
A Review of the System-Intrinsic Nonequilibrium Thermodynamics in Extended Space (MNEQT) with Applications

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Unique Macrostates in Extended State Space
1.2. Layout
2. Notation, Definitions and New Concepts
2.1. Notation
2.2. Some Definitions and New Concepts
- (a)
- Internal-equilibrium macrostates (IEQ): The nonequilibrium entropy for such a macrostate is a state function in the larger nonequilibrium state space spanned by ; is a proper subspace of : . As there is no explicit time dependence, there is no memory of the initial macrostate in IEQ macrostates.
- (b)
- Non-internal-equilibrium macrostates (NIEQ): The nonequilibrium entropy for such a macrostate is not a state function of the state variable . Accordingly, we denote it by with an explicit time dependence. The explicit time dependence gives rise to memory effects in these NEQ macrostates that lie outside the nonequilibrium state space . A NIEQ macrostate in becomes an IEQ macrostate in a larger state space , with a proper choice of .
3. Internal Variables
3.1. A Two-Level System
3.2. A Many-Level System
3.3. Disparate Degrees of Freedom
3.4. Nonuniformity
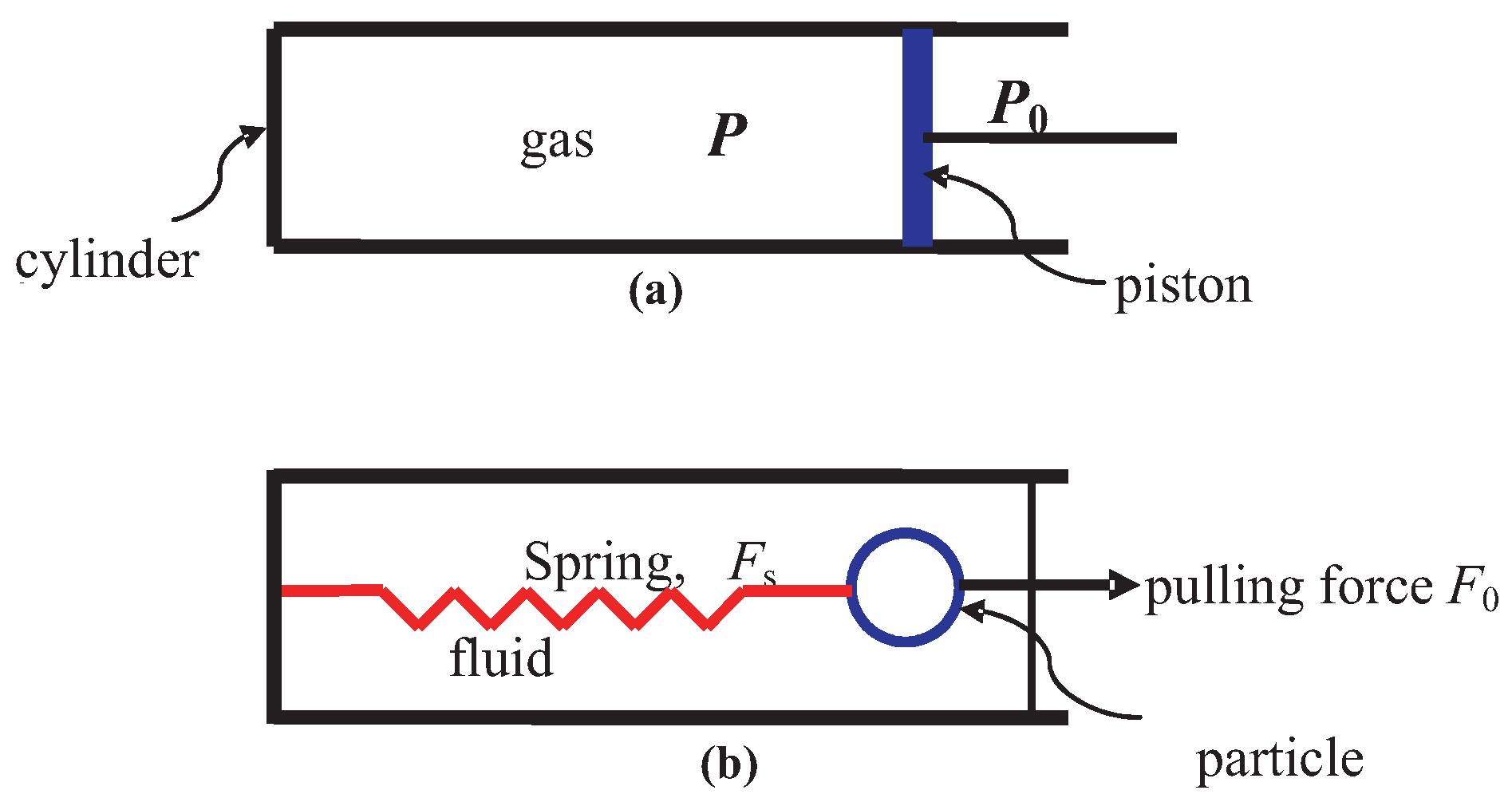
3.5. Relative Motion in Piston-Gas System
3.6. Extended State Space
4. NEQ Entropy
4.1. Determination of S
4.2. General Formulation of the Statistical Entropy
4.3. A Proof of the Second Law
5. Hamiltonian Trajectories in
5.1. Generalized Microforce and Microwork for
5.2. Statistical Significance of and
5.3. Medium
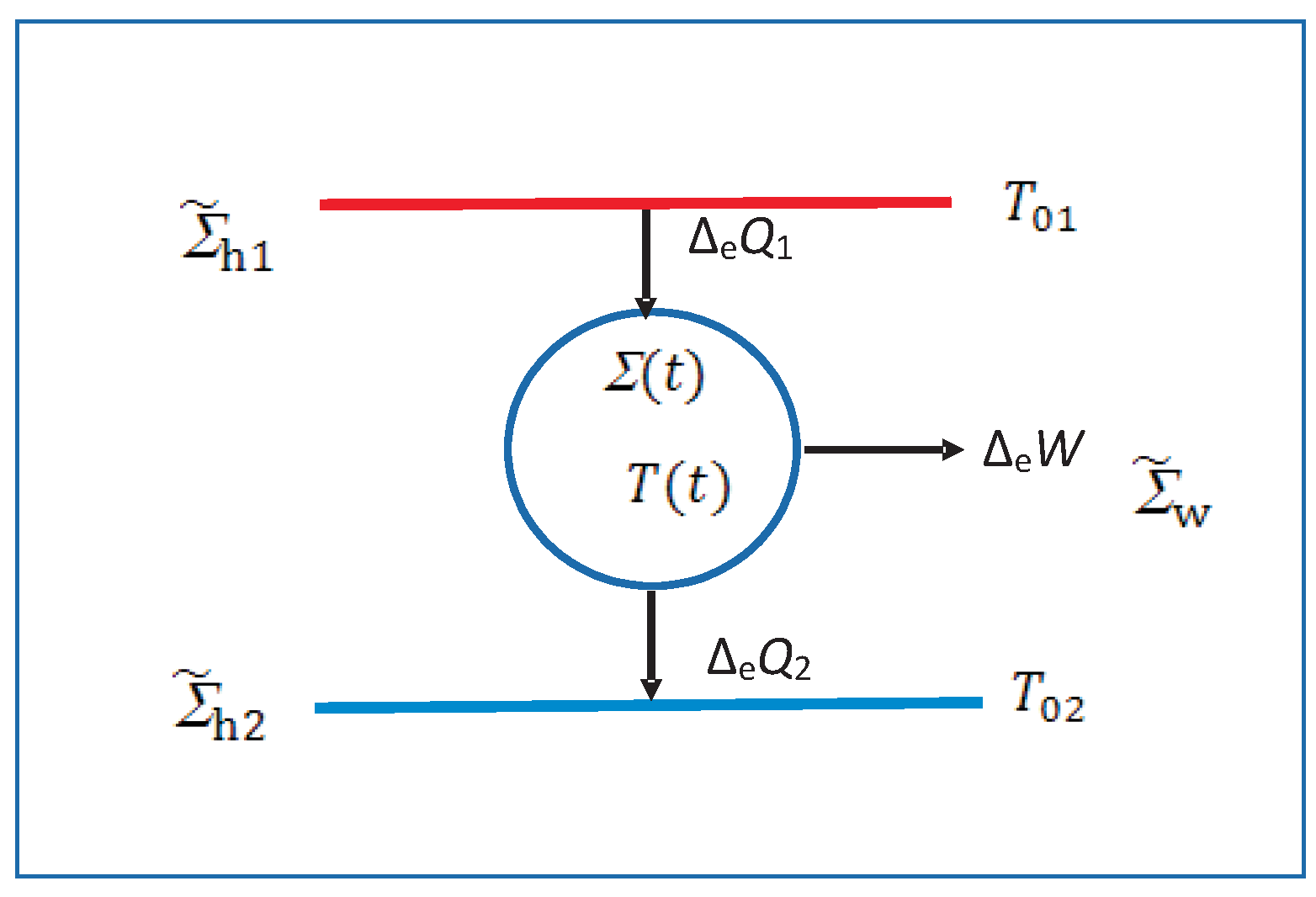
5.4. Irreversible Macrowork and Macroheat
6. Unique Macrostates
6.1. Internal Equilibrium
6.2. Gibbs Fundamental Relation
6.3. A Digression on the NEQ-Temperature
- C1
- It must be intensive and must reduce to the temperature determined by Equation (72) for and even for an isolated system.
- C2
- It must cover negative temperatures [106] that are commonly observed for some dof such as nuclear spins in a system. As these dof are not involved in any macroscopic motion ([14] (Section 73)), there is no kinetic energy involved. Most common occurrence of a negative temperature is when the above spin dof are out of equilibrium with the other dof such as lattice vibrations in the system.
- C3
- It must satisfy the Clausius statement that macroheat between two objects always flows spontaneously from hot to cold for positive temperatures. When negative temperatures are considered, macroheat must flow from a system at a negative temperature to a system at a positive temperature.
- C4
- It must be a global rather than a local property of the system so that we can differentiate hot and cold between two different systems.
- (1)
- In the LNEQT, each local volume element is in EQ so the local temperature is the EQ temperature of the volume element, and differs from T, which is a global temperature.
- (2)
- In the RNEQT, the temperature is taken as a primitive quantity along with the entropy. Because of the memory effect, the temperature at any time depends on the entire history. Thus, it is a local analog of the global temperature of in the MNEQT, but the latter is defined thermodynamically.
- (3)
- In the ENEQT, the fluxes are part of the state variables so the local temperature also depends on them. Assuming the total entropy to also depend on the fluxes ([20], (see Equation (5.66), for example)), one can identify the global analog of the temperature in the ENEQT. However, as fluxes are MI-quantities, this temperature cannot be compared with the SI-temperature in the MNEQT.
6.4. Uniqueness of and T
6.5. Irreversibility Inequalities in
6.6. Internal Variables and the Isolated System
6.7. Dissipation and Thermodynamic Forces
6.8. Cyclic Process
6.9. Steady State
6.10. Intrinsic Adiabaticity Theorem
7. Clausius Equality
8. Extended State Space and
8.1. Choice of
8.2. Microstate Probabilities for
8.3. in
8.4. External and Internal Variations of
9. A Model Entropy Calculation
9.1. 1-Dimensional Ideal Gas
9.2. Chemical Reaction Approach
10. Simple Applications
10.1. Isothermal Expansion
10.2. Intrinsic Adiabatic Expansion
10.3. Composite with Temperature Inhomogeneity
10.3.1. Isolated
10.3.2. Interacting with
10.4. Interacting with and
10.5. Interacting with and
11. Tool-Narayanaswamy Equation
12. Irreversible Carnot Cycle
13. Origin of Friction and Brownian Motion
13.1. Piston-Gas System
13.2. Particle-Spring-Fluid System
13.3. Particle-Fluid System
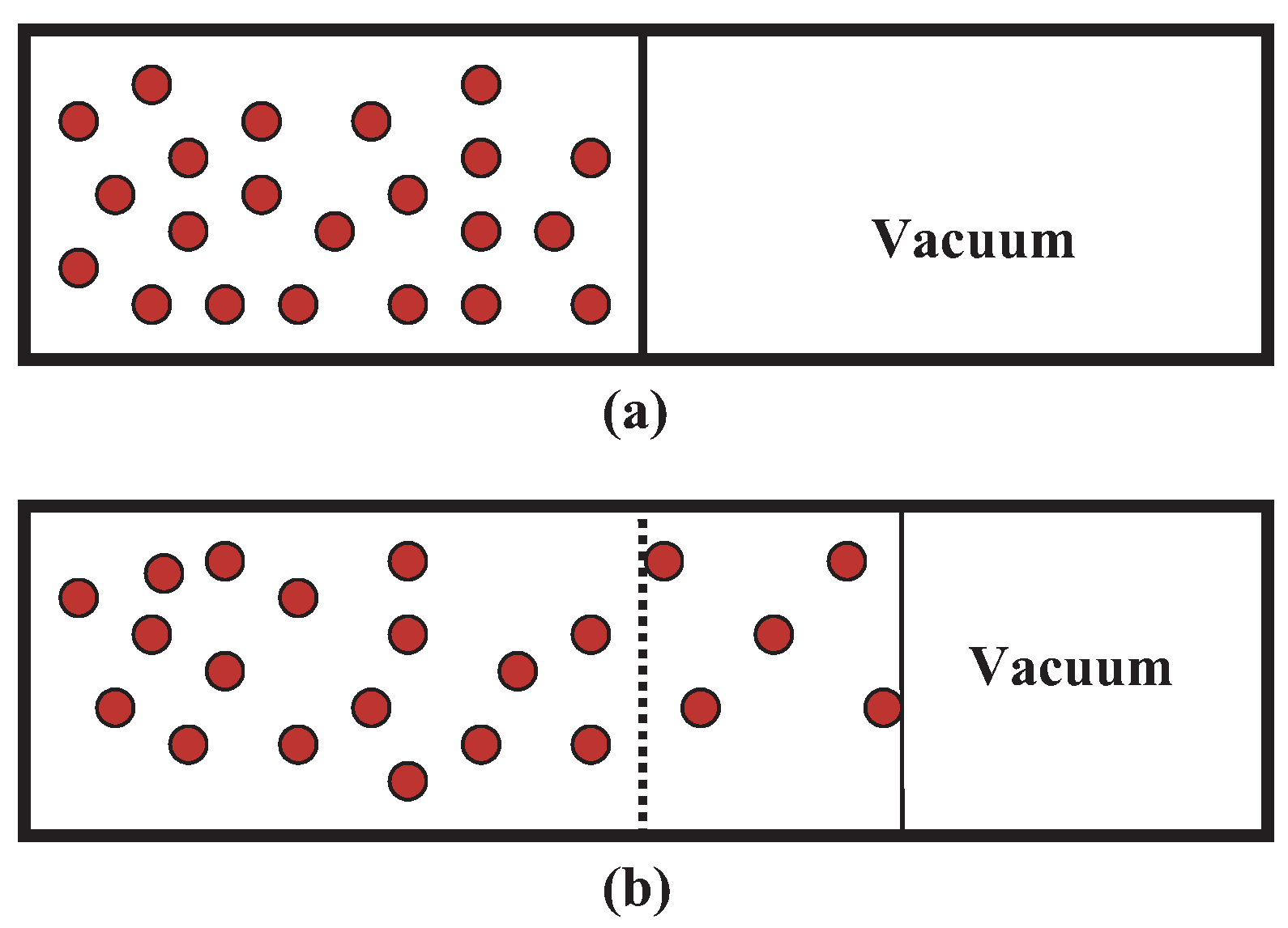
14. Free Expansion
14.1. Classical Free Expansion in
14.2. Quantum Free Expansion
15. Discussion and Conclusions
15.1. Unique NEQ S in
15.2. Unique NEQ T in
15.3. Applications
15.4. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Donder, T.D.; Rysselberghe, P.V. Thermodynamic Theory of Affinity: A Book of Principles; Oxford University Press: Oxford, UK, 1936. [Google Scholar]
- Prigogine, I. , Thermodynamics of Irreversible Processes; Wiley-Interscience: New York, NY, USA, 1971. [Google Scholar]
- de Groot, S.R.; Mazur, P. Nonequilibrium Thermodynamics, 1st ed.; Dover: New York, NY, USA, 1984. [Google Scholar]
- Eu, B.G. Kinetic Theory and Irreversible Thermodynamics; John Wiley: New York, NY, USA, 1992. [Google Scholar]
- Kuiken, G.D.C. Thermodynamics of Irreversible Processes; John Wiley: Chichester, UK, 1994. [Google Scholar]
- Ottinger, H.C. Beyond Equilibrium Thermodynamics; Wiley: Hoboken, NJ, USA, 2005. [Google Scholar]
- Kjelstrum, S.; Bedeaux, D. Nonequilibrium Thermodynamics of Heterogeneous Systems; World-Scientific: Singapore, 2008. [Google Scholar]
- Evans, D.J.; Morriss, G. Statistical Mechanics of Nonequilibrium Liquids, 2nd ed.; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]
- Fermi, E. Thermodynamics; Dover: New York, NY, USA, 1956. [Google Scholar]
- Reif, F. Fundamentals of Statistical and Thermal Physics; McGraw-Hill Inc.: New York, NY, USA, 1965. [Google Scholar]
- Woods, L.C. The Thermodynamics of Fluids Systems; Oxford University Press: Oxford, UK, 1975. [Google Scholar]
- Kestin, J. A course in Thermodynamics; McGraw-Hill Book Company: New York, NY, USA, 1979; Volume 1–2. [Google Scholar]
- Waldram, J.R. The Theory of Thermodynamics; Cambridge University: Cambridge, UK, 1985. [Google Scholar]
- Landau, L.D.; Lifshitz, E.M. Statistical Physics, 3rd ed.; Pergamon Press: Oxford, UK, 1986; Part 1. [Google Scholar]
- Balian, R. From Microphysics to Macrophysics; Springer: Berlin, Germany, 1991; Volume 1. [Google Scholar]
- Kondepudi, D.; Prigogine, I. Modern Thermodynamics; John Wiley and Sons: West Sussex, UK, 1998. [Google Scholar]
- Boltzmann, L. Lectures on Gas Theory; University of California Press: Berkeley, CA, USA, 1964. [Google Scholar]
- Boltzmann, L. Über die Beziehung zwischen dem zweiten Hauptsatze der mechanischen Wärmetheorie und der Wahrscheinlichkeitsrechnung respektive den Sätzen über das Wärmegleichgewicht. Wien Ber. 2015, 176, 1971. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, J.W. Elementary Principles in Statistical Mechanics; Scribner’s Sons: New York, NY, USA, 1902. [Google Scholar]
- Jou, D.; Casas-Vázquez, J.; Lebon, G. Extended Irreversible Thermodynamics, 2nd ed.; Springer: Berlin, Germany, 1996. [Google Scholar]
- Muschik, W. Why So Many “Schools” of Thermodynamics? Forsch Ingenieurwesen 2007, 71, 149. [Google Scholar] [CrossRef] [Green Version]
- Schottky, W. Thermodynamik; Springer: Berlin, Germany, 1929. [Google Scholar]
- Muschik, W. Discrete Systems in Thermal Physics and Engineering –A Glance from Non-Equilibrium Thermodynamics. arXiv 2020, arXiv:2010.14241v4. [Google Scholar] [CrossRef]
- Oono, Y.; Paniconi, M. Steady State Thermodynamics. Prog.Theor. Phys. Suppl. 1998, 130, 29. [Google Scholar] [CrossRef] [Green Version]
- Bejan, A. Applied Engineering Thermodynamics, 3rd ed.; John Wiley: New York, NY, USA, 2006. [Google Scholar]
- Bejan, A. Entropy generation minimization: The new thermodynamics of finite-size devices and finite-time processes. J. Appl. Phys. 1996, 79, 1191. [Google Scholar] [CrossRef] [Green Version]
- Sasa, S.; Tasaki, H. Steady State Thermodynamics. J. Stat. Phys. 2006, 125, 125. [Google Scholar] [CrossRef]
- Keizer, J. Statistical Thermodynamics of Nonequilibrium Processes; Springer: New York, NY, USA, 1987. [Google Scholar]
- Stratonovich, R.L. Nonlinear Nonequilibrium Thermodynamics I; Springer: Berlin, Germany, 1992. [Google Scholar]
- Schuss, Z. Theory and Applications of Stochastic Processes: An Analytical Approach; Springer: New York, NY, USA, 2010. [Google Scholar]
- Coffey, W.T.; Kalmykov, Y.P. The Langevin Equation, 4th ed.; World Scientific: Singapore, 2017. [Google Scholar]
- Gujrati, P.D. First-principles nonequilibrium deterministic equation of motion of a Brownian particle and microscopic viscous drag. Phys. Rev. E 2020, 102, 012140. [Google Scholar] [CrossRef] [PubMed]
- Sekimoto, K. Kinetic Characterization of Heat Bath and the Energetics of Thermal Ratchet Models. J. Phys. Soc. Jpn. 1997, 66, 1234. [Google Scholar] [CrossRef]
- Jarzynski, C. Nonequilibrium Equality for Free Energy Differences. Phys. Rev. Lett. 1997, 78, 2690. [Google Scholar] [CrossRef] [Green Version]
- Jarzynski, C. Comparison of far-from-equilibrium work relations. Comptes Rendus Phys. 2007, 8, 495. [Google Scholar] [CrossRef] [Green Version]
- Seifert, U. Stochastic thermodynamics: Principles and perspectives. Eur. Phys. J. B 2008, 64, 423. [Google Scholar] [CrossRef]
- Bochkov, G.N.; Kuzovlev, Y.E. General theory of thermal fluctuations in nonlinear systems. Sov. Phys. JETP 1977, 45, 125. [Google Scholar]
- Bochkov, G.N.; Kuzovlev, Y.E. Fluctuation-dissipation relations for nonequilibrium processes in open systems. Sov. Phys. JETP 1979, 49, 543. [Google Scholar]
- Coleman, B.D. Thermodynamics with Internal State Variables. J. Chem. Phys. 1967, 47, 597. [Google Scholar] [CrossRef]
- Maugin, G.A. The Thermodynamics of Nonlinear Irreversible Behaviors; World Scientific: Singapore, 1999. [Google Scholar]
- Gujrati, P.D. Nonequilibrium thermodynamics. II. Application to inhomogeneous systems. Phys. Rev. E 2012, 85, 041128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchbinder, E.; Langer, J.S. Nonequilibrium thermodynamics of driven amorphous materials. I. Internal degrees of freedom and volume deformation. Phys. Rev. E 2009, 80, 031131. [Google Scholar] [CrossRef] [Green Version]
- Bouchbinder, E.; Langer, J.S. Nonequilibrium thermodynamics of driven amorphous materials. II. Effective-temperature theory. Phys. Rev. E 2009, 80, 031132. [Google Scholar] [CrossRef] [Green Version]
- Pokrovskii, V.N. A Derivation of the Main Relations of Nonequilibrium Thermodynamics. Int. Sch. Res. Not. 2013, 2013, 906136. [Google Scholar] [CrossRef]
- Gujrati, P.D. Hierarchy of Relaxation Times and Residual Entropy: A Nonequilibrium Approach. Entropy 2018, 20, 149. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, M.; Simha, R. (Eds.) The Glass Transition and the Nature of the Glassy State; Academy of Sciences: New York, NY, USA, 1976. [Google Scholar]
- Davies, R.O.; Jones, G.O. Thermodynamic and kinetic properties of glasses. Adv. Phys. 1953, 2, 370–410. [Google Scholar] [CrossRef]
- Gutzow, I.S.; Schmelzer, J.W.P. The Vitreous State: Thermodynamics, Structure, Rheology, and Crystallization, 2nd ed.; Springer: Berlin, Germany, 2013. [Google Scholar]
- Nemilov, S.V. Thermodynamic and Kinetic Aspects of the Vitreous State; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Prigogine, I. Introduction to Thermodynamics of Irreversible Processes, 3rd ed.; Wiley Interscience: New York, NY, USA, 1967. [Google Scholar]
- Gujrati, P.D. Nonequilibrium Entropy. arXiv 2013, arXiv:1304.3768. [Google Scholar]
- Gujrati, P.D. On Equivalence of Nonequilibrium Thermodynamic and Statistical Entropies. Entropy 2015, 17, 710. [Google Scholar] [CrossRef] [Green Version]
- Gujrati, P.D. Loss of Temporal Homogeneity and Symmetry in Statistical Systems: Deterministic Versus Stochastic Dynamics. Symmetry 2010, 2, 1201. [Google Scholar] [CrossRef]
- Gujrati, P.D. Nonequilibrium Work and its Hamiltonian Connection for a Microstate in Nonequilibrium Statistical Thermodynamics: A Case of Mistaken Identity. arXiv 2017, arXiv:1702.00455. [Google Scholar]
- Gujrati, P.D. Nonequilibrium thermodynamics: Structural relaxation, fictive temperature, and Tool-Narayanaswamy phenomenology in glasses. Phys. Rev. E 2010, 81, 051130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilar, J.M.G.; Rubi, J.M. Thermodynamics “beyond” local equilibrium. Proc. Natl. Acad. Sci. USA 2001, 98, 11081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379. [Google Scholar] [CrossRef] [Green Version]
- Gujrati, P.D. Where is the residual entropy of a glass hiding? arXiv 2009, arXiv:0908.1075. [Google Scholar]
- Tolman, R.C. The Principles of Statistical Mechanics; Oxford University: London, UK, 1959. [Google Scholar]
- Rice, S.A.; Gray, P. The Statistical Mechanics of Simple Liquids; Interscience Publishers: New York, NY, USA, 1965. [Google Scholar]
- Jaynes, E.T. Gibbs vs Boltzmann Entropies. Am. J. Phys. 1965, 33, 391. [Google Scholar] [CrossRef]
- Crooks, G.E. Entropy production fluctuation theorem and the nonequilibrium work relation for free energy differences. Phys. Rev. E 1999, 60, 2721. [Google Scholar] [CrossRef] [Green Version]
- Pitaevskii, L.P. Rigorous results of nonequilibrium statistical physics and their experimental verification. Phys.-Uspekhi 2011, 54, 625. [Google Scholar] [CrossRef]
- Sekimoto, K. Stochastic Energetics; Springer: Berlin, Germany, 2010. [Google Scholar]
- Spohn, H.; Lebowitz, J.L. Irreversible Thermodynamics for Quantum Systems Weakly Coupled to Thermal Reservoirs. Adv. Chem. Phys. 1978, 38, 109. [Google Scholar]
- Alicki, R. The quantum open system as a model of the heat engine. J. Phys. A 1979, 12, L103. [Google Scholar] [CrossRef]
- Maruyama, K.; Nori, F.; Vedral, V. Colloquium: The physics of Maxwell’s demon and information. Rev. Mod. Phys. 2009, 81, 1. [Google Scholar] [CrossRef] [Green Version]
- Seifert, U. Stochastic thermodynamics, fluctuation theorems and molecular machines. Rep. Prog. Phys. 2012, 75, 126001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Broeck, C.V.; Esposito, M. Ensemble and trajectory thermodynamics: A brief introduction. Physica A 2015, 418, 6. [Google Scholar] [CrossRef] [Green Version]
- Gislason, E.A.; Craig, N.C. Pressure—Volume Integral Expressions for Work in Irreversible Processes. J. Chem. Educ. 2007, 84, 499. [Google Scholar] [CrossRef]
- Bertrand, G.L. Thermodynamic Calculation of Work for Some Irreversible Processes. J. Chem. Educ. 2005, 82, 874. [Google Scholar] [CrossRef]
- Bauman, R.P. Work of compressing an ideal gas. J. Chem. Educ. 1964, 41, 102. [Google Scholar] [CrossRef]
- Kivelson, D.; Oppenheim, I. Work in irreversible expansions. J. Chem. Educ. 1966, 43, 233. [Google Scholar] [CrossRef]
- Nieuwenhuizen, T.M. Thermodynamics of the Glassy State: Effective Temperature as an Additional System Parameter. Phys. Rev. Lett. 1998, 80, 5580. [Google Scholar] [CrossRef] [Green Version]
- Allahverdyan, A.E.; Nieuwenhuizen, T.M. Steady adiabatic state: Its thermodynamics, entropy production, energy dissipation, and violation of Onsager relations. Phys. Rev. E 2000, 62, 845. [Google Scholar] [CrossRef]
- Cohen, E.G.D.; Mauzerall, D. A note on the Jarzynski equality. J. Stat. Mech. 2004, 2004, P07006. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.G.D.; Mauzerall, D. The Jarzynski equality and the Boltzmann factor. Mol. Phys. 2005, 103, 21. [Google Scholar] [CrossRef]
- Jarzynski, C. Nonequilibrium work theorem for a system strongly coupled to a thermal environment. J. Stat. Mech. 2004, 2004, P09005. [Google Scholar] [CrossRef]
- Sung, J. Validity condition of the Jarzynski relation for a classical mechanical system. arXiv 2005, arXiv:cond-mat/0506214v4. [Google Scholar]
- Gross, D.H.E. Flaw of Jarzynski’s equality when applied to systems with several degrees of freedom. arXiv 2005, arXiv:cond-mat/0508721v1. [Google Scholar]
- Jarzynski, C. Reply to comments by D.H.E. Gross. arXiv 2005, arXiv:cond-mat/0509344v1. [Google Scholar]
- Peliti, L. On the work–Hamiltonian connection in manipulated systems. J. Stat. Mech. 2008, 2008, P05002. [Google Scholar]
- Vilar, J.M.G.; Rubi, J.M. Failure of the Work-Hamiltonian Connection for Free-Energy Calculations. Phys. Rev. Lett. 2008, 101, 020601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horowitz, J.; Jarzynski, C. Comment on “Failure of the Work-Hamiltonian Connection for Free-Energy Calculations”. Phys. Rev. Lett. 2008, 101, 098901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilar, J.M.G.; Rubi, J.M. Vilar and Rubi Reply. Phys. Rev. Lett. 2008, 101, 098902. [Google Scholar] [CrossRef]
- Peliti, L. Comment on “Failure of the Work-Hamiltonian Connection for Free-Energy Calculations”. Phys. Rev. Lett. 2008, 101, 098903. [Google Scholar] [CrossRef] [Green Version]
- Amotz, D.B.; Honig, J.M. Rectification of thermodynamic inequalities. J.Chem. Phys. 2003, 118, 5932. [Google Scholar] [CrossRef]
- Amotz, D.B.; Honig, J.M. Average Entropy Dissipation in Irreversible Mesoscopic Processes. Phys. Rev. Lett. 2006, 96, 020602. [Google Scholar] [CrossRef] [PubMed]
- Honig, J.M. Thermodynamics, 4th ed.; Academic Press: Oxford, UK, 2014. [Google Scholar]
- Bizarro, J.P.S. Entropy production in irreversible processes with friction. Phys. Rev. E 2008, 78, 021137. [Google Scholar] [CrossRef]
- Gujrati, P.D. Generalized Non-equilibrium Heat and Work and the Fate of the Clausius Inequality. arXiv 2011, arXiv:1105.5549. [Google Scholar]
- Gujrati, P.D. Nonequilibrium Thermodynamics. Symmetric and Unique Formulation of the First Law, Statistical Definition of Heat and Work, Adiabatic Theorem and the Fate of the Clausius Inequality: A Microscopic View. arXiv 2012, arXiv:1206.0702. [Google Scholar]
- Ruelle, D. Boltzmann’s Legacy; Gallavotti, G., Reiter, W.L., Yngvason, J., Eds.; European Mathematical Society: Zürich, Switzerland, 2008. [Google Scholar]
- Edwards, S.F.; Grinev, D.V. Granular materials: Towards the statistical mechanics of jammed configurations. Adv. Phys. 2002, 51, 1669. [Google Scholar] [CrossRef]
- Bekenstein, J.D. Black Holes and Entropy. Phys. Rev. D 1973, 7, 2333. [Google Scholar] [CrossRef]
- Hawking, S.W. Particle creation by black holes. Commun. Math. Phys. 1975, 43, 199. [Google Scholar] [CrossRef]
- Planck, M. Festschrift Ludwig Boltzmann; Meyer, S., Ed.; Barth: Leipzig, Germany, 1904; p. 113. [Google Scholar]
- Landau, L.D. The transport equation in the case of Coulomb interactions. Zh. Eksp.Teor. Fiz. 1937, 7, 203. [Google Scholar]
- Keizer, J. Heat, work, and the thermodynamic temperature at nonequilibrium steady states. J. Chem. Phys. 1985, 82, 2751. [Google Scholar] [CrossRef]
- Muschik, W. Aspects of Non-Equilibrium Thermodynamics; World Scientific: Singapore, 1990. [Google Scholar]
- Muschik, W.; Brunk, G. A concept of non-equilibrium temperature. Int. J. Engng Sci. 1977, 15, 377. [Google Scholar] [CrossRef]
- Muschik, W.; Papenfuss, C.; Ehrentraut, H. A sketch of continuum thermodynamics. J. Non-Newton. Fluid Mech. 2001, 96, 255. [Google Scholar] [CrossRef]
- Morris, G.P.; Rondoni, L. Definition of temperature in equilibrium and nonequilibrium systems. Phys. Rev. E 1999, 59, R5. [Google Scholar] [CrossRef] [Green Version]
- Casas-Vázquez, J.; Jou, D. Temperature in non-equilibrium states: A review of open problems and current proposals. Rep. Prog. Phys. 2003, 66, 1937. [Google Scholar] [CrossRef]
- Hoover, W.G.; Hoover, C.G. Nonequilibrium temperature and thermometry in heat-conducting ϕ4 models. Phys. Rev. E 2008, 77, 041104. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, N.F. Thermodynamics and Statistical Mechanics at Negative Absolute Temperatures. Phys. Rev. 1956, 103, 20. [Google Scholar] [CrossRef]
- Coleman, B.D. Thermodynamics of materials with memory. Arch. Rat. Mech. Anal. 1964, 17, 1. [Google Scholar] [CrossRef]
- Lucia, U.; Grisolia, G. Nonequilibrium Temperature: An Approach from Irreversibility. Materials 2021, 14, 2004. [Google Scholar] [CrossRef] [PubMed]
- Eu, B.C.; Garcia-Colin, L.S. Irreversible processes and temperature. Phys. Rev. E 1996, 54, 2501. [Google Scholar] [CrossRef] [PubMed]
- Gujrati, P.D. Determination of Nonequilibrium Temperature and Pressure using Clausius Equality in a State with Memory: A Simple Model Calculation. arXiv 2015, arXiv:1512.08744. [Google Scholar]
- Gujrati, P.D. Jensen inequality and the second law. Phys. Lett. A 2020, 384, 126460. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Chen, L.; Wu, S.; Sun, F.; Wu, C. Performance of an irreversible quantum Carnot engine with spin 1/2. J. Chem. Phys. 2006, 124, 214702. [Google Scholar] [CrossRef]
- Bender, C.M.; Brody, D.C.; Meister, B.K. Quantum mechanical Carnot engine. J. Phys. A 2000, 33, 4427. [Google Scholar] [CrossRef] [Green Version]
- Bender, C.M.; Brody, D.C. Meister, Unusual Quantum States: Non-Locality, Entropy, Maxwell’s Demon and Fractals. Proc. R. Soc. A 2005, 461, 733. [Google Scholar] [CrossRef] [Green Version]
- Doescher, S.W.; Rice, M.H. Infinite Square-Well Potential with a Moving Wall. Am. J. Phys. 1969, 37, 1246. [Google Scholar] [CrossRef]
- Schlitt, D.W.; Stutz, C. An Instructive Example of the Sudden Approximation in Quantum Mechanics. Am. J. Phys. 1970, 38, 70. [Google Scholar] [CrossRef]
- Stutz, C.; Schlitt, D.W. Temporal Evolution and the Approach to Equilibrium of a Quantum Particle in a Suddenly Expanded Box. Phys. Rev. A 1970, 2, 897. [Google Scholar] [CrossRef]
- Landau, L.D.; Lifshitz, E.M. Quantum Mechanics, 3rd ed.; Pergamon Press: Oxford, UK, 1977. [Google Scholar]
- Landau, L.D.; Lifshitz, E.M. Fluid Mechanics; Pergamon Press: Oxford, UK, 1982. [Google Scholar]
- Casas-Vazquez, J.; Criado-Sancho, M.; Jou, D. Comparison of three thermodynamic descriptions of nonlocal effects in viscoelasticity. Physica A 2002, 311, 353. [Google Scholar] [CrossRef]
- Hutter, M.; Brader, J.M. Nonlocal effects in nonisothermal hydrodynamics from the perspective of beyond-equilibrium thermodynamics. J. Chem. Phys. 2009, 130, 214908. [Google Scholar] [CrossRef] [Green Version]
- Reguera, D.; Rubi, J.M.; Vilar, J.M.G. The Mesoscopic Dynamics of Thermodynamic Systems. J. Phys. Chem. B 2005, 109, 21502. [Google Scholar] [CrossRef] [Green Version]
- Prigogine, I.; Mazur, P. Sur l’extension de la thermodynamique aux phénomènes irreversibles liés aux degrés de liberté internes. Physica 1953, 19, 241. [Google Scholar] [CrossRef]
- Ono, I.K.; O’Hern, C.S.; Durian, D.J.; Langer, S.A.; Liu, A.J.; Nagel, S.R. Effective Temperatures of a Driven System Near Jamming. Phys. Rev. Lett. 2002, 89, 095703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haxton, T.K.; Liu, A.J. Activated Dynamics and Effective Temperature in a Steady State Sheared Glass. Phys. Rev. Lett. 2007, 99, 195701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Kampen, N.G. Stochastic Processes in Physics and Chemistry; Elsevier Science: Amsterdam, The Netherlands, 1992. [Google Scholar]
- Muschik, W. Non-equilibrium thermodynamics and stochasticity: A phenomenological look on Jarzynski’s equality. Contin. Mech. Thermodyn. 2016, 28, 1887. [Google Scholar] [CrossRef] [Green Version]
- Zurek, W.H. Maxwell’s Demon, Szilard’s Engine and Quantum Measurements. arXiv 2003, arXiv:quant-ph/0301076v1. [Google Scholar]
- Zurek, W.H. Frontiers of Nonequilibrium Statistical Physics; Moore, G.T., Scully, M.O., Eds.; Plenum: New York, NY, USA, 1984. [Google Scholar]
- Marathe, R.; Parrondo, J.M.R. Cooling Classical Particles with a Microcanonical Szilard Engine. Phys. Rev. Lett. 2010, 104, 245704. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.W.; Sagawa, T.; Libertato, S.D.; Ueda, M. Quantum Szilard Engine. Phys. Rev. Lett. 2011, 106, 070401. [Google Scholar] [CrossRef] [Green Version]
- Wiener, N. Cybernetics, or Control and Communication in the Animal and the Machine; John Wiley and Sons: New York, NY, USA, 1948. [Google Scholar]
- Brillouin, L. Maxwell’s Demon Cannot Operate: Information and Entropy. I. J. Appl. Phys. 1951, 22, 334. [Google Scholar] [CrossRef]
- Hunt, K.L.C.; Hunt, P.M.; Ross, J. Nonlinear Dynamics and Thermodynamics of Chemical Reactions Far From Equilibrium. Annu. Rev. Phys. Chem. 1990, 41, 409. [Google Scholar] [CrossRef]
- Horn, K.; Scheffler, M. (Eds.) Handbook of Surface Science; Electronic Structure; Elsevier: Amsterdam, The Netherlands, 2000. [Google Scholar]
- Førland, K.S.; Førland, T.; Kjelstrup, S. Irreversible Thermodynamics: Theory and Application, 3rd ed.; Tapir: Trondheim, Norway, 2001. [Google Scholar]
- Hohenberg, P.C.; Swift, J.B. Hexagons and rolls in periodically modulated Rayleigh-Bénard convection. Phys. Rev. A 1987, 35, 3855. [Google Scholar] [CrossRef] [PubMed]
- Yaditi, Y.; Mears, N.; Chatterjee, A. Spatio-temporal characterization of thermal fluctuations in a non-turbulent Rayleigh–Bénard convection at steady state. Physica A 2020, 547, 123867. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Ban, T.; Iannacchione, G. Evidence of local equilibrium in a non-turbulent Rayleigh-Bénard convection at steady-state. arXiv 2021, arXiv:2107.03678v2. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gujrati, P.D. A Review of the System-Intrinsic Nonequilibrium Thermodynamics in Extended Space (MNEQT) with Applications. Entropy 2021, 23, 1584. https://doi.org/10.3390/e23121584
Gujrati PD. A Review of the System-Intrinsic Nonequilibrium Thermodynamics in Extended Space (MNEQT) with Applications. Entropy. 2021; 23(12):1584. https://doi.org/10.3390/e23121584
Chicago/Turabian StyleGujrati, Purushottam D. 2021. "A Review of the System-Intrinsic Nonequilibrium Thermodynamics in Extended Space (MNEQT) with Applications" Entropy 23, no. 12: 1584. https://doi.org/10.3390/e23121584