
Use of 6 Nucleotide Length Words to Study the Complexity of Gene Sequences from Different Organisms


Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Sequences
2.2. W Calculation Algorithm
3. Results
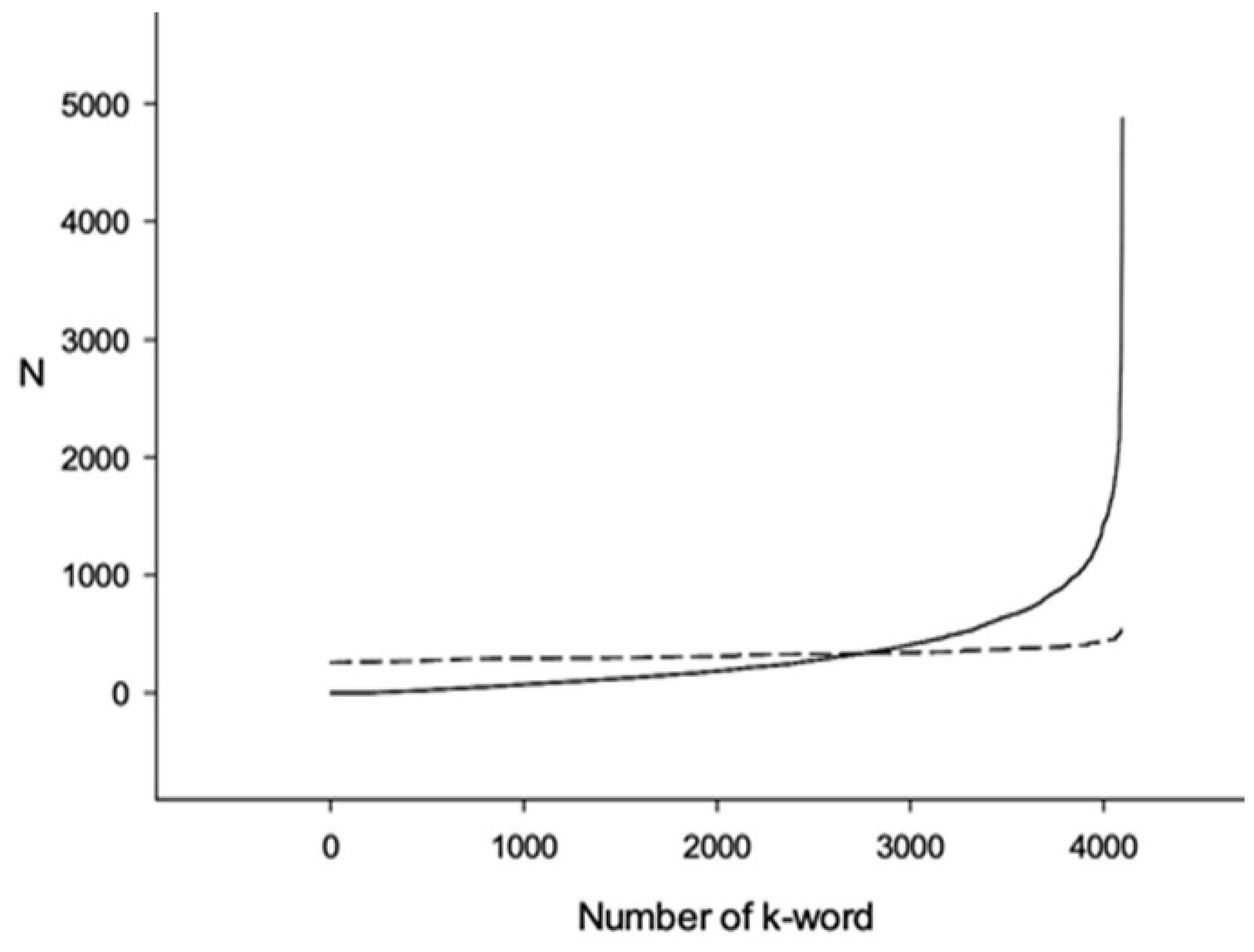
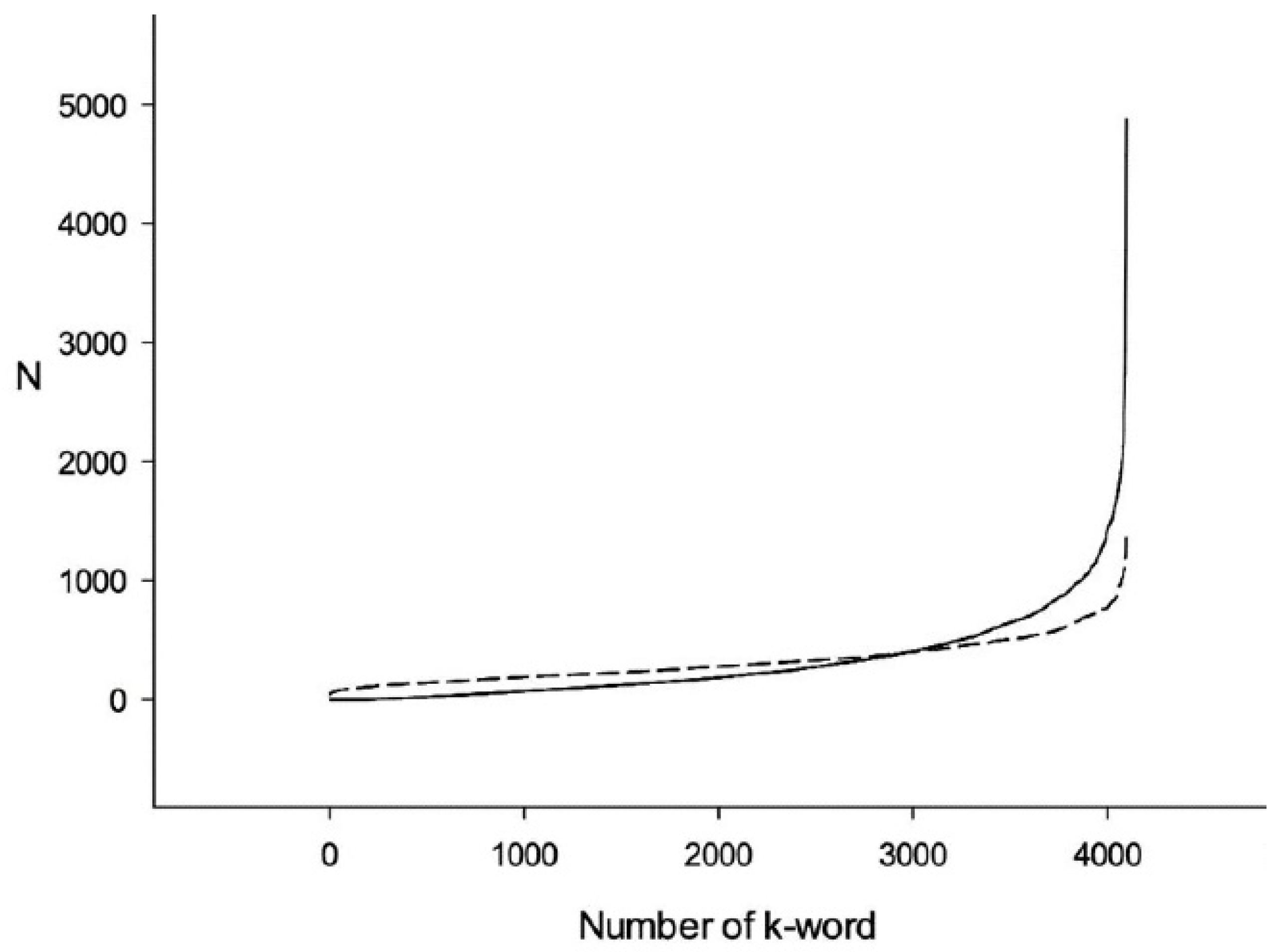
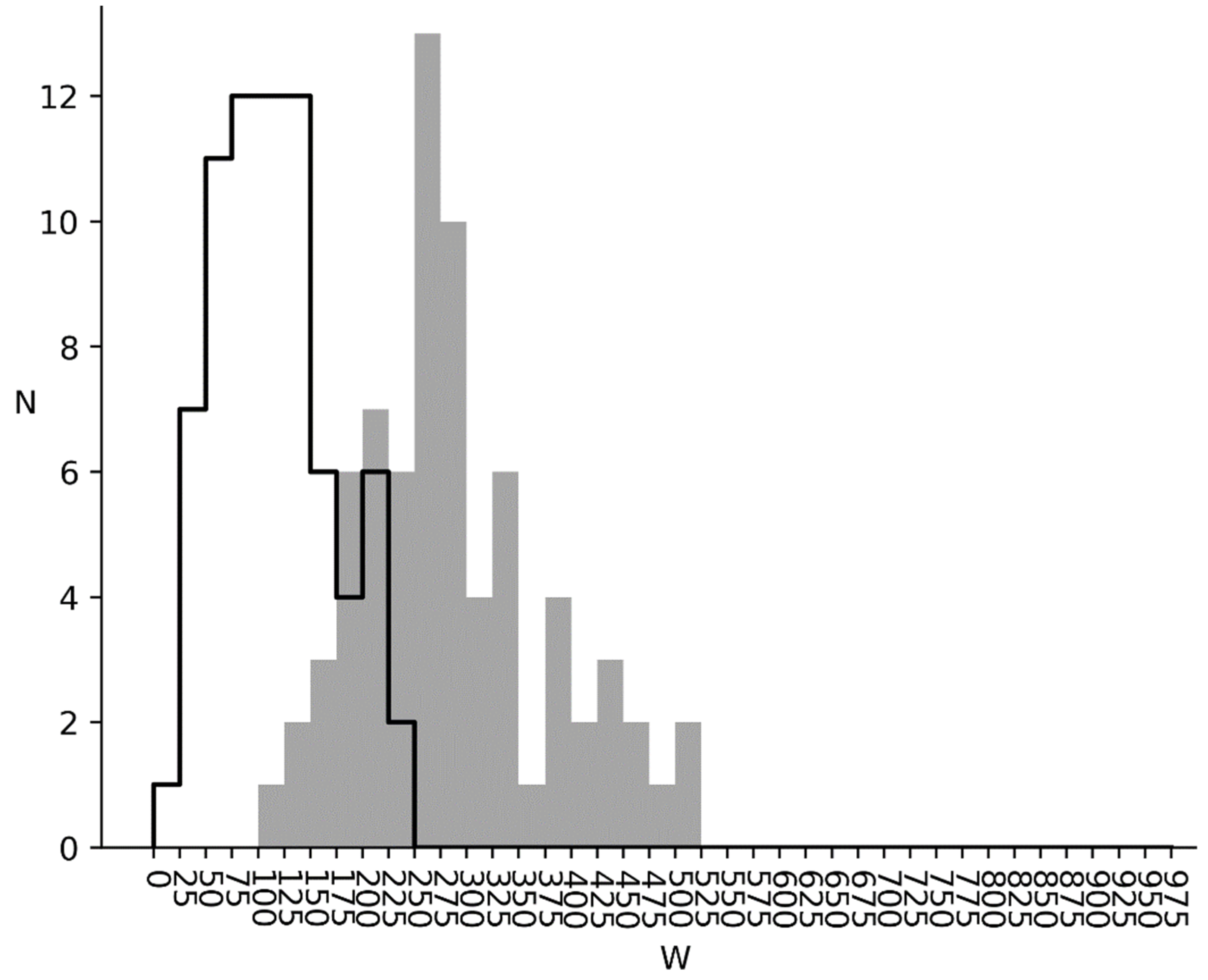
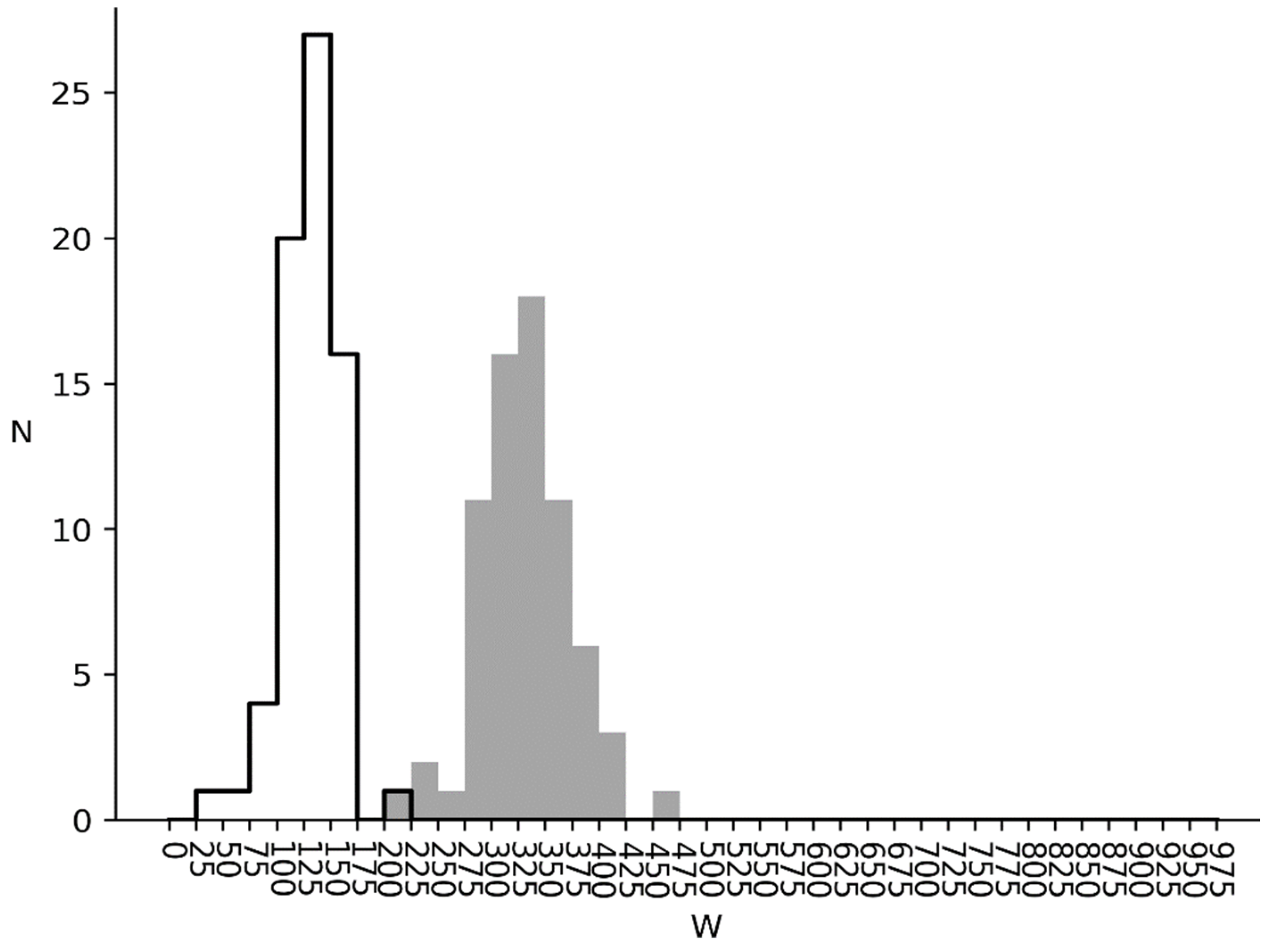
3.1. Comparison of Q1 and R1 Arrays with Q1 and T1 Arrays
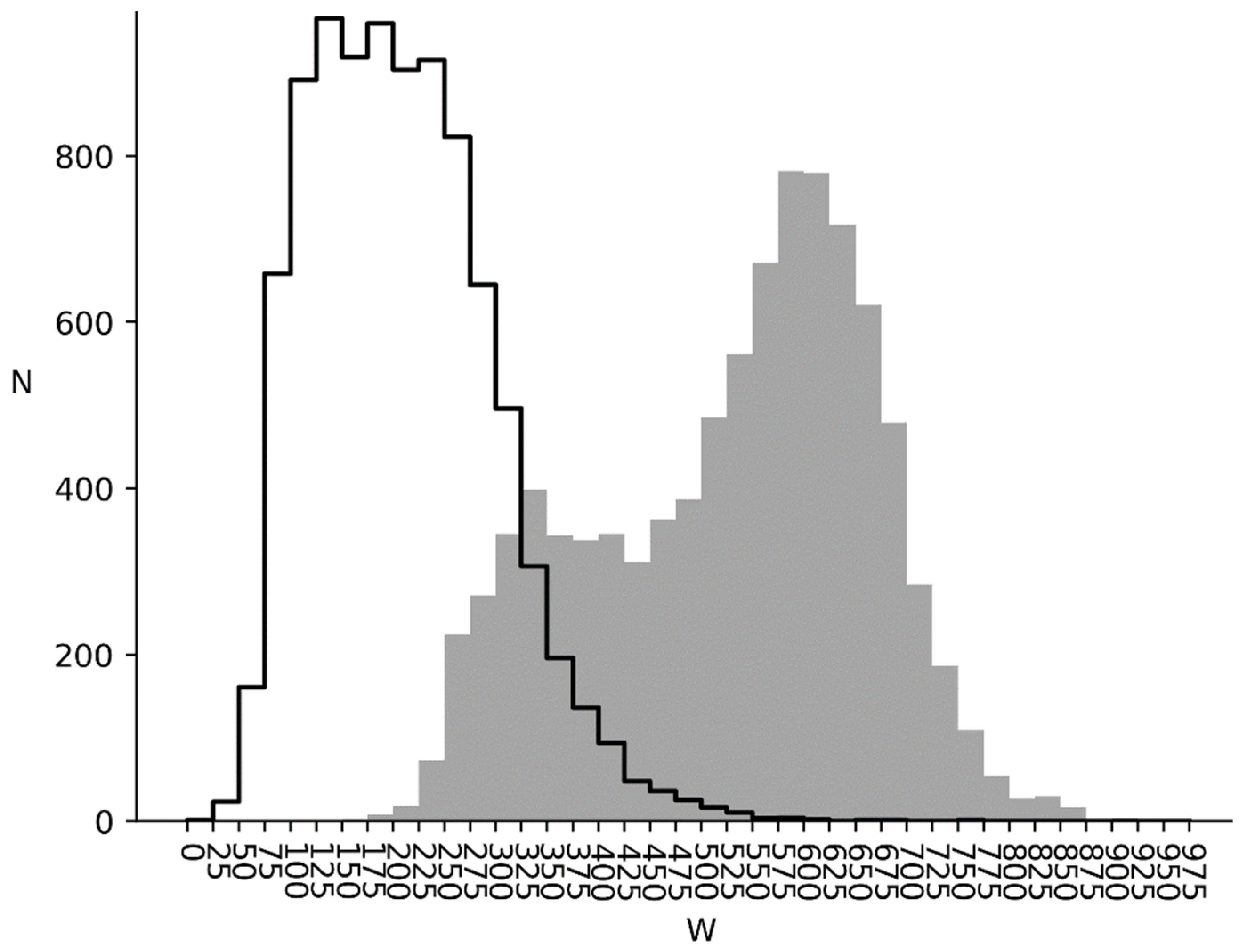
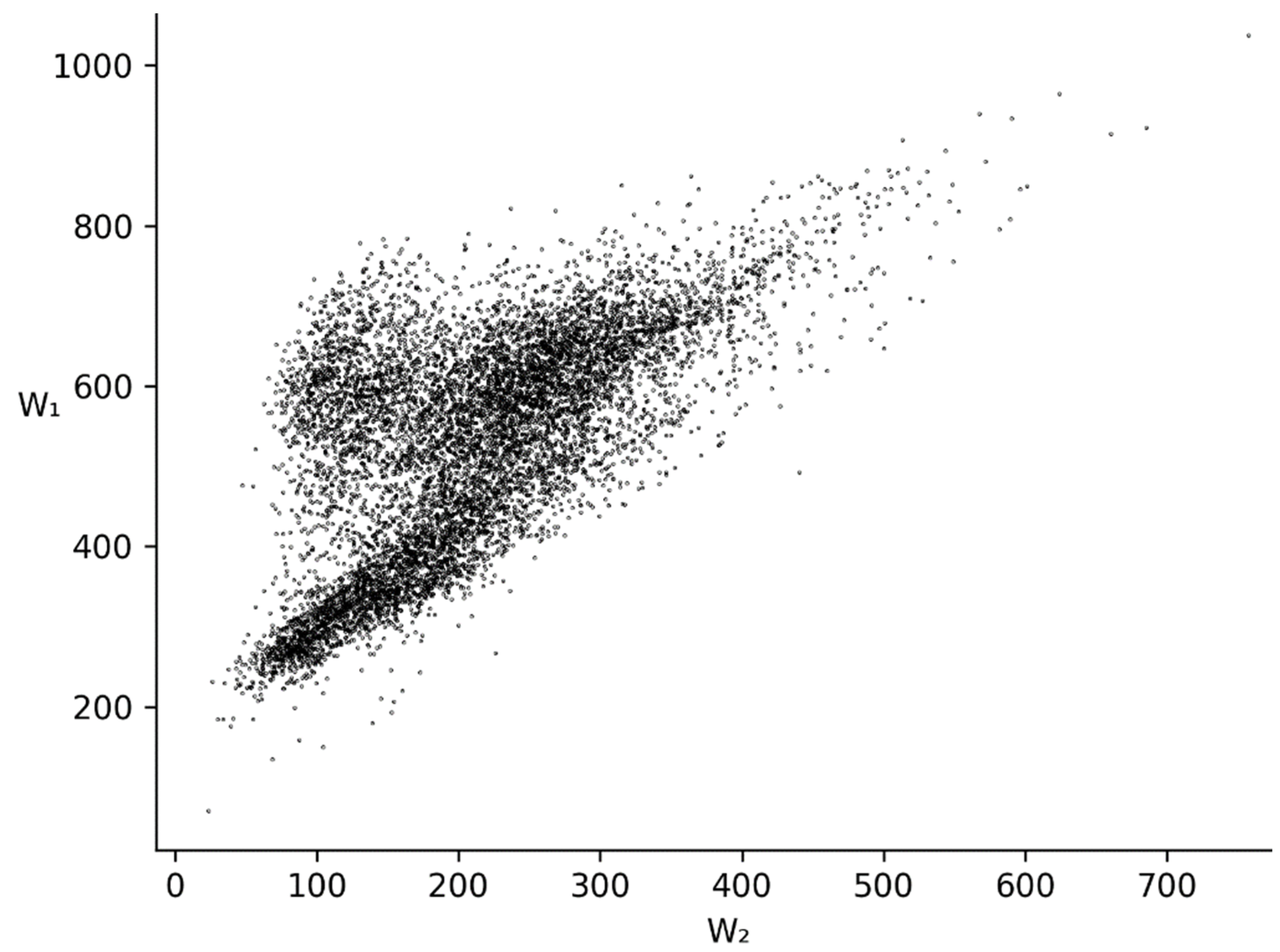
3.2. and Distributions for Bacterial, Metazoan and Virus Cds
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Remec, Z.I.; Podkrajsek, K.T.; Lampret, B.R.; Kovac, J.; Groselj, U.; Tesovnik, T.; Battelino, T.; Debeljak, M. Next-Generation Sequencing in Newborn Screening: A Review of Current State. Front. Genet. 2021, 12, 710. [Google Scholar] [CrossRef] [PubMed]
- Makeev, V.J.; Tumanyan, V.G. Search of periodicities in primary structure of biopolymers: A general Fourier approach. Comput. Appl. Bioinf. Cabios 1996, 12, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Lobzin, V.V.; Chechetkin, V.R. Order and correlations in genomic DNA sequences. The spectral approach. Uspekhi Fizicheskih Nauk 2000, 170, 57. [Google Scholar] [CrossRef]
- Sharma, D.; Issac, B.; Raghava, G.P.S.; Ramaswamy, R. Spectral Repeat Finder (SRF): Identification of repetitive sequences using Fourier transformation. Bioinformatics 2004, 20, 1405–1412. [Google Scholar] [CrossRef] [Green Version]
- Machado, J.A.T.; Costa, A.C.; Quelhas, M.D. Wavelet analysis of human DNA. Genomics 2011, 98, 155–163. [Google Scholar] [CrossRef] [Green Version]
- Korotkov, E.; Korotkova, M.; Kudryashov, N. Information decomposition method to analyze symbolical sequences. Phys. Lett. Sect. A Gen. At. Solid State Phys. 2003, 312, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Korotkov, E.V.; Suvorova, Y.M.; Kostenko, D.O.; Korotkova, M.A. Multiple alignment of promoter sequences from the arabidopsis thaliana l. Genome Genes 2021, 12, 1–21. [Google Scholar] [CrossRef]
- Suvorova, Y.M.; Korotkova, M.A.; Korotkov, E.V. Comparative analysis of periodicity search methods in DNA sequences. Comput. Biol. Chem. 2014, 53, 43–48. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Kolpakov, R.M.; Bana, G.; Kucherov, G. mreps: Efficient and flexible detection of tandem repeats in DNA. Nucleic Acids Res. 2003, 31, 3672–3678. [Google Scholar] [CrossRef]
- Pellegrini, M.; Renda, M.E.; Vecchio, A. TRStalker: An efficient heuristic for finding fuzzy tandem repeats. Bioinformatics 2010, 26, i358–i366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wexler, Y.; Yakhini, Z.; Kashi, Y.; Geiger, D. Finding Approximate Tandem Repeats in Genomic Sequences. J. Comput. Biol. 2005, 12, 928–942. [Google Scholar] [CrossRef] [Green Version]
- Jorda, J.; Kajava, A.V. T-REKS: Identification of Tandem REpeats in sequences with a K-meanS based algorithm. Bioinformatics 2009, 25, 2632–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudunuri, S.B.; Kumar, P.; Rao, A.A.; Pallamsetty, S.; Nagarajaram, H.A. G-IMEx: A comprehensive software tool for detection of microsatellites from genome sequences. Bioinformation 2010, 5, 221–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeva, V.; Regnier, M.; Papatsenko, D.; Makeev, V. Short fuzzy tandem repeats in genomic sequences, identification, and possible role in regulation of gene expression. Bioinformatics 2006, 22, 676–684. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.G.; Kwoh, C.K.; Hsu, L.Y.; Wirawan, A. Review of tandem repeat search tools: A systematic approach to evaluating algorithmic performance. Briefings Bioinform. 2013, 14, 67–81. [Google Scholar] [CrossRef] [Green Version]
- Li, W. The study of correlation structures of DNA sequences: A critical review. Comput. Chem. 1997, 21, 257–271. [Google Scholar] [CrossRef] [Green Version]
- Korotkov, E.V.; Kamionskya, A.M.; Korotkova, M.A. Detection of Highly Divergent Tandem Repeats in the Rice Genome. Genes 2021, 12, 473. [Google Scholar] [CrossRef]
- Frenkel, F.E.; Korotkov, E.V. Classification analysis of triplet periodicity in protein-coding regions of genes. Gene 2008, 421, 52–60. [Google Scholar] [CrossRef]
- Frenkel, F.E.; Korotkova, M.A.; Korotkov, E.V. Database of Periodic DNA Regions in Major Genomes. BioMed Res. Int. 2017, 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Suvorova, Y.M.; Korotkov, E.V. Study of triplet periodicity differences inside and between genomes. Stat. Appl. Genet. Mol. Biol. 2015, 14. [Google Scholar] [CrossRef] [PubMed]
- Dorfman, R. A Formula for the Gini Coefficient. Rev. Econ. Stat. 1979, 61, 146. [Google Scholar] [CrossRef]
- Kolmogorov, A.N. Three approaches to the definition of the concept “quantity of information. Probl. Peredachi Inf. 1965, 1, 3–11. [Google Scholar]
- Li, M.; Vitányi, P. An Introduction to Kolmogorov Complexity and Its Applications. Springer: Berlin/Heidelberg, Germany, 1997; p. 637. [Google Scholar]
- Li, W.; Freudenberg, J.; Miramontes, P. Diminishing return for increased Mappability with longer sequencing reads: Implications of the k-mer distributions in the human genome. BMC Bioinform. 2014, 15, 2. [Google Scholar] [CrossRef] [Green Version]
- Sheinman, M.; Ramisch, A.; Massip, F.; Arndt, P.F. Evolutionary dynamics of selfish DNA explains the abundance distribution of genomic subsequences. Sci. Rep. 2016, 6, 30851. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Yang, Y. Zipf's Law in Importance of Genes for Cancer Classification Using Microarray Data. J. Theor. Biol. 2002, 219, 539–551. [Google Scholar] [CrossRef] [Green Version]
- Kullback, S. Information Theory and Statistics; Kullback, S., Ed.; Dover Publications: New York, NY, USA, 1997. [Google Scholar]
- Piantadosi, S.T. Zipf’s word frequency law in natural language: A critical review and future directions. Psychon. Bull. Rev. 2014, 21, 1112–1130. [Google Scholar] [CrossRef] [Green Version]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 623–656. [Google Scholar] [CrossRef]
- Oates, F.H.C. Probability and information, by A. M. Yaglom and I. M. Yaglom. Pp 421. $69. 1983. 90-277-1522-X (Reidel). Math. Gaz. 1984, 68, 300–302. [Google Scholar] [CrossRef]
- Brillouin, L.; Hellwarth, R.W. Science and Information Theory. Phys. Today 2019, 9, 39. [Google Scholar] [CrossRef]
- Shannon, C.E. Prediction and Entropy of Printed English. Bell Syst. Tech. J. 1951, 30, 50–64. [Google Scholar] [CrossRef]
- Kaiser, G.E.; Starzyk, M.J. Ultrastructure and cell division of an oral bacterium resembling Alysiella filiformis. Can. J. Microbiol. 1973, 19, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Medvecky, M.; Cejkova, D.; Polansky, O.; Karasova, D.; Kubasova, T.; Cizek, A.; Rychlik, I. Whole genome sequencing and function prediction of 133 gut anaerobes isolated from chicken caecum in pure cultures. BMC Genom. 2018, 19, 561. [Google Scholar] [CrossRef] [Green Version]
- Rossau, R.; Van Landschoot, A.; Gillis, M.; De Ley, J. Taxonomy of Moraxellaceae fam. nov., a New Bacterial Family To Accommodate the Genera Moraxella, Acinetobacter, and Psychrobacter and Related Organisms. Int. J. Syst. Bacteriol. 1991, 41, 310–319. [Google Scholar] [CrossRef]
- Bacteria Collection: NCTC 10283 Alysiella Crassa. Available online: https://www.phe-culturecollections.org.uk/products/bacteria/detail.jsp?collection=nctc&refId=NCTC+10283 (accessed on 7 December 2021).
- Williams, N.; Cooper, C.; Cundy, P. Kingella kingae septic arthritis in children: Recognising an elusive pathogen. J. Child. Orthop. 2014, 8, 91–95. [Google Scholar] [CrossRef]
- Jumas-Bilak, E.; Carlier, J.-P.; Jean-Pierre, H.; Mory, F.; Teyssier, C.; Gay, B.; Campos, J.; Marchandin, H. Acidaminococcus intestini sp. nov., isolated from human clinical samples. Int. J. Syst. Evol. Microbiol. 2007, 57, 2314–2319. [Google Scholar] [CrossRef] [Green Version]
- Matos, R.; De Witte, C.; Smet, A.; Berlamont, H.; De Bruyckere, S.; Amorim, I.; Gärtner, F.; Haesebrouck, F. Antimicrobial Susceptibility Pattern of Helicobacter heilmannii and Helicobacter ailurogastricus Isolates. Microorganisms 2020, 8, 957. [Google Scholar] [CrossRef]
- Vela, A.I.; Collins, M.D.; Lawson, P.A.; García, N.; Domínguez, L.; Fernandez-Garayzabal, J.F. Uruburuella suis gen. nov., sp. nov., isolated from clinical specimens of pigs. Int. J. Syst. Evol. Microbiol. 2005, 55, 643–647. [Google Scholar] [CrossRef] [Green Version]
- De Baere, T.; Muylaert, A.; Everaert, E.; Wauters, G.; Claeys, G.; Verschraegen, G.; Vaneechoutte, M. Bacteremia due to Moraxella atlantae in a cancer patient. J. Clin. Microbiol. 2002, 40, 2693–2695. [Google Scholar] [CrossRef] [Green Version]
- Rothschild, L.J.; Mancinelli, R.L. Life in extreme environments. Nature 2001, 409, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.C.; Mukhopadhyay, R.; Wen, B.; Gitai, Z.; Wingreen, N.S. Cell shape and cell-wall organization in Gram-negative bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 19282–19287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darby, A.C.; Cho, N.-H.; Fuxelius, H.H.; Westberg, J.; Andersson, S.G.E. Intracellular pathogens go extreme: Genome evolution in the Rickettsiales. Trends Genet. 2007, 23, 511–520. [Google Scholar] [CrossRef]
- Spang, A.; Saw, J.; Jørgensen, S.L.; Zaremba-Niedzwiedzka, K.; Martijn, J.; Lind, A.E.; Van Eijk, R.; Schleper, C.; Guy, L.; Ettema, T.J.G. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 2015, 521, 173–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitrosopumilales archaeon CG15_BIG_FIL_POST_REV_8_(ID 64741)-Genome-NCBI. Available online: https://www.ncbi.nlm.nih.gov/genome/?term=txid2022694[Organism:noexp] (accessed on 7 December 2021).
- Nicol, G.W.; Hink, L.; Gubry-Rangin, C.; Prosser, J.I.; Lehtovirta-Morley, L.E. Genome Sequence of “ Candidatus Nitrosocosmicus franklandus” C13, a Terrestrial Ammonia-Oxidizing Archaeon. Microbiol. Resour. Announc. 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, F.; Takai, K.; Nealson, K.H.; Horikoshi, K. Sulfurovum lithotrophicum gen. nov., sp. nov., a novel sulfur-oxidizing chemolithoautotroph within the E-Proteobacteria isolated from Okinawa Trough hydrothermal sediments. Int. J. Syst. Evol. Microbiol. 2004, 54, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.; Nichols, C.M.; Gibson, J.A.E. Algoriphagus ratkowskyi gen. nov., sp. nov., Brumimicrobium glaciale gen. nov., sp. nov., Cryomorpha ignava gen. nov., sp. nov. and Crocinitomix catalasitica gen. nov., sp. nov., novel flavobacteria isolated from various polar habitats. Int. J. Syst. Evol. Microbiol. 2003, 53, 1343–1355. [Google Scholar] [CrossRef]
- Zakharyuk, A.; Kozyreva, L.; Ariskina, E.; Troshina, O.; Kopitsyn, D.; Shcherbakova, V. Alkaliphilus namsaraevii sp. nov., an alkaliphilic iron- and sulfur-reducing bacterium isolated from a steppe soda lake. Int. J. Syst. Evol. Microbiol. 2017, 67, 1990–1995. [Google Scholar] [CrossRef]
- ENA Browser. Available online: https://www.ebi.ac.uk/ena/browser/view/PAHQ01 (accessed on 8 December 2021).
- Tully, B.J.; Graham, E.D.; Heidelberg, J.F. The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci. Data 2018, 5, 170203. [Google Scholar] [CrossRef] [Green Version]
- Choo, Y.-J.; Lee, K.; Song, J.; Cho, J.-C. Puniceicoccus vermicola gen. nov., sp. nov., a novel marine bacterium, and description of Puniceicoccaceae fam. nov., Puniceicoccales ord. nov., Opitutaceae fam. nov., Opitutales ord. nov. and Opitutae classis nov. in the phylum ‘Verrucomicrobia’. Int. J. Syst. Evol. Microbiol. 2007, 57, 532–537. [Google Scholar] [CrossRef]
- Weymark, J.A. Generalized Gini Indices of Equality of Opportunity. J. Econ. Inequal. 2003, 1, 5–24. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| № | Bacterial Name | Accession Number | W1 |
|---|---|---|---|
| 1 | Alysiella_filiformis dsm_16848 | gca_900230205 | 1037.38 |
| 2 | Uruburuella_suis | gca_004341385 | 964.25 |
| 3 | Elusimicrobium_sp an273 | gca_002159705 | 940.01 |
| 4 | Moraxella_caviae | gca_002014985 | 933.66 |
| 5 | Alysiella_crassa | gca_900445245 | 922.0 |
| 6 | Kingella_kingae | gca_001458475 | 914.74 |
| 7 | Acidaminococcus_sp cag_542 | gca_000437815 | 907.44 |
| 8 | Moraxella_atlantae | gca_001678995 | 893.4 |
| 9 | Herpetosiphon_geysericola | gca_001306135 | 880.38 |
| 10 | Helicobacter_ailurogastricus | gca_001282985 | 871.84 |
| № | Bacterial Name | Accession Number | W1 |
|---|---|---|---|
| 1 | Rickettsiales_bacterium | gca_002691145 | 134.34 |
| 2 | Sulfurovum_sp | gca_002733355 | 158.68 |
| 3 | Lokiarchaeum_sp gc14_75 | gca_000986845 | 176.05 |
| 4 | Cryomorphaceae_bacterium | gca_002682945 | 179.68 |
| 5 | Nitrosopumilales_archaeon | gca_003856905 | 183.85 |
| 6 | Alkaliphilus_sp | gca_002733545 | 184.78 |
| 7 | Candidatus_nitrosocosmicus_franklandus | gca_900696045 | 185.51 |
| 8 | Verrucomicrobiales_bacterium | gca_002705125 | 192.75 |
| 9 | Legionellales_bacterium | gca_002719415 | 198.52 |
| 10 | Puniceicoccaceae_bacterium | gca_002690565 | 206.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korotkov, E.; Zaytsev, K.; Fedorov, A. Use of 6 Nucleotide Length Words to Study the Complexity of Gene Sequences from Different Organisms. Entropy 2022, 24, 632. https://doi.org/10.3390/e24050632
Korotkov E, Zaytsev K, Fedorov A. Use of 6 Nucleotide Length Words to Study the Complexity of Gene Sequences from Different Organisms. Entropy. 2022; 24(5):632. https://doi.org/10.3390/e24050632
Chicago/Turabian StyleKorotkov, Eugene, Konstantin Zaytsev, and Alexey Fedorov. 2022. "Use of 6 Nucleotide Length Words to Study the Complexity of Gene Sequences from Different Organisms" Entropy 24, no. 5: 632. https://doi.org/10.3390/e24050632