Genomics and Cytogenetics of Mosquitoes
Share This Topical Collection
Editors
 Prof. Dr. Igor Sharakhov
Prof. Dr. Igor Sharakhov
Prof. Dr. Igor Sharakhov
E-Mail
Website
Guest Editor
Department of Entomology and Fralin Life Science Institute, Virginia Tech, Blacksburg, VA 24061, USA
Interests: mosquitoes; genomics; cytogenetics; evolution; chromosomes; nuclear architecture
 Prof. Dr. Robert M. Waterhouse
Prof. Dr. Robert M. Waterhouse
Prof. Dr. Robert M. Waterhouse
E-Mail
Website
Guest Editor
Department of Ecology and Evolution, University of Lausanne, and Swiss Institute of Bioinformatics, CH-1015 Lausanne, Switzerland
Interests: arthropods; comparative genomics; evolutionary biology; bioinformatics
Topical Collection Information
Dear Colleagues,
Mosquitoes are vectors of numerous devastating infectious diseases. Traits relevant to the vectorial capacity of mosquitoes are determined or influenced by their genomes. Genome sequences for more than two dozen mosquito species are now available to the research community. A growing number of mosquito species have chromosome-level genome assemblies. Cytogenetics is playing an important role in developing physical maps that anchor genomic scaffolds to specific regions of chromosomes. Genomics can now be used to address questions about chromosome structure, function, and evolution. The marriage of genomics and cytogenetics raises studies to the next level—studies of mosquito population structure, genomic diversity, phylogeny, vectorial capacity, insecticide resistance, sex chromosomes, genome evolution, gene expression, and chromatin organization. Chromosome-scale assemblies facilitate the development of CRISPR-Cas9 gene drive systems for mosquitoes. For this Topical Collection, we are inviting research articles, reviews, concept papers, and technical notes on any aspect of the genomics and cytogenetics of mosquitoes.
Prof. Igor Sharakhov
Prof. Robert M. Waterhouse
Guest Editors
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. Manuscripts can be submitted until the deadline. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 100 words) can be sent to the Editorial Office for announcement on this website.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Insects is an international peer-reviewed open access monthly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 2600 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.
Keywords
- Mosquitoes
- Genomics
- Cytogenetics
- Evolution
- Chromosomes
Published Papers (9 papers)
Open AccessArticle
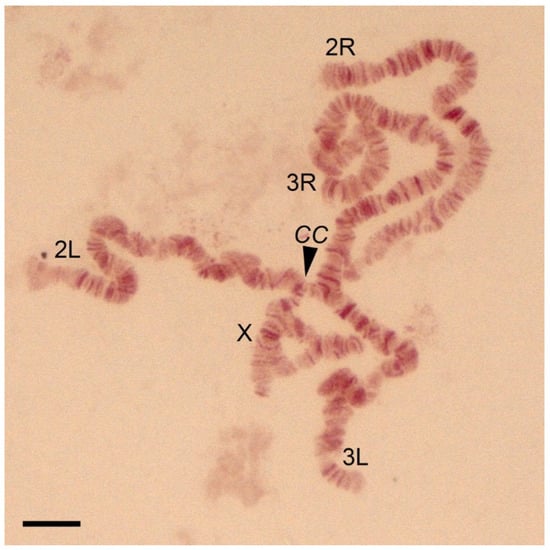
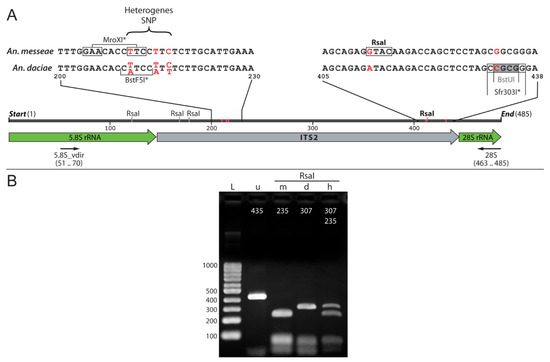
New Cytogenetic Photomap and Molecular Diagnostics for the Cryptic Species of the Malaria Mosquitoes Anopheles messeae and Anopheles daciae from Eurasia
by
Gleb N. Artemov, Valentina S. Fedorova, Dmitriy A. Karagodin, Ilya I. Brusentsov, Elina M. Baricheva, Igor V. Sharakhov, Mikhail I. Gordeev and Maria V. Sharakhova
Cited by 5 | Viewed by 2482
Abstract
The Eurasian malaria vector
Anopheles messeae is a widely spread and genetically diverse species. Five widespread polymorphic chromosomal inversions were found in natural populations of this mosquito. A cryptic species,
Anopheles daciae, was differentiated from
An. messeae by the presence of several nucleotide
[...] Read more.
The Eurasian malaria vector
Anopheles messeae is a widely spread and genetically diverse species. Five widespread polymorphic chromosomal inversions were found in natural populations of this mosquito. A cryptic species,
Anopheles daciae, was differentiated from
An. messeae by the presence of several nucleotide substitutions in the Internal Transcribed Spacer 2 (ITS2) region of ribosomal DNA. However, because of the absence of a high-quality reference cytogenetic map, the inversion polymorphisms in
An. daciae and
An. messeae remain poorly understood. Moreover, a recently determined heterogeneity in ITS2 in
An. daciae questioned the accuracy of the previously used Restriction Fragment Length Polymorphism (RFLP) assay for species diagnostics. In this study, a standard-universal cytogenetic map was constructed based on orcein stained images of chromosomes from salivary glands for population studies of the chromosomal inversions that can be used for both
An. messeae and
An. daciae. In addition, a new ITS2-RFLP approach for species diagnostics was developed. Both methods were applied to characterize inversion polymorphism in populations of
An. messeae and
An. daciae from a single location in Western Siberia in Russia. The analysis demonstrates that cryptic species are remarkably different in their frequencies of chromosomal inversion variants. Our study supports previous observations that
An. messeae has higher inversion polymorphism in all autosomes than the cryptic species
An. daciae.
Full article
►▼
Show Figures
Open AccessArticle
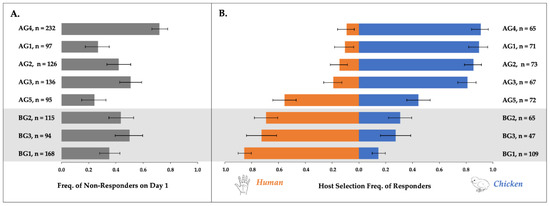
Differential Gene Expression in the Heads of Behaviorally Divergent Culex pipiens Mosquitoes
by
Anna Noreuil and Megan L. Fritz
Cited by 5 | Viewed by 2809
Abstract
Host preferences of
Cx. pipiens, a bridge vector for West Nile virus to humans, have the potential to drive pathogen transmission dynamics. Yet much remains unknown about the extent of variation in these preferences and their molecular basis. We conducted host choice
[...] Read more.
Host preferences of
Cx. pipiens, a bridge vector for West Nile virus to humans, have the potential to drive pathogen transmission dynamics. Yet much remains unknown about the extent of variation in these preferences and their molecular basis. We conducted host choice assays in a laboratory setting to quantify multi-day human and avian landing rates for
Cx. pipiens females. Assayed populations originated from five above-ground and three below-ground breeding and overwintering habitats. All three below-ground populations were biased toward human landings, with rates of human landing ranging from 69–85%. Of the five above-ground populations, four had avian landing rates of >80%, while one landed on the avian host only 44% of the time. Overall response rates and willingness to alternate landing on the human and avian hosts across multiple days of testing also varied by population. For one human- and one avian-preferring population, we examined patterns of differential expression and splice site variation at genes expressed in female heads. We also compared gene expression and splice site variation within human-seeking females in either gravid or host-seeking physiological states to identify genes that may regulate blood feeding behaviors. Overall, we identified genes with metabolic and regulatory function that were differentially expressed in our comparison of gravid and host-seeking females. Differentially expressed genes in our comparison of avian- and human-seeking females were enriched for those involved in sensory perception. We conclude with a discussion of specific sensory genes and their potential influence on the divergent behaviors of avian- and human-seeking
Cx. pipiens.
Full article
►▼
Show Figures
Open AccessReview
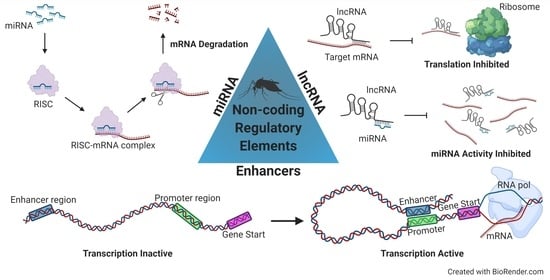
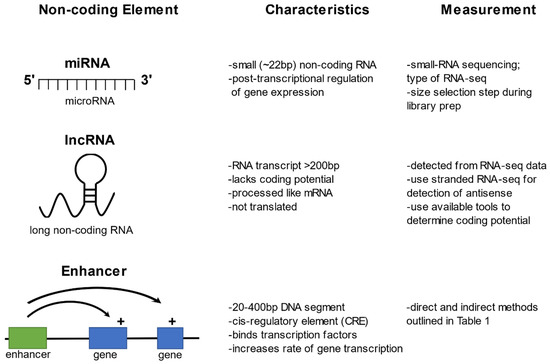
Filtering the Junk: Assigning Function to the Mosquito Non-Coding Genome
by
Elise J. Farley, Heather Eggleston and Michelle M. Riehle
Cited by 6 | Viewed by 2828
Abstract
The portion of the mosquito genome that does not code for proteins contains regulatory elements that likely underlie variation for important phenotypes including resistance and susceptibility to infection with arboviruses and Apicomplexan parasites. Filtering the non-coding genome to uncover these functional elements is
[...] Read more.
The portion of the mosquito genome that does not code for proteins contains regulatory elements that likely underlie variation for important phenotypes including resistance and susceptibility to infection with arboviruses and Apicomplexan parasites. Filtering the non-coding genome to uncover these functional elements is an expanding area of research, though identification of non-coding regulatory elements is challenging due to the lack of an amino acid-like code for the non-coding genome and a lack of sequence conservation across species. This review focuses on three types of non-coding regulatory elements: (1) microRNAs (miRNAs), (2) long non-coding RNAs (lncRNAs), and (3) enhancers, and summarizes current advances in technical and analytical approaches for measurement of each of these elements on a genome-wide scale. The review also summarizes and highlights novel findings following application of these techniques in mosquito-borne disease research. Looking beyond the protein-coding genome is essential for understanding the complexities that underlie differential gene expression in response to arboviral or parasite infection in mosquito disease vectors. A comprehensive understanding of the regulation of gene and protein expression will inform transgenic and other vector control methods rooted in naturally segregating genetic variation.
Full article
►▼
Show Figures
Open AccessArticle
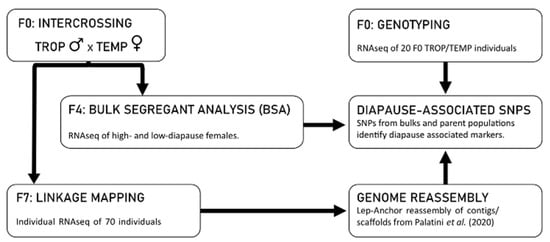
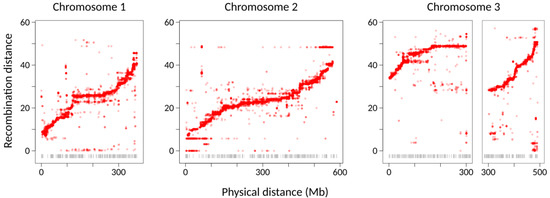
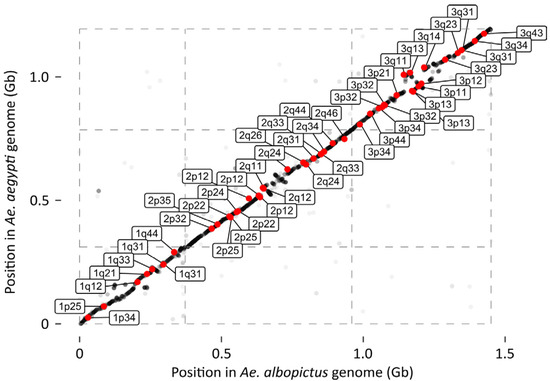
A Linkage-Based Genome Assembly for the Mosquito Aedes albopictus and Identification of Chromosomal Regions Affecting Diapause
by
John H. Boyle, Pasi M. A. Rastas, Xin Huang, Austin G. Garner, Indra Vythilingam and Peter A. Armbruster
Cited by 13 | Viewed by 2769
Abstract
The Asian tiger mosquito,
Aedes albopictus, is an invasive vector mosquito of substantial public health concern. The large genome size (~1.19–1.28 Gb by cytofluorometric estimates), comprised of ~68% repetitive DNA sequences, has made it difficult to produce a high-quality genome assembly for
[...] Read more.
The Asian tiger mosquito,
Aedes albopictus, is an invasive vector mosquito of substantial public health concern. The large genome size (~1.19–1.28 Gb by cytofluorometric estimates), comprised of ~68% repetitive DNA sequences, has made it difficult to produce a high-quality genome assembly for this species. We constructed a high-density linkage map for
Ae. albopictus based on 111,328 informative SNPs obtained by RNAseq. We then performed a linkage-map anchored reassembly of AalbF2, the genome assembly produced by Palatini et al. (2020). Our reassembled genome sequence, AalbF3, represents several improvements relative to AalbF2. First, the size of the AalbF3 assembly is 1.45 Gb, almost half the size of AalbF2. Furthermore, relative to AalbF2, AalbF3 contains a higher proportion of complete and single-copy BUSCO genes (84.3%) and a higher proportion of aligned RNAseq reads that map concordantly to a single location of the genome (46%). We demonstrate the utility of AalbF3 by using it as a reference for a bulk-segregant-based comparative genomics analysis that identifies chromosomal regions with clusters of candidate SNPs putatively associated with photoperiodic diapause, a crucial ecological adaptation underpinning the rapid range expansion and climatic adaptation of
A. albopictus.
Full article
►▼
Show Figures
Open AccessArticle
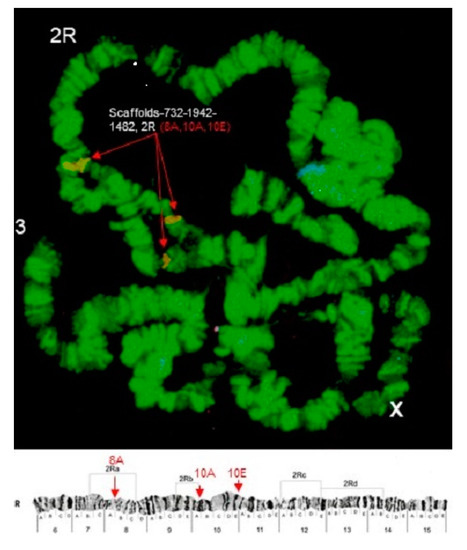
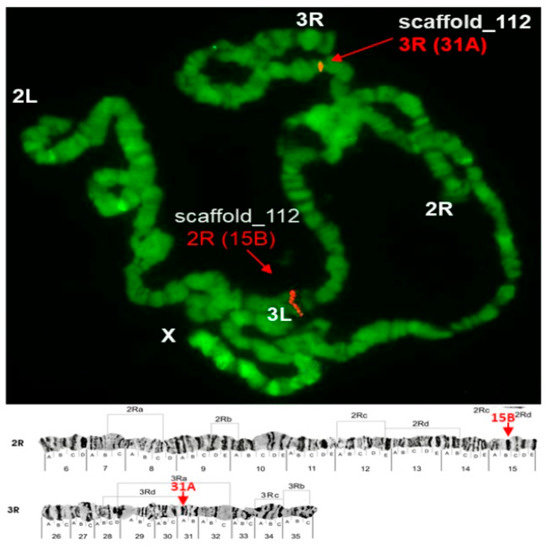
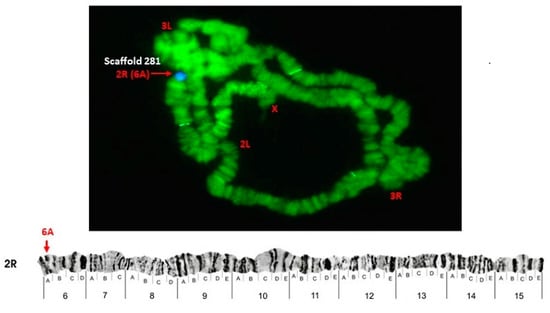
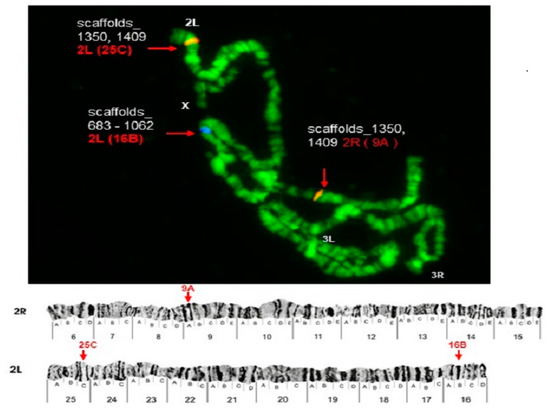
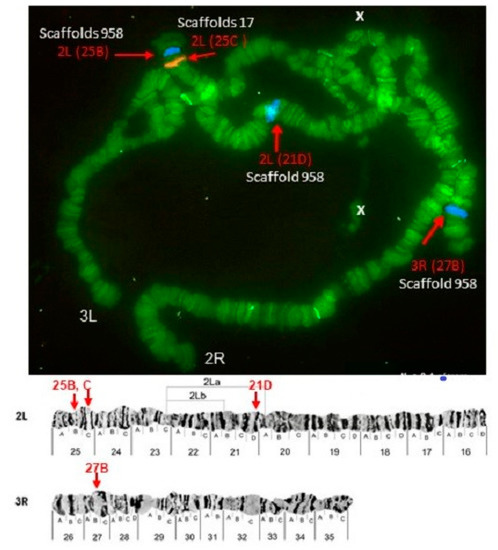
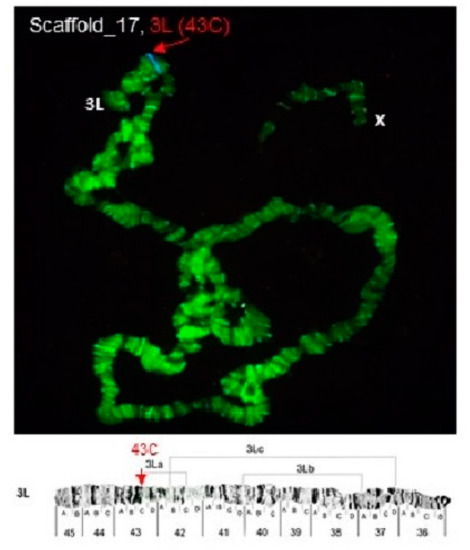
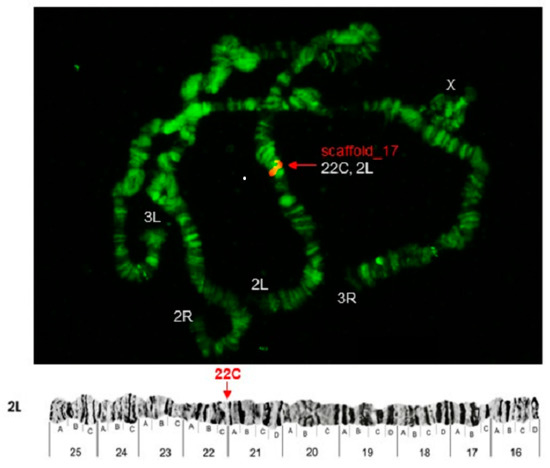
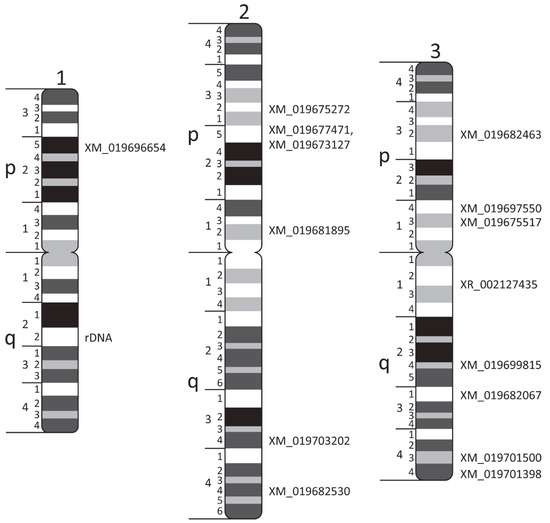
Physical Mapping of the Anopheles (Nyssorhynchus) darlingi Genomic Scaffolds
by
Míriam Silva Rafael, Leticia Cegatti Bridi, Igor V. Sharakhov, Osvaldo Marinotti, Maria V. Sharakhova, Vladimir Timoshevskiy, Giselle Moura Guimarães-Marques, Valéria Silva Santos, Carlos Gustavo Nunes da Silva, Spartaco Astolfi-Filho and Wanderli Pedro Tadei
Cited by 1 | Viewed by 2374
Abstract
The genome assembly of
Anopheles darlingi consists of 2221 scaffolds (N50 = 115,072 bp) and has a size spanning 136.94 Mbp. This assembly represents one of the smallest genomes among
Anopheles species.
Anopheles darlingi genomic DNA fragments of ~37 Kb were cloned, end-sequenced,
[...] Read more.
The genome assembly of
Anopheles darlingi consists of 2221 scaffolds (N50 = 115,072 bp) and has a size spanning 136.94 Mbp. This assembly represents one of the smallest genomes among
Anopheles species.
Anopheles darlingi genomic DNA fragments of ~37 Kb were cloned, end-sequenced, and used as probes for fluorescence in situ hybridization (FISH) with salivary gland polytene chromosomes. In total, we mapped nine DNA probes to scaffolds and autosomal arms. Comparative analysis of the
An. darlingi scaffolds with homologous sequences of the
Anopheles albimanus and
Anopheles gambiae genomes identified chromosomal rearrangements among these species. Our results confirmed that physical mapping is a useful tool for anchoring genome assemblies to mosquito chromosomes.
Full article
►▼
Show Figures
Open AccessTechnical Note
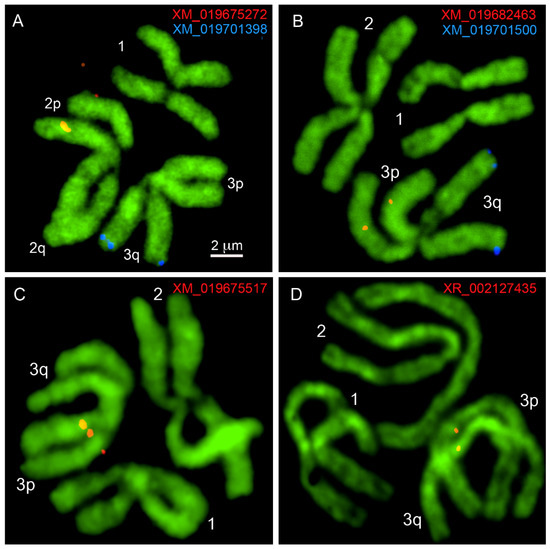
A Gene-Based Method for Cytogenetic Mapping of Repeat-Rich Mosquito Genomes
by
Reem A. Masri, Dmitriy A. Karagodin, Atashi Sharma and Maria V. Sharakhova
Cited by 1 | Viewed by 2072
Abstract
Long-read sequencing technologies have opened up new avenues of research on the mosquito genome biology, enabling scientists to better understand the remarkable abilities of vectors for transmitting pathogens. Although new genome mapping technologies such as Hi-C scaffolding and optical mapping may significantly improve
[...] Read more.
Long-read sequencing technologies have opened up new avenues of research on the mosquito genome biology, enabling scientists to better understand the remarkable abilities of vectors for transmitting pathogens. Although new genome mapping technologies such as Hi-C scaffolding and optical mapping may significantly improve the quality of genomes, only cytogenetic mapping, with the help of fluorescence in situ hybridization (FISH), connects genomic scaffolds to a particular chromosome and chromosome band. This mapping approach is important for creating and validating chromosome-scale genome assemblies for mosquitoes with repeat-rich genomes, which can potentially be misassembled. In this study, we describe a new gene-based physical mapping approach that was optimized using the newly assembled
Aedes albopictus genome, which is enriched with transposable elements. To avoid amplification of the repetitive DNA, 15 protein-coding gene transcripts were used for the probe design. Instead of using genomic DNA, complementary DNA was utilized as a template for development of the PCR-amplified probes for FISH. All probes were successfully amplified and mapped to specific chromosome bands. The genome-unique probes allowed to perform unambiguous mapping of genomic scaffolds to chromosome regions. The method described in detail here can be used for physical genome mapping in other insects.
Full article
►▼
Show Figures
Open AccessArticle
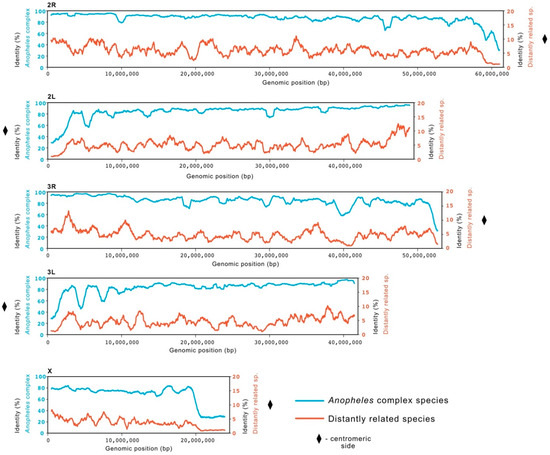
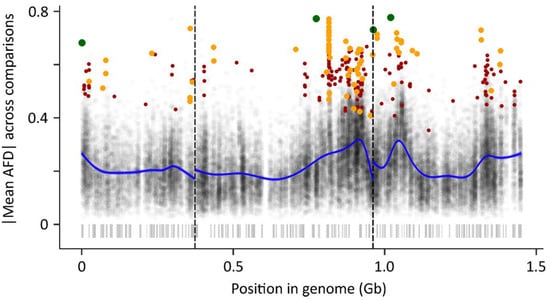
Anopheles gambiae Genome Conservation as a Resource for Rational Gene Drive Target Site Selection
by
Nace Kranjc, Andrea Crisanti, Tony Nolan and Federica Bernardini
Cited by 7 | Viewed by 3947
Abstract
The increase in molecular tools for the genetic engineering of insect pests and disease vectors, such as
Anopheles mosquitoes that transmit malaria, has led to an unprecedented investigation of the genomic landscape of these organisms. The understanding of genome variability in wild mosquito
[...] Read more.
The increase in molecular tools for the genetic engineering of insect pests and disease vectors, such as
Anopheles mosquitoes that transmit malaria, has led to an unprecedented investigation of the genomic landscape of these organisms. The understanding of genome variability in wild mosquito populations is of primary importance for vector control strategies. This is particularly the case for gene drive systems, which look to introduce genetic traits into a population by targeting specific genomic regions. Gene drive targets with functional or structural constraints are highly desirable as they are less likely to tolerate mutations that prevent targeting by the gene drive and consequent failure of the technology. In this study we describe a bioinformatic pipeline that allows the analysis of whole genome data for the identification of highly conserved regions that can point at potential functional or structural constraints. The analysis was conducted across the genomes of 22 insect species separated by more than hundred million years of evolution and includes the observed genomic variation within field caught samples of
Anopheles gambiae and
Anopheles coluzzii, the two most dominant malaria vectors. This study offers insight into the level of conservation at a genome-wide scale as well as at per base-pair resolution. The results of this analysis are gathered in a data storage system that allows for flexible extraction and bioinformatic manipulation. Furthermore, it represents a valuable resource that could provide insight into population structure and dynamics of the species in the complex and benefit the development and implementation of genetic strategies to tackle malaria.
Full article
►▼
Show Figures
Open AccessArticle
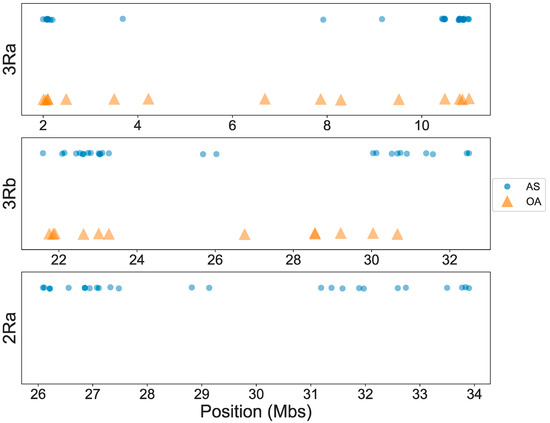
High-Throughput Genotyping of Common Chromosomal Inversions in the Afrotropical Malaria Mosquito Anopheles Funestus
by
Martin Lukindu, R. Rebecca Love, Moussa W. Guelbeogo, Scott T. Small, Melissa T. Stephens, Nathan R. Campbell, N’Fale Sagnon, Carlo Costantini and Nora J. Besansky
Cited by 6 | Viewed by 2524
Abstract
Polymorphic chromosomal inversions have been implicated in local adaptation. In anopheline mosquitoes, inversions also contribute to epidemiologically relevant phenotypes such as resting behavior. Progress in understanding these phenotypes and their mechanistic basis has been hindered because the only available method for inversion genotyping
[...] Read more.
Polymorphic chromosomal inversions have been implicated in local adaptation. In anopheline mosquitoes, inversions also contribute to epidemiologically relevant phenotypes such as resting behavior. Progress in understanding these phenotypes and their mechanistic basis has been hindered because the only available method for inversion genotyping relies on traditional cytogenetic karyotyping, a rate-limiting and technically difficult approach that is possible only for the fraction of the adult female population at the correct gonotrophic stage. Here, we focus on an understudied malaria vector of major importance in sub-Saharan Africa,
Anopheles funestus. We ascertain and validate tag single nucleotide polymorphisms (SNPs) using high throughput molecular assays that allow rapid inversion genotyping of the three most common
An. funestus inversions at scale, overcoming the cytogenetic karyotyping barrier. These same inversions are the only available markers for distinguishing two
An. funestus ecotypes that differ in indoor resting behavior, Folonzo and Kiribina. Our new inversion genotyping tools will facilitate studies of ecotypic differentiation in
An. funestus and provide a means to improve our understanding of the roles of Folonzo and Kiribina in malaria transmission.
Full article
►▼
Show Figures
Open AccessArticle
Analysis of the Metaphase Chromosome Karyotypes in Imaginal Discs of Aedes communis, Ae. punctor, Ae. intrudens, and Ae. rossicus (Diptera: Culicidae) Mosquitoes
by
Svetlana S. Alekseeva, Yulia V. Andreeva, Irina E. Wasserlauf, Anuarbek K. Sibataev and Vladimir N. Stegniy
Cited by 1 | Viewed by 2770
Abstract
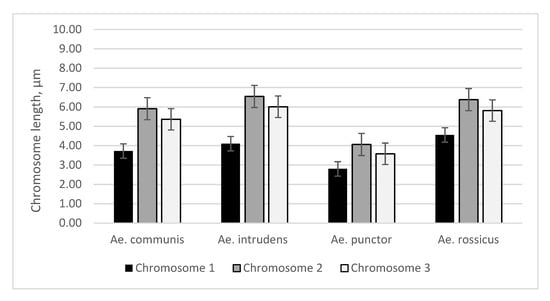
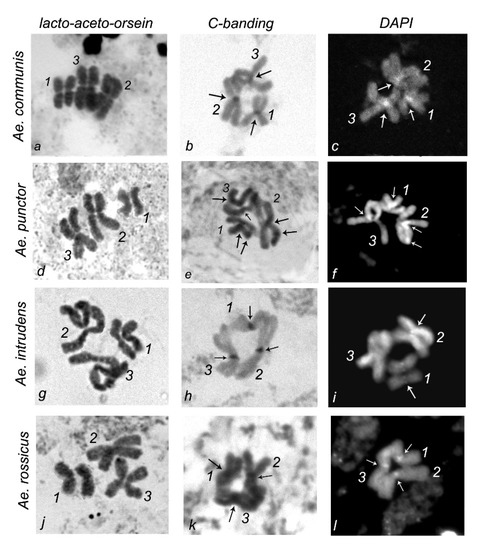
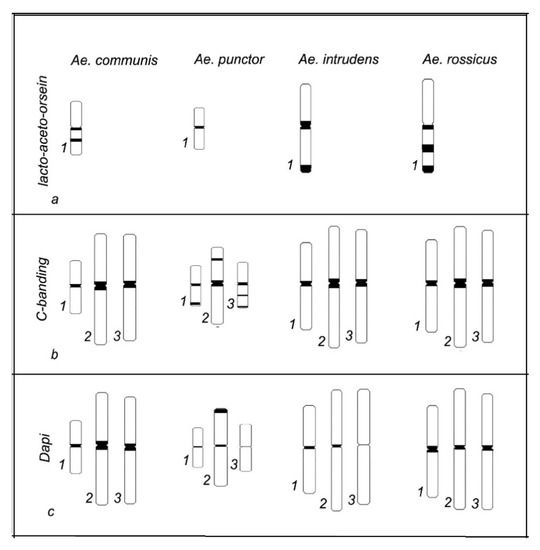
In this study, cytogenetic analysis of the metaphase chromosomes from imaginal discs of
Aedes (Diptera: Culicidae) mosquitoes—
Aedes communis,
Ae. punctor,
Ae. intrudens, and
Ae. rossicus—was performed. The patterns of C-banding and DAPI staining of the heteroсhromatin and the
[...] Read more.
In this study, cytogenetic analysis of the metaphase chromosomes from imaginal discs of
Aedes (Diptera: Culicidae) mosquitoes—
Aedes communis,
Ae. punctor,
Ae. intrudens, and
Ae. rossicus—was performed. The patterns of C-banding and DAPI staining of the heteroсhromatin and the length of the chromosomes demonstrate species specificity. In particular, the
Ae. punctor chromosomes are the shortest compared with
Ae. communis,
Ae. intrudens, and
Ae. rossicus, and they also carry additional C and DAPI bands in intercalary regions. The
Ae. intrudens chromosomes are the longest, they have pericentromeric C bands, and they almost lack any DAPI bands near the centromere of chromosome 3 versus
Ae. communis, which has the largest pericentromeric DAPI blocks in all three chromosome pairs.
Ae. rossicus also possesses DAPI bands in the centromeric regions of all chromosomes, but their staining is weaker compared with those of
Ae. communis. Therefore, the analysis of karyotypes is a tool for species-level identification of these mosquitoes.
Full article
►▼
Show Figures




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}