
Regulation of Ras-GTPase Signaling and Localization by Post-Translational Modifications


Abstract
:1. Introduction
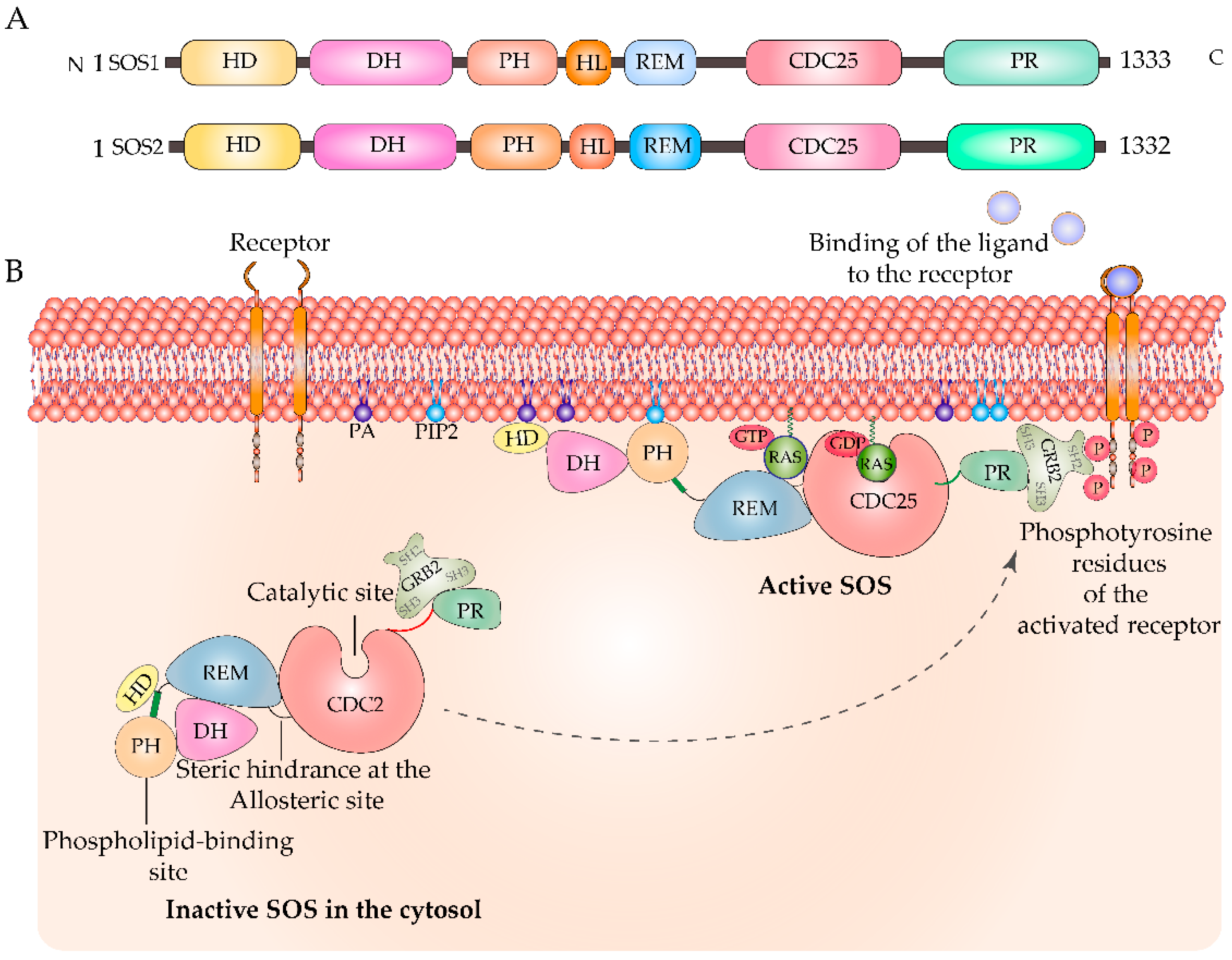
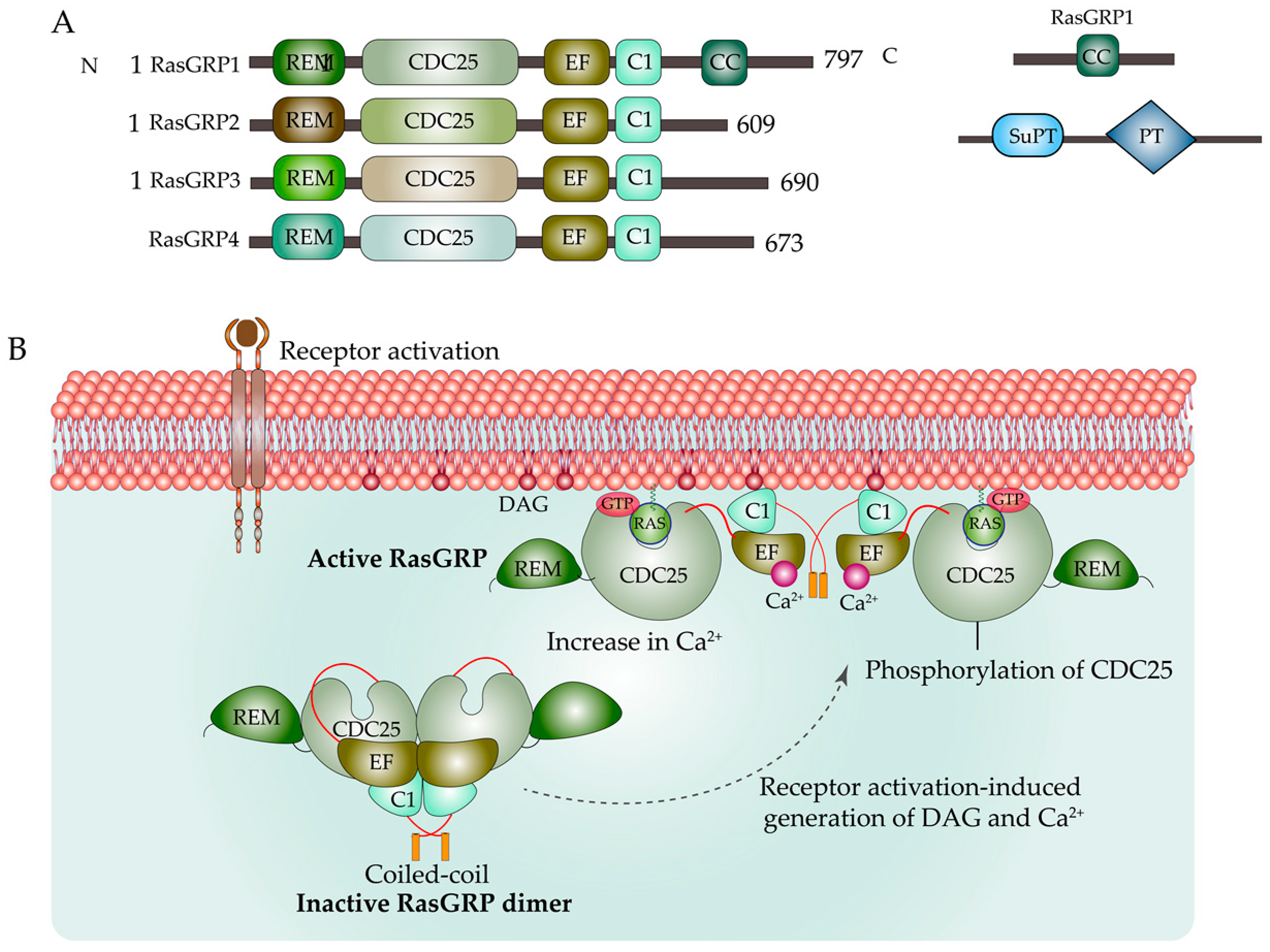
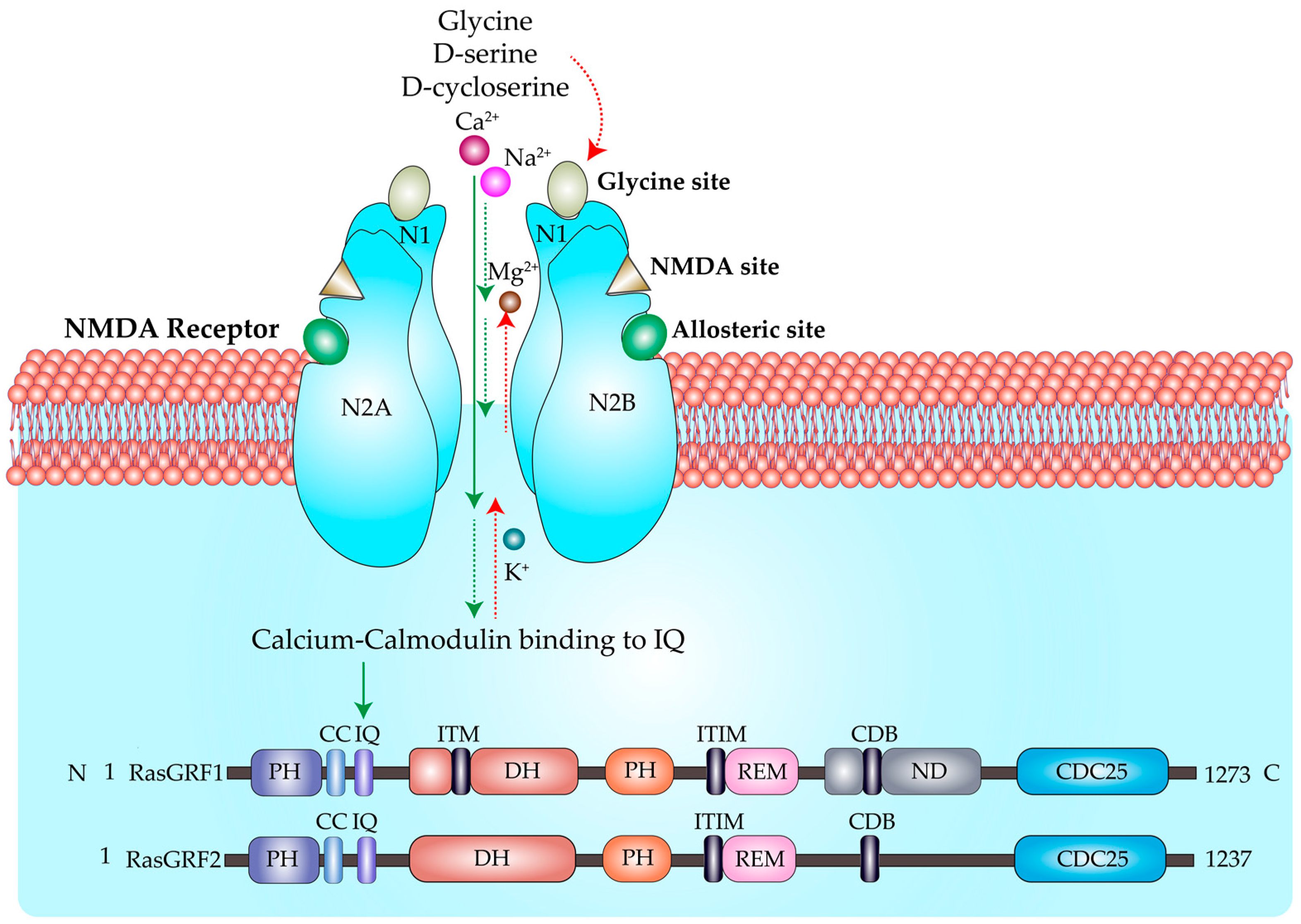
2. Regulation of Ras by GEFs and GAPs
3. Ras Post-Translational Modifications
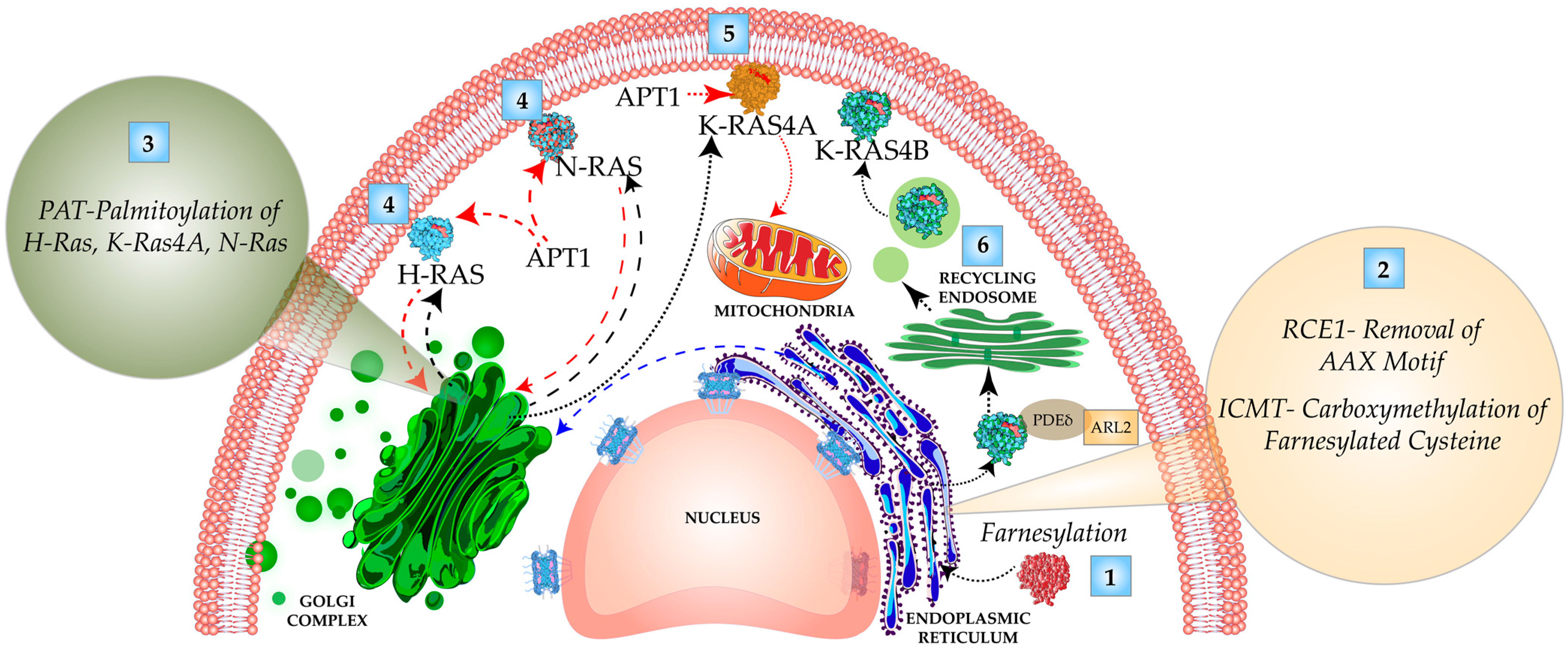
3.1. C-Terminal Carboxy-Terminal Modifications and Membrane Targeting of Ras Isoforms
3.2. Microregulation of Ras Isoforms by Ubiquitination, Acetylation, and Nitrosylation
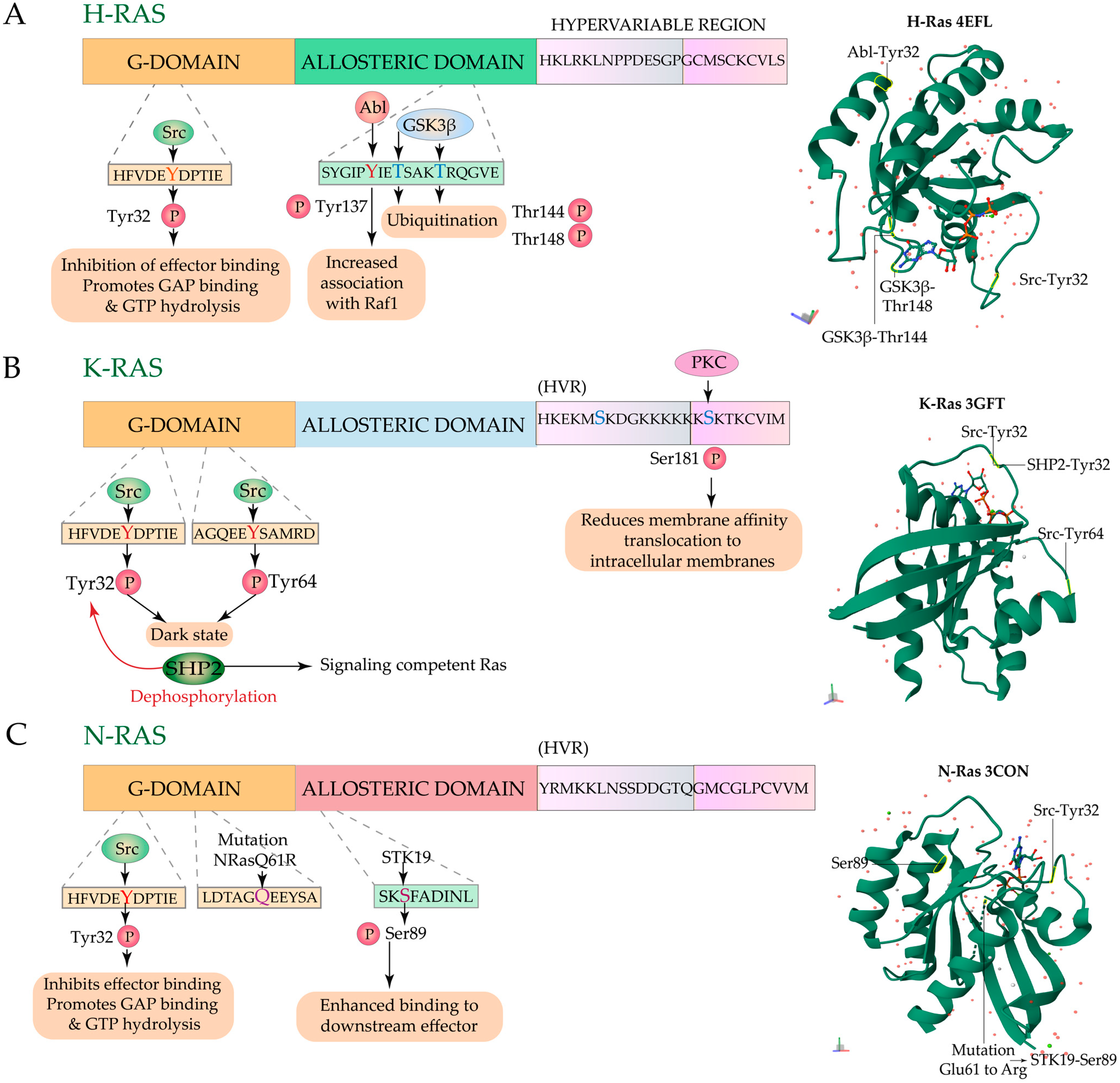
3.3. Regulation of Ras Signaling and Localization by Phosphorylation-Dephosphorylation
3.3.1. Regulation by Phosphorylation
3.3.2. Regulation by Dephosphorylation
4. Therapeutic Approaches Targeting Enzymatic Regulation of Ras
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ahearn, I.M.; Haigis, K.; Bar-Sagi, D.; Philips, M.R. Regulating the Regulator: Post-Translational Modification of RAS. Nat. Rev. Mol. Cell Biol. 2012, 13, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Dower, N.A.; Stang, S.L.; Bottorff, D.A.; Ebinu, J.O.; Dickie, P.; Ostergaard, H.L.; Stone, J.C. RasGRP Is Essential for Mouse Thymocyte Differentiation and TCR Signaling. Nat. Immunol. 2000, 1, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Kortum, R.L.; Sommers, C.L.; Alexander, C.P.; Pinski, J.M.; Li, W.; Grinberg, A.; Lee, J.; Love, P.E.; Samelson, L.E. Targeted Sos1 Deletion Reveals Its Critical Role in Early T-Cell Development. Proc. Natl. Acad. Sci. USA 2011, 108, 12407–12412. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.E.; Rubio, I.; Roose, J.P. Regulation of Ras Exchange Factors and Cellular Localization of Ras Activation by Lipid Messengers in T Cells. Front. Immunol. 2013, 4, 239. [Google Scholar] [CrossRef]
- Sondermann, H.; Soisson, S.M.; Boykevisch, S.; Yang, S.-S.; Bar-Sagi, D.; Kuriyan, J. Structural Analysis of Autoinhibition in the Ras Activator Son of Sevenless. Cell 2004, 119, 393–405. [Google Scholar] [CrossRef]
- Groves, J.T.; Kuriyan, J. Molecular Mechanisms in Signal Transduction at the Membrane. Nat. Struct. Mol. Biol. 2010, 17, 659–665. [Google Scholar] [CrossRef]
- Gureasko, J.; Kuchment, O.; Makino, D.L.; Sondermann, H.; Bar-Sagi, D.; Kuriyan, J. Role of the Histone Domain in the Autoinhibition and Activation of the Ras Activator Son of Sevenless. Proc. Natl. Acad. Sci. USA 2010, 107, 3430–3435. [Google Scholar] [CrossRef]
- Corbalan-Garcia, S.; Yang, S.S.; Degenhardt, K.R.; Bar-Sagi, D. Identification of the Mitogen-Activated Protein Kinase Phosphorylation Sites on Human Sos1 That Regulate Interaction with Grb2. Mol. Cell. Biol. 1996, 16, 5674–5682. [Google Scholar] [CrossRef]
- Baschieri, F.; Farhan, H. Crosstalk of Small GTPases at the Golgi Apparatus. Small GTPases 2012, 3, 80–90. [Google Scholar] [CrossRef]
- Arozarena, I.; Matallanas, D.; Berciano, M.T.; Sanz-Moreno, V.; Calvo, F.; Muñoz, M.T.; Egea, G.; Lafarga, M.; Crespo, P. Activation of H-Ras in the Endoplasmic Reticulum by the RasGRF Family Guanine Nucleotide Exchange Factors. Mol. Cell. Biol. 2004, 24, 1516–1530. [Google Scholar] [CrossRef]
- Irie, K.; Masuda, A.; Shindo, M.; Nakagawa, Y.; Ohigashi, H. Tumor Promoter Binding of the Protein Kinase C C1 Homology Domain Peptides of RasGRPs, Chimaerins, and Unc13s. Bioorganic. Med. Chem. 2004, 12, 4575–4583. [Google Scholar] [CrossRef] [PubMed]
- Ebinu, J.O.; Bottorff, D.A.; Chan, E.Y.; Stang, S.L.; Dunn, R.J.; Stone, J.C. RasGRP, a Ras guanyl nucleotide-releasing protein with calcium-and diacylglycerol-binding motifs. Science 1998, 280, 1082–1086. [Google Scholar] [CrossRef] [PubMed]
- Iwig, J.S.; Vercoulen, Y.; Das, R.; Barros, T.; Limnander, A.; Che, Y.; Pelton, J.G.; Wemmer, D.E.; Roose, J.P.; Kuriyan, J. Structural Analysis of Autoinhibition in the Ras-Specific Exchange Factor RasGRP1. eLife 2013, 2, e00813. [Google Scholar] [CrossRef] [PubMed]
- Roose, J.P.; Mollenauer, M.; Gupta, V.A.; Stone, J.; Weiss, A. A Diacylglycerol-Protein Kinase C-RasGRP1 Pathway Directs Ras Activation upon Antigen Receptor Stimulation of T Cells. Mol. Cell. Biol. 2005, 25, 4426–4441. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, H.; Coughlin, J.; Zheng, J.; Li, L.; Stone, J.C. Phosphorylation of RasGRP3 on Threonine 133 Provides a Mechanistic Link between PKC and Ras Signaling Systems in B Cells. Blood 2005, 105, 3648–3654. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, G.F.; Manganaro, D.; Consonni, A.; Canobbio, I.; Balduini, C.; Torti, M. Phosphorylation of the Guanine-Nucleotide-Exchange Factor CalDAG-GEFI by Protein Kinase A Regulates Ca2+-Dependent Activation of Platelet Rap1b GTPase. Biochem. J. 2013, 453, 115–123. [Google Scholar] [CrossRef]
- Nagai, T.; Nakamuta, S.; Kuroda, K.; Nakauchi, S.; Nishioka, T.; Takano, T.; Zhang, X.; Tsuboi, D.; Funahashi, Y.; Nakano, T. Phosphoproteomics of the Dopamine Pathway Enables Discovery of Rap1 Activation as a Reward Signal in Vivo. Neuron 2016, 89, 550–565. [Google Scholar] [CrossRef]
- Ren, J.; Cook, A.A.; Bergmeier, W.; Sondek, J. A Negative-Feedback Loop Regulating ERK1/2 Activation and Mediated by RasGPR2 Phosphorylation. Biochem. Biophys. Res. Commun. 2016, 474, 193–198. [Google Scholar] [CrossRef]
- Liao, F.; Shin, H.S.; Rhee, S.G. In Vitro Tyrosine Phosphorylation of PLC-Γ1 and PLC-Γ2 by Src-Family Protein Tyrosine Kinases. Biochem. Biophys. Res. Commun. 1993, 191, 1028–1033. [Google Scholar] [CrossRef]
- Bivona, T.G.; Pérez de Castro, I.; Ahearn, I.M.; Grana, T.M.; Chiu, V.K.; Lockyer, P.J.; Cullen, P.J.; Pellicer, A.; Cox, A.D.; Philips, M.R. Phospholipase Cγ Activates Ras on the Golgi Apparatus by Means of RasGRP1. Nature 2003, 424, 694–698. [Google Scholar] [CrossRef]
- Lorenzo, P.S.; Kung, J.W.; Bottorff, D.A.; Garfield, S.H.; Stone, J.C.; Blumberg, P.M. Phorbol Esters Modulate the Ras Exchange Factor RasGRP3. Cancer Res. 2001, 61, 943–949. [Google Scholar] [PubMed]
- Freshney, N.W.; Goonesekera, S.D.; Feig, L.A. Activation of the Exchange Factor Ras-GRF by Calcium Requires an Intact Dbl Homology Domain. FEBS Lett. 1997, 407, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, N.R.S. Immunoreceptor Tyrosine-Based Inhibitory Motifs on Activating Molecules. Crit. Rev. Immunol. 2000, 20, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Kesavapany, S.; Pareek, T.K.; Zheng, Y.-L.; Amin, N.; Gutkind, J.S.; Ma, W.; Kulkarni, A.B.; Grant, P.; Pant, H.C. Neuronal Nuclear Organization Is Controlled by Cyclin-Dependent Kinase 5 Phosphorylation of Ras Guanine Nucleotide Releasing Factor-1. Neurosignals 2006, 15, 157–173. [Google Scholar] [CrossRef]
- Bähler, M.; Rhoads, A. Calmodulin Signaling via the IQ Motif. FEBS Lett. 2002, 513, 107–113. [Google Scholar] [CrossRef]
- Krapivinsky, G.; Krapivinsky, L.; Manasian, Y.; Ivanov, A.; Tyzio, R.; Pellegrino, C.; Ben-Ari, Y.; Clapham, D.E.; Medina, I. The NMDA Receptor Is Coupled to the ERK Pathway by a Direct Interaction between NR2B and RasGRF1. Neuron 2003, 40, 775–784. [Google Scholar] [CrossRef]
- Feig, L.A. Regulation of Neuronal Function by Ras-GRF Exchange Factors. Genes Cancer 2011, 2, 306–319. [Google Scholar] [CrossRef]
- Wei, W.; Schreiber, S.S.; Baudry, M.; Tocco, G.; Broek, D. Localization of the Cellular Expression Pattern of Cdc25NEF and Ras in the Juvenile Rat Brain. Mol. Brain Res. 1993, 19, 339–344. [Google Scholar] [CrossRef]
- Yang, H.; Cooley, D.; Legakis, J.E.; Ge, Q.; Andrade, R.; Mattingly, R.R. Phosphorylation of the Ras-GRF1 Exchange Factor at Ser916/898 Reveals Activation of Ras Signaling in the Cerebral Cortex. J. Biol. Chem. 2003, 278, 13278–13285. [Google Scholar] [CrossRef]
- Jones, M.K.; Jackson, J.H. Ras-Grf Activates Ha-Ras, but Not n-Ras or k-Ras 4b, Protein in Vivo. J. Biol. Chem. 1998, 273, 1782–1787. [Google Scholar] [CrossRef]
- Mattingly, R.R.; Saini, V.; Macara, I.G. Activation of the Ras-GRF/CDC25Mm Exchange Factor by Lysophosphatidic Acid. Cell. Signal. 1999, 11, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Resat, H.; Straatsma, T.P.; Dixon, D.A.; Miller, J.H. The Arginine Finger of RasGAP Helps Gln-61 Align the Nucleophilic Water in GAP-Stimulated Hydrolysis of GTP. Proc. Natl. Acad. Sci. USA 2001, 98, 6033–6038. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Pots, H.; Gilsbach, B.K.; Parsons, D.; Veltman, D.M.; Ramachandra, S.G.; Li, H.; Kortholt, A.; Jin, T. C2GAP2 Is a Common Regulator of Ras Signaling for Chemotaxis, Phagocytosis, and Macropinocytosis. Front. Immunol. 2022, 13, 1075386. [Google Scholar] [CrossRef] [PubMed]
- Mangoura, D.; Sun, Y.; Li, C.; Singh, D.; Gutmann, D.H.; Flores, A.; Ahmed, M.; Vallianatos, G. Phosphorylation of Neurofibromin by PKC Is a Possible Molecular Switch in EGF Receptor Signaling in Neural Cells. Oncogene 2006, 25, 735–745. [Google Scholar] [CrossRef]
- Feng, L.; Yunoue, S.; Tokuo, H.; Ozawa, T.; Zhang, D.; Patrakitkomjorn, S.; Ichimura, T.; Saya, H.; Araki, N. PKA Phosphorylation and 14-3-3 Interaction Regulate the Function of Neurofibromatosis Type I Tumor Suppressor, Neurofibromin. FEBS Lett. 2004, 557, 275–282. [Google Scholar] [CrossRef]
- Skorski, T.; Kanakaraj, P.; Ku, D.-H.; Nieborowska-Skorska, M.; Canaani, E.; Zon, G.; Perussia, B.; Calabretta, B. Negative Regulation of P120GAP GTPase Promoting Activity by P210bcr/Abl: Implication for RAS-Dependent Philadelphia Chromosome Positive Cell Growth. J. Exp. Med. 1994, 179, 1855–1865. [Google Scholar] [CrossRef]
- Torti, M.; Marti, K.B.; Altschuler, D.; Yamamoto, K.; Lapetina, E.G. Erythropoietin Induces P21ras Activation and P120GAP Tyrosine Phosphorylation in Human Erythroleukemia Cells. J. Biol. Chem. 1992, 267, 8293–8298. [Google Scholar] [CrossRef]
- Chong, H.; Lee, J.; Guan, K.-L. Positive and Negative Regulation of Raf Kinase Activity and Function by Phosphorylation. EMBO J. 2001, 20, 3716–3727. [Google Scholar] [CrossRef]
- Wu, J.; Dent, P.; Jelinek, T.; Wolfman, A.; Weber, M.J.; Sturgill, T.W. Inhibition of the EGF-Activated MAP Kinase Signaling Pathway by Adenosine 3′, 5′-Monophosphate. Science 1993, 262, 1065–1069. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Pollock, C.; Steen, H.; Shaw, P.E.; Mischak, H.; Kolch, W. Cyclic AMP-Dependent Kinase Regulates Raf-1 Kinase Mainly by Phosphorylation of Serine 259. Mol. Cell. Biol. 2002, 22, 3237–3246. [Google Scholar] [CrossRef]
- Mischak, H.; Seitz, T.; Janosch, P.; Eulitz, M.; Steen, H.; Schellerer, M.; Philipp, A.; Kolch, W. Negative Regulation of Raf-1 by Phosphorylation of Serine 621. Mol. Cell. Biol. 1996, 16, 5409–5418. [Google Scholar] [CrossRef] [PubMed]
- Dutchak, K.; Garnett, S.; Nicoll, M.; de Bruyns, A.; Dankort, D. MOB3A Bypasses BRAF and RAS Oncogene-Induced Senescence by Engaging the Hippo Pathway. Mol. Cancer Res. 2022, 20, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Dangelmaier, C.; Manne, B.K.; Liverani, E.; Jin, J.; Bray, P.; Kunapuli, S.P. PDK1 Selectively Phosphorylates Thr (308) on Akt and Contributes to Human Platelet Functional Responses. Thromb. Haemost. 2014, 112, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-MTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Ugi, S.; Imamura, T.; Maegawa, H.; Egawa, K.; Yoshizaki, T.; Shi, K.; Obata, T.; Ebina, Y.; Kashiwagi, A.; Olefsky, J.M. Protein Phosphatase 2A Negatively Regulates Insulin’s Metabolic Signaling Pathway by Inhibiting Akt (Protein Kinase B) Activity in 3T3-L1 Adipocytes. Mol. Cell. Biol. 2004, 24, 8778–8789. [Google Scholar] [CrossRef]
- Brognard, J.; Sierecki, E.; Gao, T.; Newton, A.C. PHLPP and a Second Isoform, PHLPP2, Differentially Attenuate the Amplitude of Akt Signaling by Regulating Distinct Akt Isoforms. Mol. Cell 2007, 25, 917–931. [Google Scholar] [CrossRef]
- Catozzi, S.; Ternet, C.; Gourrege, A.; Wynne, K.; Oliviero, G.; Kiel, C. Reconstruction and Analysis of a Large-Scale Binary Ras-Effector Signaling Network. Cell Commun. Signal. 2022, 20, 24. [Google Scholar] [CrossRef]
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.-K. K-and N-Ras Are Geranylgeranylated in Cells Treated with Farnesyl Protein Transferase Inhibitors. J. Biol. Chem. 1997, 272, 14459–14464. [Google Scholar] [CrossRef]
- Nair, A.; Kubatzky, K.F.; Saha, B. Ras Isoforms from Lab Benches to Lives—What Are We Missing and How Far Are We? Int. J. Mol. Sci. 2021, 22, 6508. [Google Scholar] [CrossRef]
- Washington, C.; Chernet, R.; Gokhale, R.H.; Martino-Cortez, Y.; Liu, H.-Y.; Rosenberg, A.M.; Shahar, S.; Pfleger, C.M. A Conserved, N-Terminal Tyrosine Signal Directs Ras for Inhibition by Rabex-5. PLoS Genet. 2020, 16, e1008715. [Google Scholar] [CrossRef]
- Xu, L.; Lubkov, V.; Taylor, L.J.; Bar-Sagi, D. Feedback Regulation of Ras Signaling by Rabex-5-Mediated Ubiquitination. Curr. Biol. 2010, 20, 1372–1377. [Google Scholar] [CrossRef] [PubMed]
- Jura, N.; Scotto-Lavino, E.; Sobczyk, A.; Bar-Sagi, D. Differential Modification of Ras Proteins by Ubiquitination. Molecular Cell 2006, 21, 679–687. [Google Scholar] [CrossRef]
- Baker, R.; Wilkerson, E.M.; Sumita, K.; Isom, D.G.; Sasaki, A.T.; Dohlman, H.G.; Campbell, S.L. Differences in the Regulation of K-Ras and H-Ras Isoforms by Monoubiquitination. J. Biol. Chem. 2013, 288, 36856–36862. [Google Scholar] [CrossRef] [PubMed]
- Baietti, M.F.; Simicek, M.; Abbasi Asbagh, L.; Radaelli, E.; Lievens, S.; Crowther, J.; Steklov, M.; Aushev, V.N.; Martínez García, D.; Tavernier, J. OTUB 1 Triggers Lung Cancer Development by Inhibiting RAS Monoubiquitination. EMBO Mol. Med. 2016, 8, 288–303. [Google Scholar] [CrossRef]
- Sasaki, A.T.; Carracedo, A.; Locasale, J.W.; Anastasiou, D.; Takeuchi, K.; Kahoud, E.R.; Haviv, S.; Asara, J.M.; Pandolfi, P.P.; Cantley, L.C. Ubiquitination of K-Ras Enhances Activation and Facilitates Binding to Select Downstream Effectors. Sci. Signal. 2011, 4, ra13. [Google Scholar] [CrossRef]
- Bigenzahn, J.W.; Collu, G.M.; Kartnig, F.; Pieraks, M.; Vladimer, G.I.; Heinz, L.X.; Sedlyarov, V.; Schischlik, F.; Fauster, A.; Rebsamen, M. LZTR1 Is a Regulator of RAS Ubiquitination and Signaling. Science 2018, 362, 1171–1177. [Google Scholar] [CrossRef]
- Yang, M.H.; Nickerson, S.; Kim, E.T.; Liot, C.; Laurent, G.; Spang, R.; Philips, M.R.; Shan, Y.; Shaw, D.E.; Bar-Sagi, D. Regulation of RAS Oncogenicity by Acetylation. Proc. Natl. Acad. Sci. USA 2012, 109, 10843–10848. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Laurent, G.; Bause, A.S.; Spang, R.; German, N.; Haigis, M.C.; Haigis, K.M. HDAC6 and SIRT2 Regulate the Acetylation State and Oncogenic Activity of Mutant K-RASRegulation of K-RAS Acetylation. Mol. Cancer Res. 2013, 11, 1072–1077. [Google Scholar] [CrossRef]
- Santos, A.I.; Carreira, B.P.; Izquierdo-Alvarez, A.; Ramos, E.; Lourenço, A.S.; Filipa Santos, D.; Morte, M.I.; Ribeiro, L.F.; Marreiros, A.; Sánchez-López, N. S-Nitrosylation of Ras Mediates Nitric Oxide-Dependent Post-Injury Neurogenesis in a Seizure Model. Antioxid. Redox Signal. 2018, 28, 15–30. [Google Scholar] [CrossRef]
- Marshall, H.E.; Foster, M.W. S-Nitrosylation of Ras in Breast Cancer. Breast Cancer Res. 2012, 14, 1–2. [Google Scholar] [CrossRef]
- Ibiza, S.; Pérez-Rodríguez, A.; Ortega, Á.; Martínez-Ruiz, A.; Barreiro, O.; García-Domínguez, C.A.; Víctor, V.M.; Esplugues, J.V.; Rojas, J.M.; Sánchez-Madrid, F. Endothelial Nitric Oxide Synthase Regulates N-Ras Activation on the Golgi Complex of Antigen-Stimulated T Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 10507–10512. [Google Scholar] [CrossRef] [PubMed]
- Basu, T.; Warne, P.H.; Downward, J. Role of Shc in the Activation of Ras in Response to Epidermal Growth Factor and Nerve Growth Factor. Oncogene 1994, 9, 3483–3491. [Google Scholar] [PubMed]
- van der Geer, P.; Wiley, S.; Gish, G.D.; Pawson, T. The Shc Adaptor Protein Is Highly Phosphorylated at Conserved, Twin Tyrosine Residues (Y239/240) That Mediate Protein–Protein Interactions. Curr. Biol. 1996, 6, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Couvillon, A.D.; Brasher, B.B.; Van Etten, R.A. Tyrosine Phosphorylation of Grb2 by Bcr/Abl and Epidermal Growth Factor Receptor: A Novel Regulatory Mechanism for Tyrosine Kinase Signaling. EMBO J. 2001, 20, 6793–6804. [Google Scholar] [CrossRef]
- Bunda, S.; Heir, P.; Srikumar, T.; Cook, J.D.; Burrell, K.; Kano, Y.; Lee, J.E.; Zadeh, G.; Raught, B.; Ohh, M. Src Promotes GTPase Activity of Ras via Tyrosine 32 Phosphorylation. Proc. Natl. Acad. Sci. USA 2014, 111, E3785–E3794. [Google Scholar] [CrossRef]
- Kano, Y.; Gebregiworgis, T.; Marshall, C.B.; Radulovich, N.; Poon, B.P.; St-Germain, J.; Cook, J.D.; Valencia-Sama, I.; Grant, B.M.; Herrera, S.G. Tyrosyl Phosphorylation of KRAS Stalls GTPase Cycle via Alteration of Switch I and II Conformation. Nat. Commun. 2019, 10, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Fujita-Yamaguchi, Y.; Kathuria, S.; Xu, Q.-Y.; McDonald, J.M.; Nakano, H.; Kamata, T. In Vitro Tyrosine Phosphorylation Studies on RAS Proteins and Calmodulin Suggest That Polylysine-like Basic Peptides or Domains May Be Involved in Interactions between Insulin Receptor Kinase and Its Substrate. Proc. Natl. Acad. Sci. USA 1989, 86, 7306–7310. [Google Scholar] [CrossRef]
- Leventis, R.; Silvius, J.R. Lipid-Binding Characteristics of the Polybasic Carboxy-Terminal Sequence of K-Ras 4B. Biochemistry 1998, 37, 7640–7648. [Google Scholar] [CrossRef]
- Ballester, R.; Furth, M.E.; Rosen, O.M. Phorbol Ester-and Protein Kinase C-Mediated Phosphorylation of the Cellular Kirsten Ras Gene Product. J. Biol. Chem. 1987, 262, 2688–2695. [Google Scholar] [CrossRef]
- Bivona, T.G.; Quatela, S.E.; Bodemann, B.O.; Ahearn, I.M.; Soskis, M.J.; Mor, A.; Miura, J.; Wiener, H.H.; Wright, L.; Saba, S.G. PKC Regulates a Farnesyl-Electrostatic Switch on K-Ras That Promotes Its Association with Bcl-XL on Mitochondria and Induces Apoptosis. Molecular Cell 2006, 21, 481–493. [Google Scholar] [CrossRef]
- Ting, P.Y.; Johnson, C.W.; Fang, C.; Cao, X.; Graeber, T.G.; Mattos, C.; Colicelli, J. Tyrosine Phosphorylation of RAS by ABL Allosterically Enhances Effector Binding. FASEB J. 2015, 29, 3750–3761. [Google Scholar] [CrossRef]
- Yin, C.; Zhu, B.; Zhang, T.; Liu, T.; Chen, S.; Liu, Y.; Li, X.; Miao, X.; Li, S.; Mi, X. Pharmacological Targeting of STK19 Inhibits Oncogenic NRAS-Driven Melanomagenesis. Cell 2019, 176, 1113–1127.e16. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.-J.; Yoon, J.; Park, J.-C.; Lee, S.-H.; Lee, S.-H.; Kaduwal, S.; Kim, H.; Yoon, J.-B.; Choi, K.-Y. Ras Stabilization through Aberrant Activation of Wnt/β-Catenin Signaling Promotes Intestinal Tumorigenesis. Sci. Signal. 2012, 5, ra30. [Google Scholar] [CrossRef] [PubMed]
- Kazi, A.; Xiang, S.; Yang, H.; Delitto, D.; Trevino, J.; Jiang, R.H.; Ayaz, M.; Lawrence, H.R.; Kennedy, P.; Sebti, S.M. GSK3 Suppression Upregulates β-Catenin and c-Myc to Abrogate KRas-Dependent Tumors. Nat. Commun. 2018, 9, 5154–5162. [Google Scholar] [CrossRef] [PubMed]
- Bunda, S.; Burrell, K.; Heir, P.; Zeng, L.; Alamsahebpour, A.; Kano, Y.; Raught, B.; Zhang, Z.-Y.; Zadeh, G.; Ohh, M. Inhibition of SHP2-Mediated Dephosphorylation of Ras Suppresses Oncogenesis. Nat. Commun. 2015, 6, 8859–8870. [Google Scholar] [CrossRef] [PubMed]
- Ritt, D.A.; Daar, I.O.; Morrison, D.K. KSR Regulation of the Raf-MEK-ERK Cascade. Methods Enzymol. 2006, 407, 224–237. [Google Scholar] [PubMed]
- Müller, J.; Ory, S.; Copeland, T.; Piwnica-Worms, H.; Morrison, D.K. C-TAK1 Regulates Ras Signaling by Phosphorylating the MAPK Scaffold, KSR1. Molecular Cell 2001, 8, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Cacace, A.M.; Michaud, N.R.; Therrien, M.; Mathes, K.; Copeland, T.; Rubin, G.M.; Morrison, D.K. Identification of Constitutive and Ras-Inducible Phosphorylation Sites of KSR: Implications for 14-3-3 Binding, Mitogen-Activated Protein Kinase Binding, and KSR Overexpression. Mol. Cell. Biol. 1999, 19, 229–240. [Google Scholar] [CrossRef]
- Abraham, D.; Podar, K.; Pacher, M.; Kubicek, M.; Welzel, N.; Hemmings, B.A.; Dilworth, S.M.; Mischak, H.; Kolch, W.; Baccarini, M. Raf-1-Associated Protein Phosphatase 2A as a Positive Regulator of Kinase Activation. J. Biol. Chem. 2000, 275, 22300–22304. [Google Scholar] [CrossRef] [PubMed]
- Dubé, N.; Cheng, A.; Tremblay, M.L. The Role of Protein Tyrosine Phosphatase 1B in Ras Signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 1834–1839. [Google Scholar] [CrossRef]
- Yoshida, K.; Yamashita, Y.; Miyazato, A.; Ohya, K.; Kitanaka, A.; Ikeda, U.; Shimada, K.; Yamanaka, T.; Ozawa, K.; Mano, H. Mediation by the Protein-Tyrosine Kinase Tec of Signaling between the B Cell Antigen Receptor and Dok-1. J. Biol. Chem. 2000, 275, 24945–24952. [Google Scholar] [CrossRef]
- Kashige, N.; Carpino, N.; Kobayashi, R. Tyrosine Phosphorylation of P62dok by P210bcr-Abl Inhibits RasGAP Activity. Proc. Natl. Acad. Sci. USA 2000, 97, 2093–2098. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H. BI-3406, a Potent and Selective SOS1–KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK InhibitionPan-KRAS SOS1 Protein–Protein Interaction Inhibitor BI-3406. Cancer Discov. 2021, 11, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Hillig, R.C.; Sautier, B.; Schroeder, J.; Moosmayer, D.; Hilpmann, A.; Stegmann, C.M.; Werbeck, N.D.; Briem, H.; Boemer, U.; Weiske, J. Discovery of Potent SOS1 Inhibitors That Block RAS Activation via Disruption of the RAS–SOS1 Interaction. Proc. Natl. Acad. Sci. USA 2019, 116, 2551–2560. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.; Stang, S.L.; Zheng, Y.; Beswick, N.S.; Stone, J.C. Integration of DAG Signaling Systems Mediated by PKC-Dependent Phosphorylation of RasGRP3. Blood 2003, 102, 1414–1420. [Google Scholar] [CrossRef]
- Kedei, N.; Yang, D.; Blumberg, P. Bryostatin 1 Suppresses Induction by Protein Kinase C of the Ras Activator RasGRP3 in LNCaP Prostrate Cancer Cells. Cancer Res. 2007, 67, 3954. [Google Scholar]
- Stone, J.C. Regulation and Function of the RasGRP Family of Ras Activators in Blood Cells. Genes Cancer 2011, 2, 320–334. [Google Scholar] [CrossRef]
- Schmitt, J.M.; Guire, E.S.; Saneyoshi, T.; Soderling, T.R. Calmodulin-Dependent Kinase Kinase/Calmodulin Kinase I Activity Gates Extracellular-Regulated Kinase-Dependent Long-Term Potentiation. J. Neurosci. 2005, 25, 1281–1290. [Google Scholar] [CrossRef]
- Giglione, C.; Gonfloni, S.; Parmeggiani, A. Differential Actions of P60c-Src and Lck Kinases on the Ras Regulators P120-GAP and GDP/GTP Exchange Factor CDC25Mm. Eur. J. Biochem. 2001, 268, 3275–3283. [Google Scholar] [CrossRef]
- Kiyono, M.; Kato, J.; Kataoka, T.; Kaziro, Y.; Satoh, T. Stimulation of Ras Guanine Nucleotide Exchange Activity of Ras-GRF1/CDC25Mm upon Tyrosine Phosphorylation by the Cdc42-Regulated Kinase ACK1. J. Biol. Chem. 2000, 275, 29788–29793. [Google Scholar] [CrossRef]
- Marom, M.; Haklai, R.; Ben-Baruch, G.; Marciano, D.; Egozi, Y.; Kloog, Y. Selective Inhibition of Ras-Dependent Cell Growth by Farnesylthiosalisylic Acid (∗). J. Biol. Chem. 1995, 270, 22263–22270. [Google Scholar] [CrossRef] [PubMed]
- Elad, G.; Paz, A.; Haklai, R.; Marciano, D.; Cox, A.; Kloog, Y. Targeting of K-Ras 4B by S-Trans, Trans-Farnesyl Thiosalicylic Acid. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 1999, 1452, 228–242. [Google Scholar] [CrossRef]
- Weisz, B.; Giehl, K.; Gana-Weisz, M.; Egozi, Y.; Ben-Baruch, G.; Marciano, D.; Gierschik, P.; Kloog, Y. A New Functional Ras Antagonist Inhibits Human Pancreatic Tumor Growth in Nude Mice. Oncogene 1999, 18, 2579–2588. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, L.; Israeli, R.; Kloog, Y. FTS and 2-DG Induce Pancreatic Cancer Cell Death and Tumor Shrinkage in Mice. Cell Death Dis. 2012, 3, e284. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, L.; Ocherashvilli, A.; Daniels, D.; Last, D.; Cohen, Z.R.; Tamar, G.; Kloog, Y.; Mardor, Y. Salirasib (Farnesyl Thiosalicylic Acid) for Brain Tumor Treatment: A Convection-Enhanced Drug Delivery Study in Rats. Mol. Cancer Ther. 2008, 7, 3609–3616. [Google Scholar] [CrossRef]
- Charette, N.; De Saeger, C.; Lannoy, V.; Horsmans, Y.; Leclercq, I.; Stärkel, P. Salirasib Inhibits the Growth of Hepatocarcinoma Cell Lines in Vitro and Tumor Growth in Vivo through Ras and MTOR Inhibition. Molecular Cancer 2010, 9, 256–269. [Google Scholar] [CrossRef]
- Riely, G.J.; Johnson, M.L.; Medina, C.; Rizvi, N.A.; Miller, V.A.; Kris, M.G.; Pietanza, M.C.; Azzoli, C.G.; Krug, L.M.; Pao, W. A Phase II Trial of Salirasib in Patients with Lung Adenocarcinomas with KRAS Mutations. J. Thorac. Oncol. 2011, 6, 1435–1437. [Google Scholar] [CrossRef]
- Badar, T.; Cortes, J.E.; Ravandi, F.; O’Brien, S.; Verstovsek, S.; Garcia-Manero, G.; Kantarjian, H.; Borthakur, G. Phase I Study of S-Trans, Trans-Farnesylthiosalicylic Acid (Salirasib), a Novel Oral RAS Inhibitor in Patients with Refractory Hematologic Malignancies. Clin. Lymphoma Myeloma Leuk. 2015, 15, 433–438.e2. [Google Scholar] [CrossRef]
- Laheru, D.; Rudek, M.; Taylor, G.; Goldsweig, H.; Rajeshkumar, N.V.; Linden, S.; Angenendt, M.; Le, D.; Donehower, R.; Jimeno, A. Integrated Development of S-Trans, Trans-Farnesylthiosalicyclic Acid (FTS, Salirasib) in Advanced Pancreatic Cancer. J. Clin. Oncol. 2009, 27, 4529. [Google Scholar] [CrossRef]
- Tsai, F.D.; Lopes, M.S.; Zhou, M.; Court, H.; Ponce, O.; Fiordalisi, J.J.; Gierut, J.J.; Cox, A.D.; Haigis, K.M.; Philips, M.R. K-Ras4A Splice Variant Is Widely Expressed in Cancer and Uses a Hybrid Membrane-Targeting Motif. Proc. Natl. Acad. Sci. USA 2015, 112, 779–784. [Google Scholar] [CrossRef]
- Cuiffo, B.; Ren, R. Palmitoylation of Oncogenic NRAS Is Essential for Leukemogenesis. Blood J. Am. Soc. Hematol. 2010, 115, 3598–3605. [Google Scholar] [CrossRef]
- Ducker, C.E.; Stettler, E.M.; French, K.J.; Upson, J.J.; Smith, C.D. Huntingtin Interacting Protein 14 Is an Oncogenic Human Protein: Palmitoyl Acyltransferase. Oncogene 2004, 23, 9230–9237. [Google Scholar] [CrossRef] [PubMed]
- Dekker, F.J.; Rocks, O.; Vartak, N.; Menninger, S.; Hedberg, C.; Balamurugan, R.; Wetzel, S.; Renner, S.; Gerauer, M.; Schölermann, B. Small-Molecule Inhibition of APT1 Affects Ras Localization and Signaling. Nat. Chem. Biol. 2010, 6, 449–456. [Google Scholar] [CrossRef]
- Hedberg, C.; Dekker, F.J.; Rusch, M.; Renner, S.; Wetzel, S.; Vartak, N.; Gerding-Reimers, C.; Bon, R.S.; Bastiaens, P.I.; Waldmann, H. Development of Highly Potent Inhibitors of the Ras-targeting Human Acyl Protein Thioesterases Based on Substrate Similarity Design. Angew. Chem. Int. Ed. 2011, 50, 9832–9837. [Google Scholar] [CrossRef]
- Zimmermann, T.J.; Bürger, M.; Tashiro, E.; Kondoh, Y.; Martinez, N.E.; Görmer, K.; Rosin-Steiner, S.; Shimizu, T.; Ozaki, S.; Mikoshiba, K. Boron-Based Inhibitors of Acyl Protein Thioesterases 1 and 2. Chembiochem 2013, 14, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Bachovchin, D.A.; Brown, S.J.; Rosen, H.; Cravatt, B.F. Identification of Selective Inhibitors of Uncharacterized Enzymes by High-Throughput Screening with Fluorescent Activity-Based Probes. Nat. Biotechnol. 2009, 27, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Frans, L.; Opdam, E.V. Combination Therapy of RMC-4630 and LY3214996 in Metastatic KRAS Mutant Cancers (SHERPA); National Library of Medicine (U.S.): Bethesda, MD, USA, 2022.
- Lauren Wood, S.W. SHP2 Inhibitor BBP-398 in Combination with Nivolumab in Patients with Advanced Non-Small Cell Lung Cancer With a KRAS Mutation; National Library of Medicine (U.S.): Bethesda, MD, USA, 2022.
- Susanna Wen, L.W. SHP2 Inhibitor BBP-398 in Combination with Sotorasib in Patients with Advanced Solid Tumors and a KRAS-G12C Mutation (Argonaut); National Library of Medicine (U.S.): Bethesda, MD, USA, 2022.
- Novartis Pharmaceuticals. Dose Finding Study of TNO155 in Adult Patients with Advanced Solid Tumors; National Library of Medicine (U.S.): Bethesda, MD, USA, 2022.
- Susanna Wen, L.W. First-in-Human Study of the SHP2 Inhibitor BBP-398 in Patients with Advanced Solid Tumors; National Library of Medicine (U.S.): Bethesda, MD, USA, 2022.
- Qian, L.; Chen, K.; Wang, C.; Chen, Z.; Meng, Z.; Wang, P. Targeting NRAS-Mutant Cancers with the Selective STK19 Kinase Inhibitor ChelidonineChelidonine for NRAS-Mutant Cancer Treatment. Clin. Cancer Res. 2020, 26, 3408–3419. [Google Scholar] [CrossRef]
- Rodríguez-Martínez, M.; Boissiére, T.; Gonzalez, M.N.; Litchfield, K.; Mitter, R.; Walker, J.; Kjœr, S.; Ismail, M.; Downward, J.; Swanton, C. Evidence That STK19 Is Not an NRAS-Dependent Melanoma Driver. Cell 2020, 181, 1395–1405.e11. [Google Scholar] [CrossRef]
- Dan, M.; Schmitt, A.P.M. 9-ING-41 in Patients with Advanced Cancers; National Library of Medicine (U.S.): Bethesda, MD, USA, 2022.
- Zimmermann, G.; Papke, B.; Ismail, S.; Vartak, N.; Chandra, A.; Hoffmann, M.; Hahn, S.A.; Triola, G.; Wittinghofer, A.; Bastiaens, P.I. Small Molecule Inhibition of the KRAS–PDEδ Interaction Impairs Oncogenic KRAS Signalling. Nature 2013, 497, 638–642. [Google Scholar] [CrossRef]
- Chakraborty, S.; Srivastava, A.; Jha, M.K.; Nair, A.; Pandey, S.P.; Srivastava, N.; Kumari, S.; Singh, S.; Krishnasastry, M.V.; Saha, B. Inhibition of CD40-Induced N-Ras Activation Reduces Leishmania Major Infection. J. Immunol. 2015, 194, 3852–3860. [Google Scholar] [CrossRef]
- Nair, A.; Chakraborty, S.; Banerji, L.A.; Srivastava, A.; Navare, C.; Saha, B. Ras Isoforms: Signaling Specificities in CD40 Pathway. Cell Commun. Signal. 2020, 18, 3–14. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Condition or Disease | Intervention/Treatment | Clinical Trial Status |
|---|---|---|---|
| 1 | Solid Tumors, KRAS Mutation; SOS1 | Drug: BI 1701963 Drug: Trametinib | Phase 1 |
| 2 | Non-Small-Cell Lung Carcinoma (NSCLC) | Drug: Salirasib | Phase 2 |
| 3 | Pancreatic Cancer Unresectable Pancreatic Cancer Metastatic Pancreatic Cancer KRAS P.G12C | Drug: Sotorasib Drug: Liposomal Irinotecan (nal-IRI) Drug: 5 Fluorouracil (5FU) Drug: Leucovorin (LV) Drug: Gemcitabine (GEM) Drug: Nab paclitaxel | Phase 1 Phase 2 |
| 4 | Pancreatic Cancer, Colorectal Cancer Non-Small Cell Lung Cancer KRAS Mutation-Related Tumors | Drug: RMC-4630 Drug: LY3214996 | Phase 1 |
| Advanced EGFR mutant NSCLC, KRAS G12-mutant NSCLC, Esophageal Squamous Cell Cancer (SCC), Head/Neck SCC, Melanoma | Drug: TNO155 Drug: TNO155 in combination with EGF816 (nazartinib) | Phase 1 | |
| 5 | Tumor, Solid | Drug: BBP-398 (formerly known as IACS-15509) | Phase 1 |
| 6 | Solid Tumor, Adult Metastatic Solid Tumor Metastatic NSCLC Non-Small Cell Lung Cancer | Drug: BBP-398 Drug: sotorasib | Phase 1 |
| 7 | Non-Small Cell Lung Cancer Solid Tumor | Drug: BBP-398 with nivolumab | Phase 1 |
| 8 | Cancer Pancreatic Cancer Sarcoma Renal Cancer Refractory Cancer Refractory Neoplasm Refractory Non-Hodgkin Lymphoma Pancreatic Adenocarcinoma Resistant Cancer Neoplasm Metastasis Neoplasm of Bone Neoplasm, Breast Neoplasm of Lung Neoplasms, Colorectal Neoplasms, Pancreatic Malignant Glioma Malignancies Malignancies, Multiple Bone Metastases Bone Neoplasm Bone Cancer Pancreatic Cancer Pancreatic Neoplasms Breast Neoplasms Acute T Cell Leukemia Lymphoma | Drug: 9-ING-41(GSK 3 β inhibitor) Drug: Gemcitabine—21-day cycle Drug: Doxorubicin Drug: Lomustine Drug: Carboplatin Drug: Nab paclitaxel Drug: Paclitaxel Drug: Gemcitabine—28-day cycle Drug: Irinotecan | Phase 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nair, A.; Saha, B. Regulation of Ras-GTPase Signaling and Localization by Post-Translational Modifications. Kinases Phosphatases 2023, 1, 97-116. https://doi.org/10.3390/kinasesphosphatases1020007
Nair A, Saha B. Regulation of Ras-GTPase Signaling and Localization by Post-Translational Modifications. Kinases and Phosphatases. 2023; 1(2):97-116. https://doi.org/10.3390/kinasesphosphatases1020007
Chicago/Turabian StyleNair, Arathi, and Bhaskar Saha. 2023. "Regulation of Ras-GTPase Signaling and Localization by Post-Translational Modifications" Kinases and Phosphatases 1, no. 2: 97-116. https://doi.org/10.3390/kinasesphosphatases1020007