
Epigenetic and Metabolic Regulation of Macrophages during Gout


{kind=link}
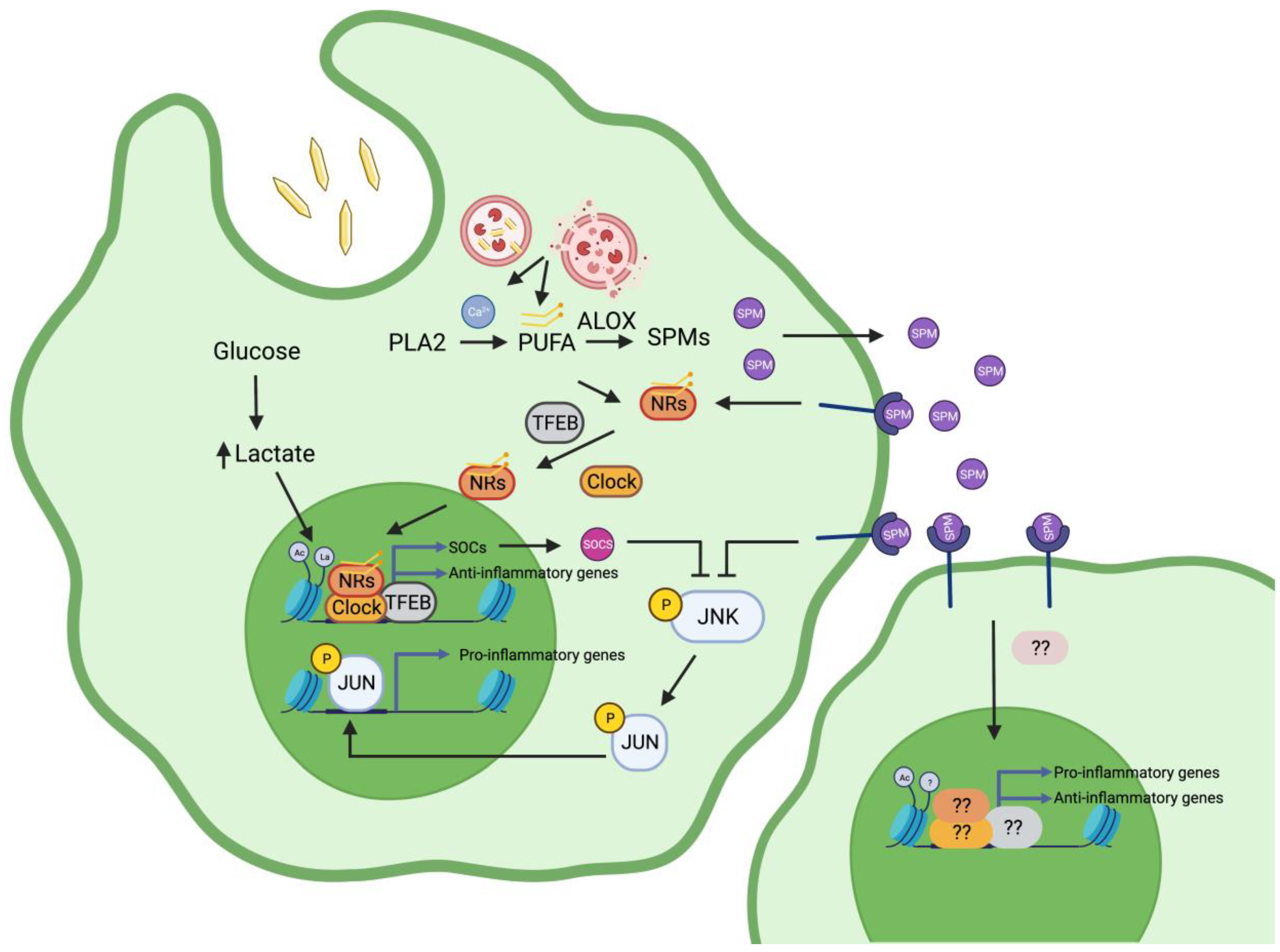
Abstract
:1. Introduction
2. From Metabolomics to Epigenetics and Transcription Factor Binding: Coupling Environmental Changes to Molecular Phenotypes
2.1. Histone Lactylation Contributes to Establishing an Inflammation Resolving Program in Macrophages
2.2. Histone Acetylation Takes Charge of the Dynamic Enhancer Activation in Macrophages in Response to External Stimuli
2.3. Differential Recruitment of Transcription Factor Binding to Genomic Regulatory Regions Regulates the Response of Macrophages to MSUc: The Case of the AP-1 Family
2.4. Epigenetic Changes by Higher Soluble Urate Levels in Myeloid Cells
3. Lipidomics and Gout, Signalling Pathways in the Resolution of Inflammation by Macrophages
3.1. Phospholipases A2
3.2. COX and ALOX5/ALOX5AP
3.3. Activation of Enzymatic Pathways by Damaged Subcellular Organelles
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Botstein, D.; Risch, N. Discovering genotypes underlying human phenotypes: Past successes for mendelian disease, future approaches for complex disease. Nat. Genet. 2003, 33, 228–237. [Google Scholar] [CrossRef]
- Stranger, B.E.; Stahl, E.A.; Raj, T. Progress and Promise of Genome-Wide Association Studies for Human Complex Trait Genetics. Genetics 2011, 187, 367–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shastry, B.S. SNPs in disease gene mapping, medicinal drug development and evolution. J. Hum. Genet. 2007, 52, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Hall, S.S. Revolution Postponed. Sci. Am. 2010, 303, 60–67. [Google Scholar] [CrossRef]
- Maher, B. Personal genomes: The case of the missing heritability. Nature 2008, 456, 18–21. [Google Scholar] [CrossRef] [Green Version]
- Rappaport, S.M.; Barupal, D.K.; Wishart, D.; Vineis, P.; Scalbert, A. The Blood Exposome and Its Role in Discovering Causes of Disease. Environ. Health Perspect. 2014, 122, 769–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhimal, M.; Neupane, T.; Dhimal, M.L. Understanding linkages between environmental risk factors and noncommunicable diseases—A review. FASEB BioAdv. 2021, 3, 287–294. [Google Scholar] [CrossRef]
- Prüss-Ustün, A.; Van Deventer, E.; Mudu, P.; Campbell-Lendrum, D.; Vickers, C.; Ivanov, I.; Forastiere, F.; Gumy, S.; Dora, C.; Adair-Rohani, H.; et al. Environmental risks and non-communicable diseases. BMJ 2019, 364, l265. [Google Scholar] [CrossRef] [Green Version]
- Dehlin, M.; Jacobsson, L.; Roddy, E. Global epidemiology of gout: Prevalence, incidence, treatment patterns and risk factors. Nat. Rev. Rheumatol. 2020, 16, 380–390. [Google Scholar] [CrossRef]
- Huang, Y.; Xiao, M.; Ou, J.; Lv, Q.; Wei, Q.; Chen, Z.; Wu, J.; Tu, L.; Jiang, Y.; Zhang, X.; et al. Identification of the urine and serum metabolomics signature of gout. Rheumatology 2020, 59, 2960–2969. [Google Scholar] [CrossRef]
- El Ridi, R.; Tallima, H. Physiological functions and pathogenic potential of uric acid: A review. J. Adv. Res. 2017, 8, 487–493. [Google Scholar] [CrossRef]
- Shipley, M. Hyperuricaemia and gout. J. R. Coll. Physicians Edinb. 2011, 41, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Oppedisano, F.; Gratteri, S.; Muscoli, C.; Mollace, V. Regulation of uric acid metabolism and excretion. Int. J. Cardiol. 2016, 213, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Dalbeth, N.; Gosling, A.L.; Gaffo, A.; Abhishek, A. Gout. Lancet 2021, 397, 1843–1855. [Google Scholar] [CrossRef] [PubMed]
- Stamp, L.K.; Farquhar, H.; Pisaniello, H.L.; Vargas-Santos, A.B.; Fisher, M.; Mount, D.B.; Choi, H.K.; Terkeltaub, R.; Hill, C.L.; Gaffo, A.L. Management of gout in chronic kidney disease: A G-CAN Consensus Statement on the research priorities. Nat. Rev. Rheumatol. 2021, 17, 633–641. [Google Scholar] [CrossRef]
- Lee, T.H.; Chen, J.-J.; Wu, C.-Y.; Yang, C.-W.; Yang, H.-Y. Hyperuricemia and Progression of Chronic Kidney Disease: A Review from Physiology and Pathogenesis to the Role of Urate-Lowering Therapy. Diagnostics 2021, 11, 1674. [Google Scholar] [CrossRef]
- Szekely, Y.; Arbel, Y. A Review of Interleukin-1 in Heart Disease: Where Do We Stand Today? Cardiol. Ther. 2018, 7, 25–44. [Google Scholar] [CrossRef] [Green Version]
- Everett, B.M.; MacFadyen, J.G.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Inhibition of Interleukin-1β and Reduction in Atherothrombotic Cardiovascular Events in the CANTOS Trial. J. Am. Coll. Cardiol. 2020, 76, 1660–1670. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, N. Anti-Interleukin-1 Therapy in the Management of Gout. Curr. Rheumatol. Rep. 2014, 16, 398. [Google Scholar] [CrossRef]
- Solomon, D.H.; Glynn, R.J.; MacFadyen, J.G.; Libby, P.; Thuren, T.; Everett, B.M.; Ridker, P.M. Relationship of Interleukin-1β Blockade With Incident Gout and Serum Uric Acid Levels: Exploratory Analysis of a Randomized Controlled Trial. Ann. Intern. Med. 2018, 169, 535–542. [Google Scholar] [CrossRef]
- Lv, Q.; Meng, X.-F.; He, F.-F.; Chen, S.; Su, H.; Xiong, J.; Gao, P.; Tian, X.-J.; Liu, J.-S.; Zhu, Z.-H.; et al. High Serum Uric Acid and Increased Risk of Type 2 Diabetes: A Systemic Review and Meta-Analysis of Prospective Cohort Studies. PLoS ONE 2013, 8, e56864. [Google Scholar] [CrossRef] [Green Version]
- Thottam, G.E.; Krasnokutsky, S.; Pillinger, M.H. Gout and Metabolic Syndrome: A Tangled Web. Curr. Rheumatol. Rep. 2017, 19, 60. [Google Scholar] [CrossRef]
- Puig, J.G.; Martínez, M.A. Hyperuricemia, gout and the metabolic syndrome. Curr. Opin. Rheumatol. 2008, 20, 187–191. [Google Scholar] [CrossRef]
- Major, T.J.; Dalbeth, N.; Stahl, E.A.; Merriman, T.R. An update on the genetics of hyperuricaemia and gout. Nat. Rev. Rheumatol. 2018, 14, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Dalbeth, N.; Stamp, L.K.; Merriman, T.R. The genetics of gout: Towards personalised medicine? BMC Med. 2017, 15, 108. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, S.; Yuan, M.; Xu, Y.; Xu, H. Gout and Diet: A Comprehensive Review of Mechanisms and Management. Nutrients 2022, 14, 3525. [Google Scholar] [CrossRef]
- Li, R.; Yu, K.; Li, C. Dietary factors and risk of gout and hyperuricemia: A meta-analysis and systematic review. Asia Pac. J. Clin. Nutr. 2018, 27, 1344–1356. [Google Scholar] [CrossRef] [PubMed]
- Vedder, D.; Walrabenstein, W.; Heslinga, M.; de Vries, R.; Nurmohamed, M.; van Schaardenburg, D.; Gerritsen, M. Dietary Interventions for Gout and Effect on Cardiovascular Risk Factors: A Systematic Review. Nutrients 2019, 11, 2955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokose, C.; McCormick, N.; Choi, H.K. The role of diet in hyperuricemia and gout. Curr. Opin. Rheumatol. 2021, 33, 135–144. [Google Scholar] [CrossRef]
- Wu, X.; You, C. The biomarkers discovery of hyperuricemia and gout: Proteomics and metabolomics. PeerJ 2023, 11, e14554. [Google Scholar] [CrossRef]
- Albrecht, E.; Waldenberger, M.; Krumsiek, J.; Evans, A.M.; Jeratsch, U.; Breier, M.; Adamski, J.; Koenig, W.; Zeilinger, S.; Fuchs, C.; et al. Metabolite profiling reveals new insights into the regulation of serum urate in humans. Metabolomics 2014, 10, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Shao, T.; Shao, L.; Li, H.; Xie, Z.; He, Z.; Wen, C. Combined Signature of the Fecal Microbiome and Metabolome in Patients with Gout. Front. Microbiol. 2017, 8, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobo, I.; Cheng, A.; Murillo-Saich, J.; Coras, R.; Torres, A.; Abe, Y.; Lana, A.J.; Schlachetzki, J.; Liu-Bryan, R.; Terkeltaub, R.; et al. Monosodium urate crystals regulate a unique JNK-dependent macrophage metabolic and inflammatory response. Cell Rep. 2022, 38, 110489. [Google Scholar] [CrossRef]
- Renaudin, F.; Orliaguet, L.; Castelli, F.; Fenaille, F.; Prignon, A.; Alzaid, F.; Combes, C.; Delvaux, A.; Adimy, Y.; Cohen-Solal, M.; et al. Gout and pseudo-gout-related crystals promote GLUT1-mediated glycolysis that governs NLRP3 and interleukin-1β activation on macrophages. Ann. Rheum. Dis. 2020, 79, 1506–1514. [Google Scholar] [CrossRef]
- Shen, X.; Wang, C.; Liang, N.; Liu, Z.; Li, X.; Zhu, Z.; Merriman, T.R.; Dalbeth, N.; Terkeltaub, R.; Li, C.; et al. Serum Metabolomics Identifies Dysregulated Pathways and Potential Metabolic Biomarkers for Hyperuricemia and Gout. Arthritis Rheumatol. 2021, 73, 1738–1748. [Google Scholar] [CrossRef]
- Luo, Y.; Wang, L.; Liu, X.-Y.; Chen, X.; Song, Y.-X.; Li, X.-H.; Jiang, C.; Peng, A.; Liu, J.-Y. Plasma profiling of amino acids distinguishes acute gout from asymptomatic hyperuricemia. Amino Acids 2018, 50, 1539–1548. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, H.; Chang, D.; Guo, F.; Pan, H.; Yang, Y. Metabolomics approach by 1H NMR spectroscopy of serum reveals progression axes for asymptomatic hyperuricemia and gout. Arthritis Res. Ther. 2018, 20, 111. [Google Scholar] [CrossRef] [Green Version]
- Guma, M.; Dadpey, B.; Coras, R.; Mikuls, T.R.; Hamilton, B.; Quehenberger, O.; Thorisdottir, H.; Bittleman, D.; Lauro, K.; Reilly, S.M.; et al. Xanthine oxidase inhibitor urate-lowering therapy titration to target decreases serum free fatty acids in gout and suppresses lipolysis by adipocytes. Arthritis Res. Ther. 2022, 24, 175. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wang, L.; Peng, A.; Liu, J.-Y. Metabolic profiling of human plasma reveals the activation of 5-lipoxygenase in the acute attack of gouty arthritis. Rheumatology 2019, 58, 345–351. [Google Scholar] [CrossRef]
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef]
- Beger, R.D. A Review of Applications of Metabolomics in Cancer. Metabolites 2013, 3, 552–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, L.M.N.; Lesner, N.P.; Sabatier, M.; Ubellacker, J.M.; Tasdogan, A. Emerging metabolomic tools to study cancer metastasis. Trends Cancer 2022, 8, 988–1001. [Google Scholar] [CrossRef]
- Pallares-Méndez, R.; Aguilar-Salinas, C.A.; Cruz-Bautista, I.; del Bosque-Plata, L. Metabolomics in diabetes, a review. Ann. Med. 2016, 48, 89–102. [Google Scholar] [CrossRef] [PubMed]
- McGranaghan, P.; Kirwan, J.A.; Garcia-Rivera, M.A.; Pieske, B.; Edelmann, F.; Blaschke, F.; Appunni, S.; Saxena, A.; Rubens, M.; Veledar, E.; et al. Lipid Metabolite Biomarkers in Cardiovascular Disease: Discovery and Biomechanism Translation from Human Studies. Metabolites 2021, 11, 621. [Google Scholar] [CrossRef] [PubMed]
- McGranaghan, P.; Saxena, A.; Rubens, M.; Radenkovic, J.; Bach, D.; Schleußner, L.; Pieske, B.; Edelmann, F.; Trippel, T.D. Predictive value of metabolomic biomarkers for cardiovascular disease risk: A systematic review and meta-analysis. Biomarkers 2020, 25, 101–111. [Google Scholar] [CrossRef]
- Ojanen, X.; Cheng, R.; Törmäkangas, T.; Rappaport, N.; Wilmanski, T.; Wu, N.; Fung, E.; Nedelec, R.; Sebert, S.; Vlachopoulos, D.; et al. Towards early risk biomarkers: Serum metabolic signature in childhood predicts cardio-metabolic risk in adulthood. eBioMedicine 2021, 72, 103611. [Google Scholar] [CrossRef]
- Hur, B.; Gupta, V.K.; Huang, H.; Wright, K.A.; Warrington, K.J.; Taneja, V.; Davis, J.M., 3rd; Sung, J. Plasma metabolomic profiling in patients with rheumatoid arthritis identifies biochemical features predictive of quantitative disease activity. Arthritis Res. Ther. 2021, 23, 164. [Google Scholar] [CrossRef]
- Bartikoski, B.J.; De Oliveira, M.S.; Santo, R.C.D.E.; Dos Santos, L.P.; Dos Santos, N.G.; Xavier, R.M. A Review of Metabolomic Profiling in Rheumatoid Arthritis: Bringing New Insights in Disease Pathogenesis, Treatment and Comorbidities. Metabolites 2022, 12, 394. [Google Scholar] [CrossRef]
- Xiao, Z.; Zhang, Z.; Huang, S.; Lon, J.R.; Xie, S. Metabolic Profiling of Serum for Osteoarthritis Biomarkers. Dis. Markers 2022, 2022, 1800812. [Google Scholar] [CrossRef]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Molecular Biological and Clinical Understanding of the Pathophysiology and Treatments of Hyperuricemia and Its Association with Metabolic Syndrome, Cardiovascular Diseases and Chronic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 9221. [Google Scholar] [CrossRef]
- Huang, Z.; Xie, N.; Illes, P.; Di Virgilio, F.; Ulrich, H.; Semyanov, A.; Verkhratsky, A.; Sperlagh, B.; Yu, S.-G.; Huang, C.; et al. From purines to purinergic signalling: Molecular functions and human diseases. Signal Transduct. Target. Ther. 2021, 6, 162. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Liebal, U.W.; Phan, A.N.T.; Sudhakar, M.; Raman, K.; Blank, L.M. Machine Learning Applications for Mass Spectrometry-Based Metabolomics. Metabolites 2020, 10, 243. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Y.; Li, X.; Deng, X.; Kong, Y.; Wang, W.; Zhou, Y. Machine learning of plasma metabolome identifies biomarker panels for metabolic syndrome: Findings from the China Suboptimal Health Cohort. Cardiovasc. Diabetol. 2022, 21, 288. [Google Scholar] [CrossRef]
- Sperber, H.; Mathieu, J.; Wang, Y.; Ferreccio, A.; Hesson, J.; Xu, Z.; Fischer, K.A.; Devi, A.; Detraux, D.; Gu, H.; et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat. Cell Biol. 2015, 17, 1523–1535. [Google Scholar] [CrossRef]
- Yanes, O.; Clark, J.; Wong, D.M.; Patti, G.J.; Sánchez-Ruiz, A.; Benton, H.P.; Trauger, S.A.; Desponts, C.; Ding, S.; Siuzdak, G. Metabolic oxidation regulates embryonic stem cell differentiation. Nat. Chem. Biol. 2010, 6, 411–417. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Rando, T.A. Interaction between epigenetic and metabolism in aging stem cells. Curr. Opin. Cell Biol. 2017, 45, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’aniello, C.; Cermola, F.; Patriarca, E.J.; Minchiotti, G. Metabolic–Epigenetic Axis in Pluripotent State Transitions. Epigenomes 2019, 3, 13. [Google Scholar] [CrossRef] [Green Version]
- Voss, K.; Hong, H.S.; Bader, J.E.; Sugiura, A.; Lyssiotis, C.A.; Rathmell, J.C. A guide to interrogating immunometabolism. Nat. Rev. Immunol. 2021, 21, 637–652. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Chavakis, T. Immunometabolism: Where Immunology and Metabolism Meet. J. Innate Immun. 2022, 14, 1–3. [Google Scholar] [CrossRef]
- Petersen, A.-K.; Zeilinger, S.; Kastenmüller, G.; Römisch-Margl, W.; Brugger, M.; Peters, A.; Meisinger, C.; Strauch, K.; Hengstenberg, C.; Pagel, P.; et al. Epigenetics meets metabolomics: An epigenome-wide association study with blood serum metabolic traits. Hum. Mol. Genet. 2014, 23, 534–545. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Zoghbi, H.Y.; Beaudet, A.L. Epigenetics and Human Disease. Cold Spring Harb. Perspect. Biol. 2016, 8, a019497. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaikkonen, M.U.; Spann, N.J.; Heinz, S.; Romanoski, C.E.; Allison, K.A.; Stender, J.D.; Chun, H.B.; Tough, D.F.; Prinjha, R.K.; Benner, C.; et al. Remodeling of the Enhancer Landscape during Macrophage Activation Is Coupled to Enhancer Transcription. Mol. Cell 2013, 51, 310–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidman, J.S.; Troutman, T.D.; Sakai, M.; Gola, A.; Spann, N.J.; Bennett, H.; Bruni, C.M.; Ouyang, Z.; Li, R.Z.; Sun, X.; et al. Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity 2020, 52, 1057–1074.e7. [Google Scholar] [CrossRef]
- Da Lin, D.; Xu, W.; Hong, P.; Wu, C.; Zhang, Z.; Zhang, S.; Xing, L.; Yang, B.; Zhou, W.; Xiao, Q.; et al. Decoding the spatial chromatin organization and dynamic epigenetic landscapes of macrophage cells during differentiation and immune activation. Nat. Commun. 2022, 13, 5857. [Google Scholar] [CrossRef]
- Zhang, P.; Amarasinghe, H.E.; Whalley, J.P.; Tay, C.; Fang, H.; Migliorini, G.; Brown, A.C.; Allcock, A.; Scozzafava, G.; Rath, P.; et al. Epigenomic analysis reveals a dynamic and context-specific macrophage enhancer landscape associated with innate immune activation and tolerance. Genome Biol. 2022, 23, 136. [Google Scholar] [CrossRef] [PubMed]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Qin, D. Next-generation sequencing and its clinical application. Cancer Biol. Med. 2019, 16, 4–10. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.E.; Wilson, R.K.; Mardis, E.R. The Next-Generation Sequencing Revolution and Its Impact on Genomics. Cell 2013, 155, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Heinz, S.; Romanoski, C.E.; Benner, C.; Glass, C.K. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 2015, 16, 144–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troutman, T.D.; Kofman, E.; Glass, C.K. Exploiting dynamic enhancer landscapes to decode macrophage and microglia phenotypes in health and disease. Mol. Cell 2021, 81, 3888–3903. [Google Scholar] [CrossRef]
- Diskin, C.; Ryan, T.A.J.; O’Neill, L.A.J. Modification of Proteins by Metabolites in Immunity. Immunity 2021, 54, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Brooks, G.A. Lactate as a fulcrum of metabolism. Redox Biol. 2020, 35, 101454. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; Enerbäck, S. Lactate: The ugly duckling of energy metabolism. Nat. Metab. 2020, 2, 566–571. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Zhang, B.; Lin, X.; Fu, X.; An, Y.; Zou, Y.; Wang, J.-X.; Wang, Z.; Yu, T. Lactate metabolism in human health and disease. Signal Transduct. Target. Ther. 2022, 7, 305. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Zhan, L.; Guo, J.Y.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Reyes, I.; Chandel, N.S. Waste Not, Want Not: Lactate Oxidation Fuels the TCA Cycle. Cell Metab. 2017, 26, 803–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltazar, F.; Afonso, J.; Costa, M.; Granja, S. Lactate Beyond a Waste Metabolite: Metabolic Affairs and Signaling in Malignancy. Front. Oncol. 2020, 10, 231. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Yang, K.; Fan, M.; Wang, X.; Xu, J.; Wang, Y.; Tu, F.; Gill, P.S.; Ha, T.; Liu, L.; Williams, D.L.; et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. 2022, 29, 133–146. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Li, W.; Zhou, X. Lactylation, an emerging hallmark of metabolic reprogramming: Current progress and open challenges. Front. Cell Dev. Biol. 2022, 10, 972020. [Google Scholar] [CrossRef]
- Karin, M.; Clevers, H. Reparative inflammation takes charge of tissue regeneration. Nature 2016, 529, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Koh, T.J.; DiPietro, L.A. Inflammation and wound healing: The role of the macrophage. Expert Rev. Mol. Med. 2011, 13, e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat. Commun. 2019, 10, 1076. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 2019, 129, 2619–2628. [Google Scholar] [CrossRef] [Green Version]
- Day, E.A.; O’Neill, L.A. Protein targeting by the itaconate family in immunity and inflammation. Biochem. J. 2022, 479, 2499–2510. [Google Scholar] [CrossRef] [PubMed]
- Peace, C.G.; O’Neill, L.A. The role of itaconate in host defense and inflammation. J. Clin. Investig. 2022, 132, e148548. [Google Scholar] [CrossRef] [PubMed]
- Hoeksema, M.A.; Shen, Z.; Holtman, I.R.; Zheng, A.; Spann, N.J.; Cobo, I.; Gymrek, M.; Glass, C.K. Mechanisms underlying divergent responses of genetically distinct macrophages to IL-4. Sci. Adv. 2021, 7, eabf9808. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef]
- Glass, C.K.; Natoli, G. Molecular control of activation and priming in macrophages. Nat. Immunol. 2016, 17, 26–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Xue, G.; Liu, J.; Li, Q.; Wang, Y. Revealing transcription factor and histone modification co-localization and dynamics across cell lines by integrating ChIP-seq and RNA-seq data. BMC Genom. 2018, 19, 914. [Google Scholar] [CrossRef]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Chinenov, Y.; Kerppola, T.K. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene 2001, 20, 2438–2452. [Google Scholar] [CrossRef] [Green Version]
- Bakiri, L.; Matsuo, K.; Wisniewska, M.; Wagner, E.F.; Yaniv, M. Promoter Specificity and Biological Activity of Tethered AP-1 Dimers. Mol. Cell. Biol. 2002, 22, 4952–4964. [Google Scholar] [CrossRef] [Green Version]
- Bakiri, L.; Hasenfuss, S.C.; Wagner, E.F. A FATal AP-1 dimer switch in hepatosteatosis. Cell Cycle 2014, 13, 1218–1219. [Google Scholar] [CrossRef] [Green Version]
- Jochum, W.; Passegué, E.; Wagner, E.F. AP-1 in mouse development and tumorigenesis. Oncogene 2001, 20, 2401–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angel, P.; Karin, M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1991, 1072, 129–157. [Google Scholar] [CrossRef]
- Badii, M.; Gaal, O.I.; Cleophas, M.C.; Klück, V.; Davar, R.; Habibi, E.; Keating, S.T.; Novakovic, B.; Helsen, M.M.; Dalbeth, N.; et al. Urate-induced epigenetic modifications in myeloid cells. Arthritis Res. Ther. 2021, 23, 202. [Google Scholar] [CrossRef]
- Cabău, G.; Crișan, T.O.; Klück, V.; Popp, R.A.; Joosten, L.A.B. Urate-induced immune programming: Consequences for gouty arthritis and hyperuricemia. Immunol. Rev. 2020, 294, 92–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tin, A.; Schlosser, P.; Matias-Garcia, P.R.; Thio, C.H.L.; Joehanes, R.; Liu, H.; Yu, Z.; Weihs, A.; Hoppmann, A.; Grundner-Culemann, F.; et al. Epigenome-wide association study of serum urate reveals insights into urate co-regulation and the SLC2A9 locus. Nat. Commun. 2021, 12, 7173. [Google Scholar] [CrossRef] [PubMed]
- Vitart, V.; Rudan, I.; Hayward, C.; Gray, N.K.; Floyd, J.; Palmer, C.N.A.; Knott, S.A.; Kolcic, I.; Polasek, O.; Graessler, J.; et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat. Genet. 2008, 40, 437–442. [Google Scholar] [CrossRef]
- Serhan, C.N.; Petasis, N.A. Resolvins and Protectins in Inflammation Resolution. Chem. Rev. 2011, 111, 5922–5943. [Google Scholar] [CrossRef] [Green Version]
- Sommer, C.; Birklein, F. Resolvins and inflammatory pain. F1000 Med. Rep. 2011, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef] [Green Version]
- Julliard, W.A.; Myo, Y.P.A.; Perelas, A.; Jackson, P.D.; Thatcher, T.H.; Sime, P.J. Specialized pro-resolving mediators as modulators of immune responses. Semin. Immunol. 2022, 59, 101605. [Google Scholar] [CrossRef]
- Basil, M.C.; Levy, B.D. Specialized pro-resolving mediators: Endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 2016, 16, 51–67. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Dalli, J.; Levy, B.D. Lipid Mediators in the Resolution of Inflammation. Cold Spring Harb. Perspect. Biol. 2014, 7, a016311. [Google Scholar] [CrossRef] [Green Version]
- Isopi, E.; Mattoscio, D.; Codagnone, M.; Mari, V.C.; Lamolinara, A.; Patruno, S.; D’aurora, M.; Cianci, E.; Nespoli, A.; Franchi, S.; et al. Resolvin D1 Reduces Lung Infection and Inflammation Activating Resolution in Cystic Fibrosis. Front. Immunol. 2020, 11, 581. [Google Scholar] [CrossRef]
- Sulciner, M.L.; Serhan, C.N.; Gilligan, M.M.; Mudge, D.K.; Chang, J.; Gartung, A.; Lehner, K.A.; Bielenberg, D.R.; Schmidt, B.; Dalli, J.; et al. Resolvins suppress tumor growth and enhance cancer therapy. J. Exp. Med. 2018, 215, 115–140. [Google Scholar] [CrossRef] [Green Version]
- Coras, R.; Murillo-Saich, J.D.; Singh, A.G.; Kavanaugh, A.; Guma, M. Lipidomic Profiling in Synovial Tissue. Front. Med. 2022, 9, 857135. [Google Scholar] [CrossRef] [PubMed]
- Schwab, J.M.; Chiang, N.; Arita, M.; Serhan, C.N. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 2007, 447, 869–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, M.; Gemperle, C.; Rimann, N.; Hersberger, M. Resolvin D1 Polarizes Primary Human Macrophages toward a Proresolution Phenotype through GPR32. J. Immunol. 2016, 196, 3429–3437. [Google Scholar] [CrossRef] [Green Version]
- Dalli, J.; Serhan, C.N. Specific lipid mediator signatures of human phagocytes: Microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood 2012, 120, e60–e72. [Google Scholar] [CrossRef] [Green Version]
- Romano, M.; Recchia, I.; Recchiuti, A. Lipoxin Receptors. Sci. World J. 2007, 7, 1393–1412. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Bianchini, F.; Aliberti, J.; Sher, A.; Chiang, N.; Hong, S.; Yang, R.; Petasis, N.A.; Serhan, C.N. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 2005, 201, 713–722. [Google Scholar] [CrossRef]
- Chiang, N.; Dalli, J.; Colas, R.A.; Serhan, C.N. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J. Exp. Med. 2015, 212, 1203–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Yacoubian, S.; Lee, C.-H.; Yang, R.; Petasis, N.A.; Serhan, C.N. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. USA 2010, 107, 1660–1665. [Google Scholar] [CrossRef]
- Margalit, A.; Duffin, K.L.; Shaffer, A.F.; Gregory, S.A.; Isakson, P.C. Altered arachidonic acid metabolism in urate crystal induced inflammation. Inflammation 1997, 21, 205–222. [Google Scholar] [CrossRef]
- Rae, S.A.; Davidson, E.M.; Smith, M.J. Leukotriene B4, an inflammatory mediator in gout. Lancet 1982, 320, 1122–1124. [Google Scholar] [CrossRef]
- Vasquez, A.M.; Mouchlis, V.D.; Dennis, E.A. Review of four major distinct types of human phospholipase A2. Adv. Biol. Regul. 2018, 67, 212–218. [Google Scholar] [CrossRef]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 2009, 50, S237–S242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, M.; Kudo, I. Phospholipase A2. J. Biochem. 2002, 131, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Leslie, C.C. Cytosolic phospholipase A2: Physiological function and role in disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef] [Green Version]
- Dabral, D.; van den Bogaart, G. The Roles of Phospholipase A2 in Phagocytes. Front. Cell Dev. Biol. 2021, 9, 673502. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.E.; Hsu, Y.-H.; Deems, R.A.; Li, S.; Woods, V.L., Jr.; Dennis, E.A. A Phospholipid Substrate Molecule Residing in the Membrane Surface Mediates Opening of the Lid Region in Group IVA Cytosolic Phospholipase A2. J. Biol. Chem. 2008, 283, 31227–31236. [Google Scholar] [CrossRef] [Green Version]
- Stahelin, R.V.; Subramanian, P.; Vora, M.; Cho, W.; Chalfant, C.E. Ceramide-1-phosphate Binds Group IVA Cytosolic Phospholipase a2 via a Novel Site in the C2 Domain. J. Biol. Chem. 2007, 282, 20467–20474. [Google Scholar] [CrossRef] [Green Version]
- Seilhamer, J.J.; Pruzanski, W.; Vadas, P.; Plant, S.; Miller, J.A.; Kloss, J.; Johnson, L.K. Cloning and Recombinant Expression of Phospholipase A2 Present in Rheumatoid Arthritic Synovial Fluid. J. Biol. Chem. 1989, 264, 5335–5338. [Google Scholar] [CrossRef]
- Kramer, R.M.; Hession, C.; Johansen, B.; Hayes, G.; McGray, P.; Chow, E.P.; Tizard, R.; Pepinsky, R.B. Structure and Properties of a Human Non-pancreatic Phospholipase A2. J. Biol. Chem. 1989, 264, 5768–5775. [Google Scholar] [CrossRef] [PubMed]
- Six, D.A.; Dennis, E.A. Essential Ca2+-independent Role of the Group IVA Cytosolic Phospholipase A2 C2 Domain for Interfacial Activity. J. Biol. Chem. 2003, 278, 23842–23850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 biochemistry. Cardiovasc. Drugs Ther. 2009, 23, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Jaulmes, A.; Janvier, B.; Andreani, M.; Raymondjean, M. Autocrine and Paracrine Transcriptional Regulation of Type IIA Secretory Phospholipase A2 Gene in Vascular Smooth Muscle Cells. Arter. Thromb. Vasc. Biol. 2005, 25, 1161–1167. [Google Scholar] [CrossRef] [Green Version]
- Antonio, V.; Brouillet, A.; Janvier, B.; Monne, C.; Bereziat, G.; Andreani, M.; Raymondjean, M. Transcriptional regulation of the rat type IIA phospholipase A2 gene by cAMP and interleukin-1β in vascular smooth muscle cells: Interplay of the CCAAT/enhancer binding protein (C/EBP), nuclear factor-κB and Ets transcription factors. Biochem. J. 2002, 368, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Pouliot, M.; James, M.J.; McColl, S.R.; Naccache, P.H.; Cleland, L.G. Monosodium urate microcrystals induce cyclooxygenase-2 in human monocytes. Blood 1998, 91, 1769–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, F.A.; Costa, V.V.; Tavares, L.D.; Sachs, D.; Coelho, F.M.; Fagundes, C.T.; Soriani, F.M.; Silveira, T.N.; Cunha, L.D.; Zamboni, D.S.; et al. NLRP3 inflammasome-mediated neutrophil recruitment and hypernociception depend on leukotriene B4 in a murine model of gout. Arthritis Rheum. 2012, 64, 474–484. [Google Scholar] [CrossRef]
- Murakami, Y.; Akahoshi, T.; Hayashi, I.; Endo, H.; Hashimoto, A.; Kono, S.; Kondo, H.; Kawai, S.; Inoue, M.; Kitasato, H. Inhibition of monosodium urate monohydrate crystal-induced acute inflammation by retrovirally transfected prostaglandin D synthase. Arthritis Rheum. 2003, 48, 2931–2941. [Google Scholar] [CrossRef]
- Ruiz-Miyazawa, K.W.; Staurengo-Ferrari, L.; Pinho-Ribeiro, F.A.; Fattori, V.; Zaninelli, T.H.; Badaro-Garcia, S.; Borghi, S.M.; Andrade, K.C.; Clemente-Napimoga, J.T.; Alves-Filho, J.C.; et al. 15d-PGJ2-loaded nanocapsules ameliorate experimental gout arthritis by reducing pain and inflammation in a PPAR-gamma-sensitive manner in mice. Sci. Rep. 2018, 8, 13979. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Andersson, A.M.C.; Semsey, S.; Sneppen, K. Structure and function of negative feedback loops at the interface of genetic and metabolic networks. Nucleic Acids Res. 2006, 34, 2455–2462. [Google Scholar] [CrossRef] [Green Version]
- Chubukov, V.; Zuleta, I.A.; Li, H. Regulatory architecture determines optimal regulation of gene expression in metabolic pathways. Proc. Natl. Acad. Sci. USA 2012, 109, 5127–5132. [Google Scholar] [CrossRef] [PubMed]
- Häfner, A.-K.; Kahnt, A.S.; Steinhilber, D. Beyond leukotriene formation—The noncanonical functions of 5-lipoxygenase. Prostaglandins Other Lipid Mediat. 2019, 142, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Kreiß, M.; Oberlis, J.H.; Seuter, S.; Bischoff-Kont, I.; Sürün, D.; Thomas, D.; Göbel, T.; Schmid, T.; Rådmark, O.; Brandes, R.P.; et al. Human 5-lipoxygenase regulates transcription by association to euchromatin. Biochem. Pharmacol. 2022, 203, 115187. [Google Scholar] [CrossRef]
- Weissmann, G.; Rita, G.A. Molecular Basis of Gouty Inflammation: Interaction of Monosodium Urate Crystals with Lysosomes and Liposomes. Nat. New Biol. 1972, 240, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Fenando, A.; Rednam, M.; Widrich, J. Gout. In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2021. [Google Scholar]
- Hoffstein, S.; Weissmann, G. Mechanisms of lysosomal enzyme release from leukocytes. IV. Interaction of monosodium urate crystals with dogfish and human leukocytes. Arthritis Rheum. 1975, 18, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Shirahama, T.; Cohen, A.S. Ultrastructural evidence for leakage of lysosomal contents after phagocytosis of monosodium urate crystals. A mechanism of gouty inflammation. Am. J. Pathol. 1974, 76, 501–520. [Google Scholar]
- Rajan, K.T. Lysosomes and Gout. Nature 1966, 210, 959–960. [Google Scholar] [CrossRef]
- Höglinger, D.; Haberkant, P.; Aguilera-Romero, A.; Riezman, H.; Porter, F.D.; Platt, F.M.; Galione, A.; Schultz, C. Intracellular sphingosine releases calcium from lysosomes. eLife 2015, 4, e10616. [Google Scholar] [CrossRef] [PubMed]
- Kiselyov, K.; Yamaguchi, S.; Lyons, C.W.; Muallem, S. Aberrant Ca2+ handling in lysosomal storage disorders. Cell Calcium 2010, 47, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, A.; Suzuki, M.; Sakaguchi, R.; Hanada, T.; Yasukawa, H. SOCS, Inflammation, and Autoimmunity. Front. Immunol. 2012, 3, 20. [Google Scholar] [CrossRef] [Green Version]
- Sobah, M.L.; Liongue, C.; Ward, A.C. SOCS Proteins in Immunity, Inflammatory Diseases, and Immune-Related Cancer. Front. Med. 2021, 8, 727987. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Hsieh, S.-C.; Chen, W.-Y.; Li, K.-J.; Wu, C.-H.; Wu, P.-C.; Tsai, C.-Y.; Yu, C.-L. Spontaneous resolution of acute gouty arthritis is associated with rapid induction of the anti-inflammatory factors TGF 1, IL-10 and soluble TNF receptors and the intracellular cytokine negative regulators CIS and SOCS3. Ann. Rheum. Dis. 2011, 70, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Orji, O.C.; López-Domínguez, M.B.; Sandoval-Plata, G.; Guetta-Baranes, T.; Valdes, A.M.; Doherty, M.; Morgan, K.; Abhishek, A. Upregulated expression of FFAR2 and SOC3 genes is associated with gout. Rheumatology 2023, 62, 977–983. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cobo, I.; Murillo-Saich, J.; Alishala, M.; Guma, M. Epigenetic and Metabolic Regulation of Macrophages during Gout. Gout Urate Cryst. Depos. Dis. 2023, 1, 137-151. https://doi.org/10.3390/gucdd1030013
Cobo I, Murillo-Saich J, Alishala M, Guma M. Epigenetic and Metabolic Regulation of Macrophages during Gout. Gout, Urate, and Crystal Deposition Disease. 2023; 1(3):137-151. https://doi.org/10.3390/gucdd1030013
Chicago/Turabian StyleCobo, Isidoro, Jessica Murillo-Saich, Mohnish Alishala, and Monica Guma. 2023. "Epigenetic and Metabolic Regulation of Macrophages during Gout" Gout, Urate, and Crystal Deposition Disease 1, no. 3: 137-151. https://doi.org/10.3390/gucdd1030013