v | is the specific volume |
| is the molar mass of the component k | |
| represents a set of mass fractions of the components within the system | |
| represents the set of mass fractions of the components in the quantum states identified by the symbol j when the quantum states have nonequilibrium population | |
| corresponds to the equilibrium population of the quantum states | |
| is the quantum state-wise mass density with respect to the mass density for the equilibrium population of quantum states | |
| is the quantum state-wise mass density with respect to the mass density for the nonequilibrium population of quantum states | |
| is the local chemical potential per unit mass of the component k when the quantum states have equilibrium population | |
| corresponds to when the quantum states have nonequilibrium population | |
| is the quantum state-wise chemical potential when the system is in equilibrium | |
| is the chemical potential when there we have nonequilibrium population of the quantum states | |
| t | is time |
| is the positional coordinate | |
| is the entropy flux density | |
| is the entropy source strength | |
| is the mass density | |
| is the mass density of the component k | |
| is the quantum statewise mass density for the equilibrium population of quantum states | |
| is the quantum statewise mass density for the nonequilibrium population of quantum states | |
| is the so-called heat flux density | |
| is the diffusion flux density of the component k | |
| is the component-wise diffusion flux density of the component k when the system has the local equilibrium population of quantum states | |
| is the component-wise diffusion flux density of the component k when the system has the local nonequilibrium population of quantum states | |
| is the dissipative stress tensor | |
| is the barycentric velocity vector | |
| is the chemical affinity as defined by Equation (8) for the -th chemical reaction when the quantum states have local equilibrium population | |
| is the chemical affinity of -th chemical reaction when system has nonequilibrium population of quantum states | |
| is the extent of the advancement of -th chemical reaction and with other subscripts it denotes the corresponding extent of advancement of the process | |
| is the affinity of internal population equilibration defined in Equation (18) | |
| is the i-th component of the intensity X | |
| is the per unit mass local Gibbs function | |
| is the effective thermal conductivity | |
| is the shear viscosity | |
| is the Giesekus parameter or is the elasticity modulus of material | |
| is the thermal conductivity | |
| l | is the mean free path of phonons |
| is the diffusion coefficient of the component k | |
| is the mole number of the component k | |
| D | is the driving force of internal process as defined in Equation (35) |
| is the conservative body force for the component k | |
| is the stoichiometric coefficient of the component k in the -th reaction | |
| or | are the nonequilibrium entropy proposed in EIT, TIV and Keizer’s version of thermodynamics of irreversible processes |
| or | are the nonequilibrium temperatures used in EIT, TIV and Keizer’s version of thermodynamics of irreversible processes, and is also used for denoting the temperature in energy units |
| and | are the nonequilibrium pressures used in EIT, TIV and Keizer’s version of thermodynamics of irreversible processes |
| f and | are the molecular velocity distribution functions in nonequilibrium and Maxwellian respectively |
| is the nonequilibrium contribution to the nonequilibrium velocity distribution function | |
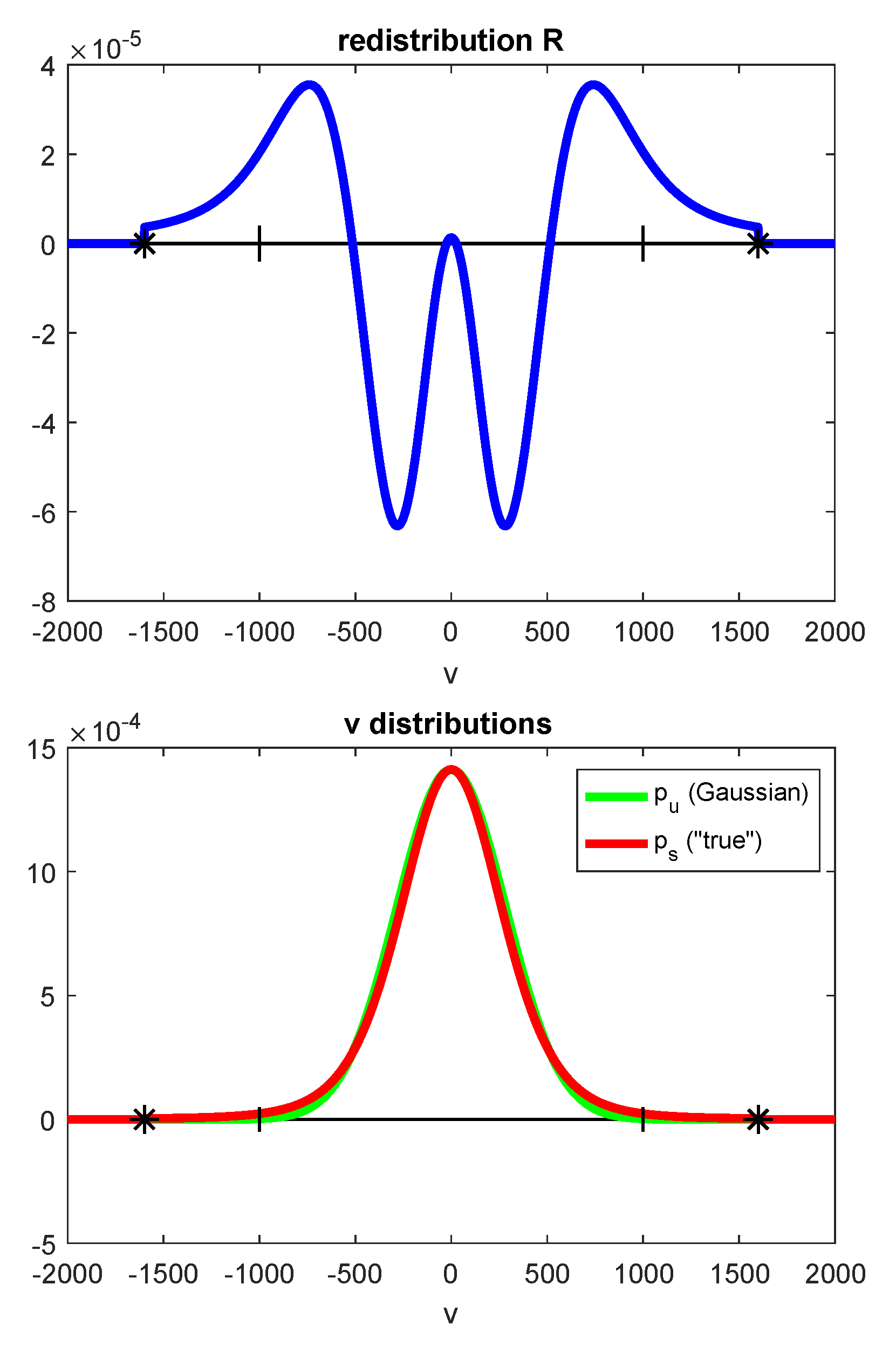
| is the redistribution function (this symbol is also used in photochemical reactions for reactants), is the true distribution and is the Gaussian | |
| is the stoichiometric coefficient of the component k in the collisional mechanism of the population equilibration process for the j-th quantum state | |
| is the extent of advancement of population equilibration in internal molecular quantum states | |
| is the molecular energy in a quantum state and the subscript j to it identifies the quantum state and the other subscripts specify which type of quantum state is referred to | |
| , , … | represent electronic singlet states: ground, first excited, … |
| , , … | represent electronic triplate states: ground, first excited, … |
| stands for molarity inverse and second (time) inverse | |
| is the stoichiometric coefficient of rotational–vibrational collisional mechanism of the population equilibration | |
| , , | are the physical coefficients those appear in Equation (55) |
| is fractionally weighted relaxation time for the time rate of change of the concentration gradient | |
| , , | are the relaxation times for the decay of heat flux density, dissipative momentum flux density and the matter diffusion flux density respectively |
| is the retardation time of the viscoelastic fluids | |
| is also been used to denote the dimensionless mobility factor in viscoelastic fluids | |
| is the chemical potential in the standard state of unit concentration of the component k at the given magnitudes of physical fluxes to which belongs | |
| R | is the universal gas constant |
| is the differential amount of heat exchanged by the system at temperature from a system at temperature | |
| is the universal heat capacity ratio | |
References
- De Groot, S.R.; Mazur, P. Non-Equilibrium Thermodynamics; North Holland: Amsterdam, The Netherlands, 1962. [Google Scholar]
- Kjelstrup, S.; Bedeaux, D. Non-Equilibrium Thermodynamics of Heterogeneous Systems. In Series on Advances in Statistical Mechanics, 1st ed.; Rasetti, M., Ed.; World Scientific: Hackensack, NJ, USA, 2008; Volume 16. [Google Scholar]
- Prigogine, I. Introduction to Thermodynamics of Irreversible Processes; John Wiley-Interscience: New York, NY, USA, 1967. [Google Scholar]
- Haase, R. Thermodynamics of Irreversible Processes; Addison-Wesley: Reading, MA, USA, 1969. [Google Scholar]
- Gyarmati, I. Non-Equilibrium Thermodynamics; Springer: Berlin, Germany, 1970. [Google Scholar]
- García-Colín, L.S. Termodinámica de Processos Irreversibles; Colección CBI-UAM: Iztapalapa, Mexico, 1990. [Google Scholar]
- Hillert, M. Phase Equilibria, Phase Diagrams and Phase Transformations. Their Thermodynamic Basis, 2nd ed.; Cambridge University Press: New York, NY, USA, 2008; ISBN 978-0-51150-620-8. [Google Scholar]
- Rubin, M.H.; Andresen, B. Optimal Staging of Endoreversible Heat Engines. J. Appl. Phys. 1982, 53, 1–7. [Google Scholar] [CrossRef]
- Andresen, B. Current Trends in Finite-Time Thermodynamics. Angew. Chem. Int. Ed. 2011, 50, 2690–2705. [Google Scholar] [CrossRef] [PubMed]
- Onsager, L. Reciprocal Relations in Irreversible Processes. I. Phys. Rev. 1931, 37, 405–426. [Google Scholar] [CrossRef]
- Onsager, L.; Machlup, S. Fluctuations and Irreversible Processes. Phys. Rev. 1953, 91, 1505–1515. [Google Scholar] [CrossRef]
- Rastogi, R.P. Introduction to Non-Equilibrium Physical Chemistry. Towards Complexity and Non-Linear Sciences; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- White, W.B.; Johnson, S.M.; Dantzig, G.B. Chemical Equilibrium in Complex Mixtures. J. Chem. Phys. 1958, 28, 751–755. [Google Scholar] [CrossRef]
- Eriksson, G. Thermodynamic Studies of High Temperature Equilibria. III. SOLGAS, a Computer Program for Calculating the Composition and Heat Condition of an Equilibrium Mixture. Acta Chem. Scand 1971, 25, 2651–2658. [Google Scholar] [CrossRef]
- Dinsdale, A.T. SGTE Data for Pure Elements. Calphad 1991, 15, 317–425. [Google Scholar] [CrossRef]
- Andersson, J.-O.; Helander, T.; Höglud, L.; Shi, P.; Sundman, B. THERMO-CALC & DICTRA, Computational Tools For Materials Science. Calphad 2002, 26, 273–312. [Google Scholar]
- Ågren, J.; Hayes, F.H.; Höglund, L.; Kattner, U.R.; Legendre, B.; Schmid-Fetzer, R. Applications of Computational Thermodynamics. Z. Metallkd. 2002, 93, 128–142. [Google Scholar] [CrossRef]
- Steinbach, I.; Böttger, B.; Eiken, J.; Warnken, N.; Fries, S.G. CALPHAD and Phase-Field Modeling: A Successful Liaison. J. Phase Equilibria Diffus. 2007, 28, 101–106. [Google Scholar] [CrossRef]
- Sundman, B.; Lu, X.G.; Ohtani, H. The Implementation of an Algorithm to Calculate Thermodynamic Equilibria for Multi-Component Systems With Non-Ideal Phases in a Free Software. Comput. Mater. Sci. 2015, 101, 127–137. [Google Scholar] [CrossRef]
- Kattner, U.R. The Calphad Method and Its Role in Material and Process Development. Tecnol. Metal. Mater. Min. 2016, 13, 3–15. [Google Scholar] [CrossRef]
- Kui Liu, Z.; Wang, Y. Computational Thermodynamics of Materials, 1st ed.; Cambridge University Press: Cambridge, UK, 2016; ISBN 978-0-52119-896-7. [Google Scholar]
- Kui Liu, Z. Computational Thermodynamics and Its Applications. Acta Mater. 2020, 200, 745–792. [Google Scholar] [CrossRef]
- Lebon, G.; Jou, D.; Casas-Vázquez, J. Understanding Non-Equilibrium Thermodynamics. Foundations, Applications, Frontiers; Springer: Berlin, Germany, 2008. [Google Scholar]
- Casas-Vázquez, J.; Jou, D.; Lebon, G. (Eds.) Recent developments in nonequilibrium thermodynamics. In Proceedings of the Meeting Held at Bellaterra School of Thermodynamics Autonomous University of Barcelona, Bellaterra, Barcelona, Spain, 26–30 September 1983. [Google Scholar]
- García-Colín, L.S.; Uribe, F.J. Extended Irreversible Thermodynamics Beyond the Linear Regime. A Critical Overview. J. Non-Equilib. Thermodyn. 1991, 16, 89–128. [Google Scholar]
- Jou, D.; Casas-Vázquez, J.; Lebon, G. Extended Irreversible Thermodynamics, 2nd ed.; Springer: Berlin, Germany, 1996. [Google Scholar]
- Müller, I.; Weiss, W. Irreversible Thermodynamics–Past and Present. Eur. Phys. J. H 2012, 37, 139–236. [Google Scholar] [CrossRef]
- Nettleton, R.E.; Sobolev, S.L. Applications of Extended Thermodynamics to Chemical, Rheological, and Transport Processes: A Special Survey Part I. Approaches and Scalar Rate Processes; and Part II. Vector Transport Processes, Shear Relaxation, and Rheology. J. Non-Equilib. Thermodyn. 1995, 20, 200–229, 297–331. [Google Scholar]
- Nettleton, R.E.; Sobolev, S.L. Applications of Extended Thermodynamics to Chemical, Rheological, and Transport Processes: A Special Survey Part III. Wave Phenomena. J. Non-Equilib. Thermodyn. 1996, 21, 1–16. [Google Scholar]
- Coleman, B.D.; Gurtin, M.E. Thermodynamics with Internal State Variables. J. Chem. Phys. 1967, 47, 597–613. [Google Scholar] [CrossRef] [Green Version]
- Šilhavy, M. A Condition Equivalent to the Existence of Non-Equilibrium Entropy and Temperature for Materials with Internal Variables. Arch. Ration. Mech. Anal. 1978, 68, 299–322. [Google Scholar] [CrossRef]
- Lype, E.F. Entropy of Systems with Internal Variables. Int. J. Thermophys. 1986, 7, 111–124. [Google Scholar] [CrossRef]
- Bampi, F.; Morro, A. Nonequilibrium Thermodynamics: A Hidden Variable Approach. In Proceedings of the Recent Developments in Nonequilibrium Thermodynamics, Barcelona, Spain, 26–30 September 1983; pp. 211–232. [Google Scholar]
- Muschik, W. Internal Variables in Non-Equilibrium Thermodynamics. In Recent Developments in Micromechanics; Springer: Berlin, Germany, 1991; Chapter 2; pp. 18–34. [Google Scholar]
- Lhuillier, D.; Grmela, M.; Lebon, G. A Comparative Study of the Coupling of Flow with Non-Fickean Thermodiffusion. Part III: Internal Variables. J. Non-Equilib. Thermodyn. 2003, 28, 51–68. [Google Scholar] [CrossRef]
- Maugin, G. The Thermodynamics of Nonlinear Irreversible Behaviors: An Introduction, 1st ed.; World Scientific Series on Nonlinear Science, Series A; World Scientific: Singapore, 1999; Volume 27. [Google Scholar]
- Maugin, G.; Muschik, W. Thermodynamics with Internal Variables. Part I. General Concepts. J. Non-Equilib. Thermodyn. 1994, 19, 217–249. [Google Scholar] [CrossRef]
- Maugin, G.; Muschik, W. Thermodynamics with Internal Variables. Part II. Applications. J. Non-Equilib. Thermodyn. 1994, 19, 250–289. [Google Scholar] [CrossRef]
- Pokrovskii, V.N. A Derivation of the Main Relations of Nonequilibrium Thermodynamics. Int. Sch. Res. Not. 2013, 2013, 906136. [Google Scholar] [CrossRef]
- Keizer, J. A Theory of Spontaneous Fluctuations in Microscopic Systems. J. Chem. Phys. 1975, 63, 398–403. [Google Scholar] [CrossRef]
- Keizer, J. Concentration Fluctuations in Chemical Reactions. J. Chem. Phys. 1975, 63, 5037–5043. [Google Scholar] [CrossRef]
- Keizer, J. Dissipation and Fluctuations in Nonequilibrium Thermodynamics. J. Chem. Phys. 1976, 64, 1679–1687. [Google Scholar] [CrossRef]
- Keizer, J. On the Kinetic Meaning of the Second Law of Thermodynamics. J. Chem. Phys. 1976, 64, 4466–4474. [Google Scholar] [CrossRef]
- Keizer, J. Fluctuations, Stability, and Generalized State Functions at Nonequilibrium Steady States. J. Chem. Phys. 1976, 65, 4431–4444. [Google Scholar] [CrossRef]
- Keizer, J. Thermodynamics at Nonequilibrium Steady States. J. Chem. Phys. 1978, 69, 2609–2620. [Google Scholar] [CrossRef]
- Keizer, J. Nonequilibrium Thermodynamics and the Stability of States Far from Equilibrium. Acc. Chem. Res. 1979, 12, 243–249. [Google Scholar] [CrossRef]
- Keizer, J. Reversibility, Work, and Heat at Nonequilibrium Steady States. Phys. Rev. A 1984, 30, 1115–1117. [Google Scholar] [CrossRef]
- Keizer, J. Heat, Work, and the Thermodynamic Temperature at Nonequilibrium Steady States. J. Chem. Phys. 1985, 82, 2751–2771. [Google Scholar] [CrossRef]
- Keizer, J. Statistical Thermodynamics of Nonequilibrium Processes; Springer: Berlin, Germany, 1987. [Google Scholar]
- Oláh, K.; Farkas, H.; Bódiss, J. The Entropy Dissipation Function. Period. Polytech. Bp. 1989, 33, 125–139. [Google Scholar]
- Lucia, U.; Grisolia, G. Nonequilibrium Temperature: An Approach from Irreversibility. Materials 2021, 14, 2004. [Google Scholar] [CrossRef]
- Bhalekar, A.A. Irreversible Thermodynamic Framework Using Compatible Equations from Thermodynamics and Fluid Dynamics. A Second Route to Generalized Phenomenological Irreversible Thermodynamic Theory (GPITT). Bull. Cal. Math. Soc. 2002, 94, 209–224. [Google Scholar]
- Bhalekar, A.A.; Andresen, B. On the Nonequilibrium Thermodynamic Roots of the Additional Facets of Chemical Interaction. In Recent Advances in Thermodynamic Research Including Nonequilibrium Thermodynamics, Proceedings of the 3rd National Conference on Thermodynamics of Chemical and Biological Systems (NCTCBS-2008), Nagpur, India, 16–17 October 2008; Natarajan, G.S., Bhalekar, A.A., Dhondge, S.S., Juneja, H.D., Eds.; Department of Chemistry, R. T. M. Nagpur University: Nagpur, India, 2008; pp. 53–62. [Google Scholar]
- Bhalekar, A.A. Thermodynamic Insight of Irreversibility. In Proceedings of the Third International Conference on Lie-Admissible Treatment of Irreversible Processes (ICLATIP-3), Dhulikhel, Nepal, 3–7 January 2011; pp. 135–162. [Google Scholar]
- Chapman, S.; Cowling, T.G. Mathematical Theory of Non-Uniform Gases: An Account of the Kinetic Theory of Viscosity, Thermal Conduction and Diffusion in Gases, 3rd ed.; Cambridge University Press: Cambridge, UK, 1970. [Google Scholar]
- Essex, C.; Andresen, B. The Ideal Gas in Slow Time. J. Non-Equilib. Thermodyn. 2021, 46, 35–43. [Google Scholar] [CrossRef]
- Liboff, R.L. Kinetic Theory: Classical, Quantum, and Relativistic Descriptions, 3rd ed.; Graduate Texts in Contemporary Physics; Springer: New York, NY, USA, 2003. [Google Scholar]
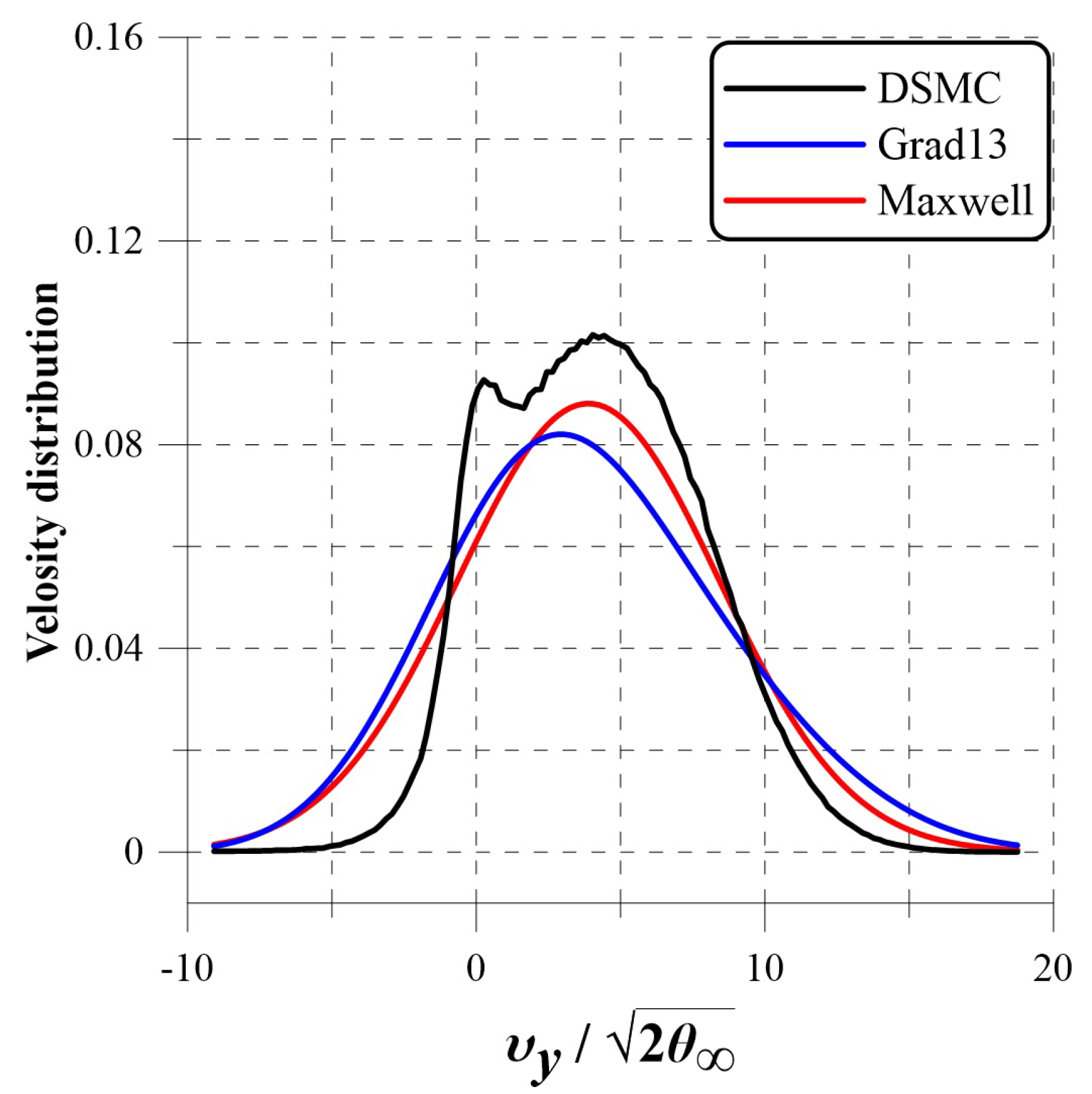
- Bondar, Y.; Shoev, G.; Kokhanchik, A.; Timokhin, M. Nonequilibrium velocity distribution in steady regular shock-wave reflection. AIP Conf. Proc. 2019, 2132, 120005-1–120005-8. [Google Scholar]
- Prigogine, I.; Defay, R. Chemical Thermodynamics; Everett, D.H., Translator; Longmans Green: London, UK, 1954. [Google Scholar]
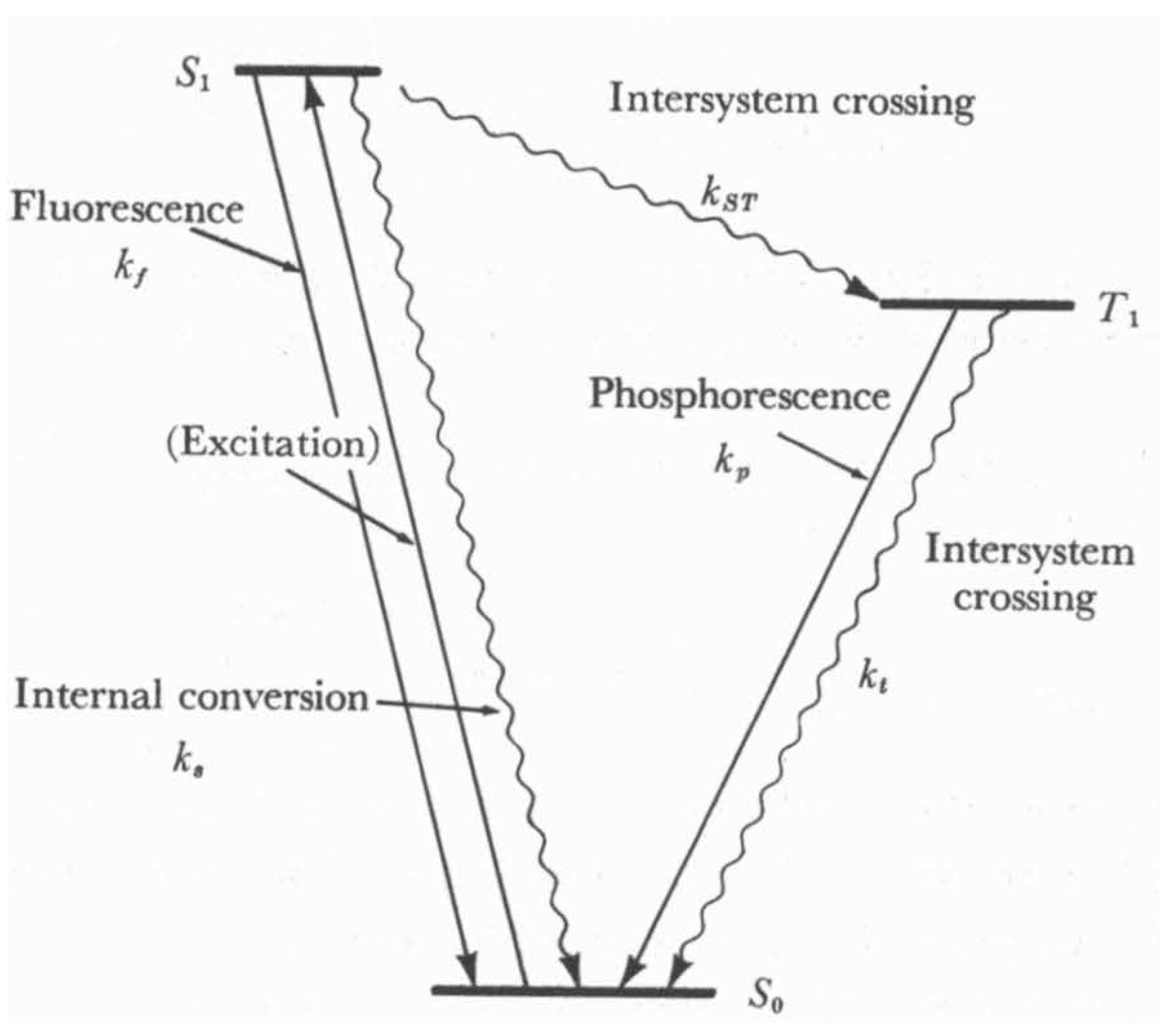
- Turro, N.J. Modern Molecular Photochemistry; University Science Books: Sausalito, CA, USA, 1991. [Google Scholar]
- Rohatgi-Mukherjee, K.K. Fundamentals of Photochemistry; New Age International: New Delhi, India, 1978. [Google Scholar]
- Turro, N.J.; Ramamurthy, V.; Scaiano, J.C. Principles of Molecular Photochemistry. An Introduction, Indian ed.; University Science Books/Viva Books Pvt. Ltd.: Mill Valley, CA, USA; New Delhi, India, 2015. [Google Scholar]
- Glasstone, S. Thermodynamics for Chemists; Princeton/D. Van Nostrand: Princeton, NJ, USA, 1967; New York, NY, USA, 1958. [Google Scholar]
- Lewis, G.N.; Randall, M. Thermodynamics, 2nd ed.; Pitzer, K.S., Brewer, L., Eds.; McGraw-Hill Book Co., Inc.: New York, NY, USA, 1961. [Google Scholar]
- Hague, D. Experimental Methods for the Study of Fast Reactions. In Comprehensive Chemical Kinetics; Elsevier: New York, NY, USA, 1969; Volume 1, Chapter 2; pp. 112–179. [Google Scholar]
- Sutin, N.; Creutz, C. Electron-Transfer Reactions of Excited States. J. Chem. Educ. 1983, 60, 809–814. [Google Scholar] [CrossRef]
- Denbigh, K.G. The Principles of Chemical Equilibrium; Cambridge University Press: Cambridge, UK, 1973. [Google Scholar]
- Jordan, R.B. Reaction Mechanisms of Inorganic and Organometallic Systems, 3rd ed.; Oxford University Press: Oxford, NY, USA, 2007. [Google Scholar]
- Saji, T.; Aoyagui, S. Electron-Transfer Kinetics of Transition-metal Complexes in Lower Oxidation States. Part I. Electron Spin Resonance Measurements of the Electron-Exchange Rate between Cr(bipy)31+ and Cr(bipy)3. Bull. Chem. Soc. Jpn. 1973, 46, 2101–2105. [Google Scholar] [CrossRef] [Green Version]
- Bhalekar, A.A. Extended Irreversible Thermodynamics and the Quality of Temperature and Pressure. Pramana-J. Phys. 1999, 53, 331–339. [Google Scholar] [CrossRef]
- Joseph, D.D.; Preziosi, L. Heat Waves. Rev. Mod. Phys. 1989, 61, 41–74. [Google Scholar] [CrossRef]
- Joseph, D.D.; Preziosi, L. Addendum to the Paper “Heat Waves”. Rev. Mod. Phys. 1989, 61, 41, Erratum in Rev. Mod. Phys.1990, 62, 375–391. [Google Scholar] [CrossRef]
- Guyer, R.A.; Krumhansl, J.A. Solution of Linearized Boltzmann Phonon Equation. Phys. Rev. 1966, 148, 766–778. [Google Scholar] [CrossRef]
- Straughan, B. Heat Waves, 1st ed.; Springer: Dordrecht, The Netherlands, 2011. [Google Scholar] [CrossRef]
- Joseph, D.D. Fluid Dynamics of Viscoelastic Liquids, 1st ed.; Springer: Berlin, Germany, 1990. [Google Scholar]
- Guillopè, C.; Talhouk, R. Steady Flows of Slightly Compressible Viscoelastic Fluids of Jeffreys’ Type Around an Obstacle. Differ. Integral Equ. 2003, 16, 1293–1320. [Google Scholar] [CrossRef]
- Rauscher, M.; Münch, A.; Wagner, B.; Blossey, R. A Thin-Film Equation for Viscoelastic Liquids of Jeffreys Type. Eur. Phys. J. E 2005, 17, 373–379. [Google Scholar] [CrossRef]
- Rukolaine, S.A.; Samsonov, A.M. Local Immobilization of Particles in Mass Transfer Described by a Jeffreys-Type Equation. Phys. Rev. E 2013, 88, 062116. [Google Scholar] [CrossRef] [Green Version]
- Balescu, R.C. Equilibrium and Nonequilibrium Statistical Mechanics, 1st ed.; Wiley-Interscience; John Wiley and Sons: New York, NY, USA, 1975; ISBN 0-47-104600-0. [Google Scholar]
- Bhalekar, A.A. The Universe of Operations of Thermodynamics vis-à-vis Boltzmann Integro-Differential Equation. Indian J. Phys. 2003, 77B, 391–397. [Google Scholar]
- García-Colín, L.S. Teoría Cinética de Los Gases; Colección CBI; Universidad Autónoma Metropolitana-Iztapalapa: Mexico City, Mexico, 1990; Volume 2, p. 65. [Google Scholar]
- Lewis, G.N.; Randall, M. Thermodynamics and the Free Energy of Chemical Substances, 1st ed.; McGraw-Hill Publishing Co.: London, UK, 1923. [Google Scholar]
- Holzwarth, J.F.; Schmidt, A.; Wolff, H.; Volk, R. Nanosecond Temperature-Jump Technique With an Iodine Laser. J. Phys. Chem. 1977, 81, 2300–2301. [Google Scholar] [CrossRef]
- Chen, S.; Lee, I.Y.S.; Tolbert, W.A.; Wen, X.; Dlott, D.D. Applicatlons of Ultrafast Temperature Jump Spectroscopy to Condensed Phase Molecular Dynamics. J. Phys. Chem. 1992, 96, 7178–7186. [Google Scholar] [CrossRef]
- Wen, X.; Tolbert, W.A.; Dlott, D.D. Multiphonon Up-Pumping and Molecular Hot Spots in Superheated Polymers Studied by Ultrafast Optical Calorimetry. Chem. Phys. Lett. 1992, 192, 315–320. [Google Scholar] [CrossRef]
- Lian, T.; Locke, B.; Kholodenko, Y.; Hochstrasser, R.M. Energy Flow from Solute to Solvent Probed by Femtosecond IR Spectroscopy: Malachite Green and Heme Protein Solutions. J. Phys. Chem. 1994, 98, 11648–11656. [Google Scholar] [CrossRef]
- Ma, H.; Wan, C.; Zewail, A.H. Ultrafast T-Jump in Water: Studies of Conformation and Reaction Dynamics at the Thermal Limit. J. Am. Chem. Soc. 2006, 128, 6338–6340. [Google Scholar] [CrossRef]
- Bhalekar, A.A.; García-Colín, L.S. On the Construction of an Extended Irreversible Thermodynamic Framework for Irreversible Processes. Pramana-J. Phys. 1998, 50, 295–305. [Google Scholar] [CrossRef]
- Bhalekar, A.A. On the Generalized Zeroth Law of Thermodynamics. Indian J. Phys. 2000, 74B, 153–157. [Google Scholar]
- Muschik, W.; Brunk, G.A. A Concept of Non-Equilibrium Temperature. Int. J. Eng. Sci. 1977, 15, 377–389. [Google Scholar] [CrossRef]
- Muschik, W. Empirical Foundation and Axiomatic Treatment of Non-Equilibrium Temperature. Arch. Ration. Mech. Anal. 1977, 66, 379–401. [Google Scholar] [CrossRef]
- Muschik, W.; Berezovski, A. Non-Equilibrium Contact Quantities and Compound Deficiency at Interfaces Between Discrete Systems. Proc.-Est. Acad. Sci. Phys. Math 2007, 56, 133–145. [Google Scholar]
- Bridgman, P.W. Reflections on Thermodynamics. Proc. Am. Acad. Arts Sci. 1953, 82, 301–309. [Google Scholar] [CrossRef]
- Bridgman, P.W. The Nature of Thermodynamics; Peter Smith: Gloucester, MA, USA, 1969. [Google Scholar]
- Lucia, U.; Grisolia, G.; Kuzemsky, A.L. Time, Irreversibility and Entropy Production in Nonequilibrium Systems. Entropy 2020, 22, 887. [Google Scholar] [CrossRef]
- Uffink, J. Bluff Your Way in the Second Law of Thermodynamics. Stud. Hist. Phil. Mod. Phys. 2001, 32, 305–394. [Google Scholar] [CrossRef] [Green Version]
- Landsberg, P.T. Entropies Galore! Braz. J. Phys. 1999, 29, 46–49. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tangde, V.M.; Bhalekar, A.A. How Flexible Is the Concept of Local Thermodynamic Equilibrium? Entropy 2023, 25, 145. https://doi.org/10.3390/e25010145
Tangde VM, Bhalekar AA. How Flexible Is the Concept of Local Thermodynamic Equilibrium? Entropy. 2023; 25(1):145. https://doi.org/10.3390/e25010145
Chicago/Turabian StyleTangde, Vijay M., and Anil A. Bhalekar. 2023. "How Flexible Is the Concept of Local Thermodynamic Equilibrium?" Entropy 25, no. 1: 145. https://doi.org/10.3390/e25010145