

Recent Advancements in Computational Drug Design Algorithms through Machine Learning and Optimization
 , and
, and
Abstract
:1. Introduction
2. Biological and Computational Terms
- Ligands: Molecules or ions that are coordinated with the central atom or ion in the coordination compound are called ligands.
- Molecular descriptors: Molecular descriptors are numerical representations of molecule attributes. Physical and chemical properties of the molecule are numerically represented by molecular descriptors [4].
- Molecular docking: Docking is a method of molecular modeling that predicts the preferred orientation of a ligand when it is bound in an active site of a molecule to form a stable complex [5].
- Molecular dynamics: Molecular dynamics (MD) is a computer simulation method for analyzing the physical movement of atoms and molecules. The atoms and molecules are allowed to interact for a fixed period of time, giving a dynamic view of the system. MD simulation is based on Newton’s second law or the equation of motion.
3. Importance of Computational Drug Discovery
4. Process of Drug Discovery
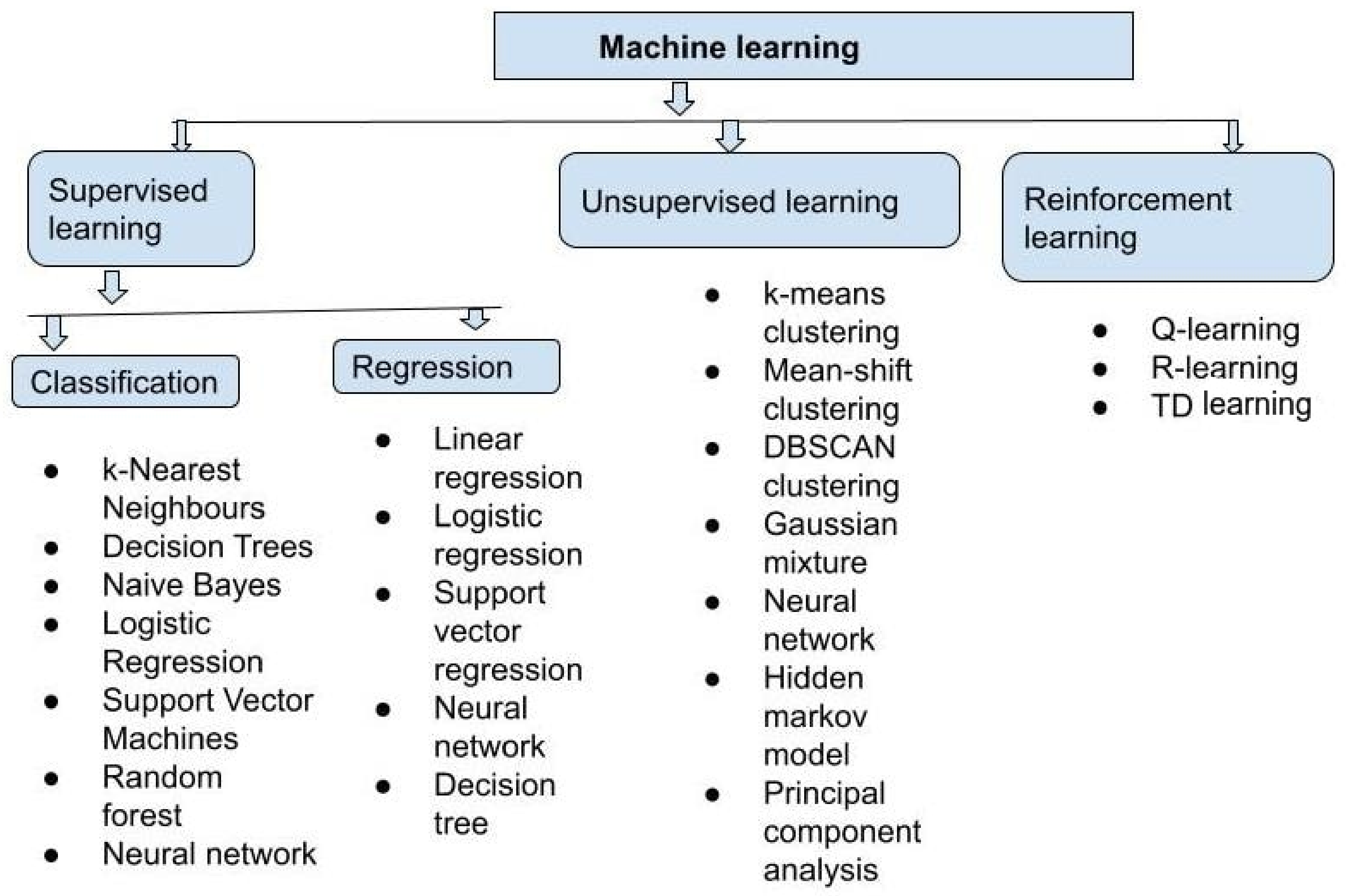
5. Machine Learning and Deep Learning Techniques Used for Drug Discovery
- We can divide supervised learning into two categories: (1) classification and (2) regression. A classification algorithm is used to classify test data and allocate it to certain groups. It recognizes certain entities in the dataset and makes educated guesses about how those entities should be labeled or defined. Linear classifiers, support vector machines (SVMs), decision trees, k-nearest neighbor, and random forest are some of the most common classification algorithms. To explore the relationship between dependent and independent variables, regression is used. It is widely used to produce predictions, such as for a company’s sales revenue. Popular regression algorithms include linear regression, logistical regression, and polynomial regression.
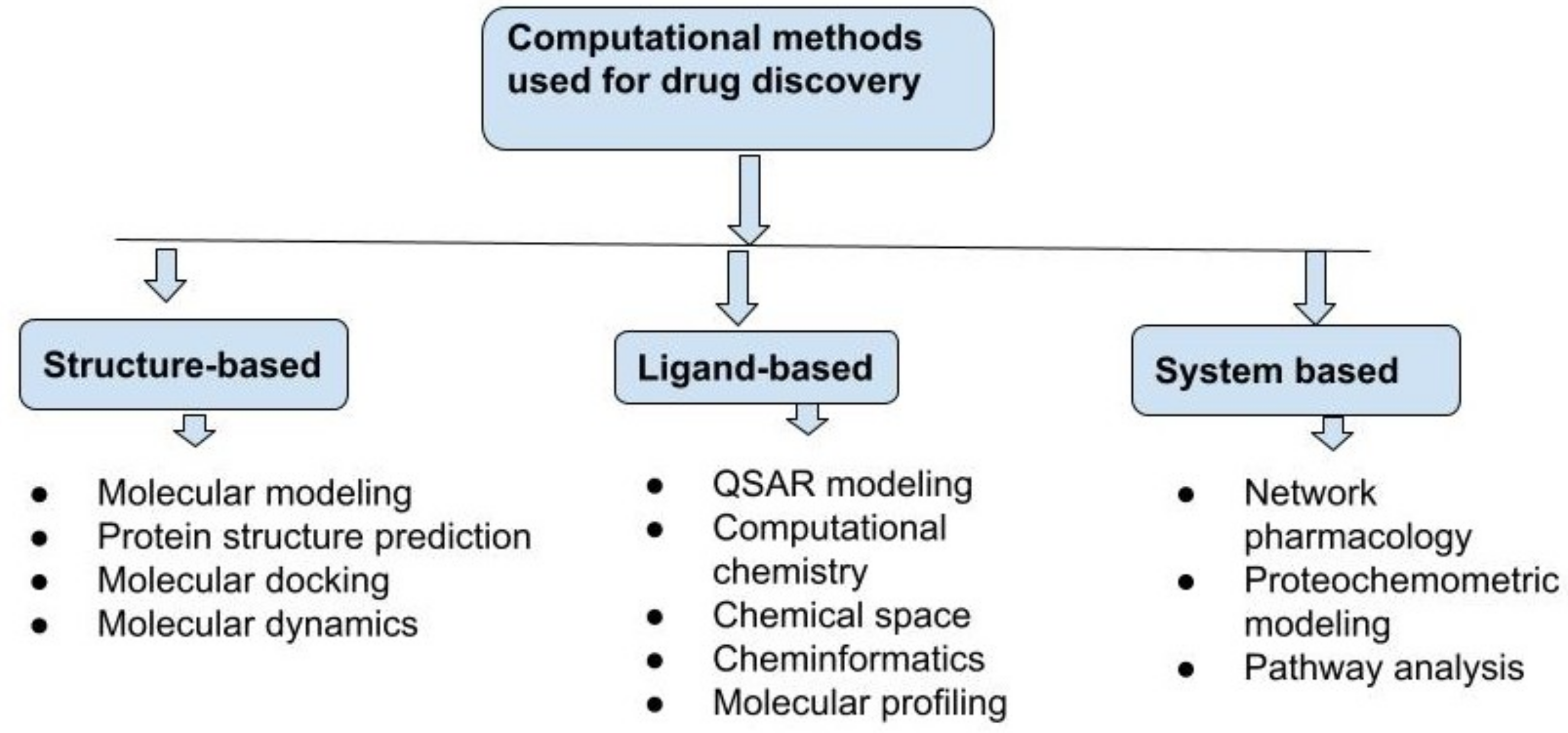
6. Different Approaches for Computational Drug Discovery
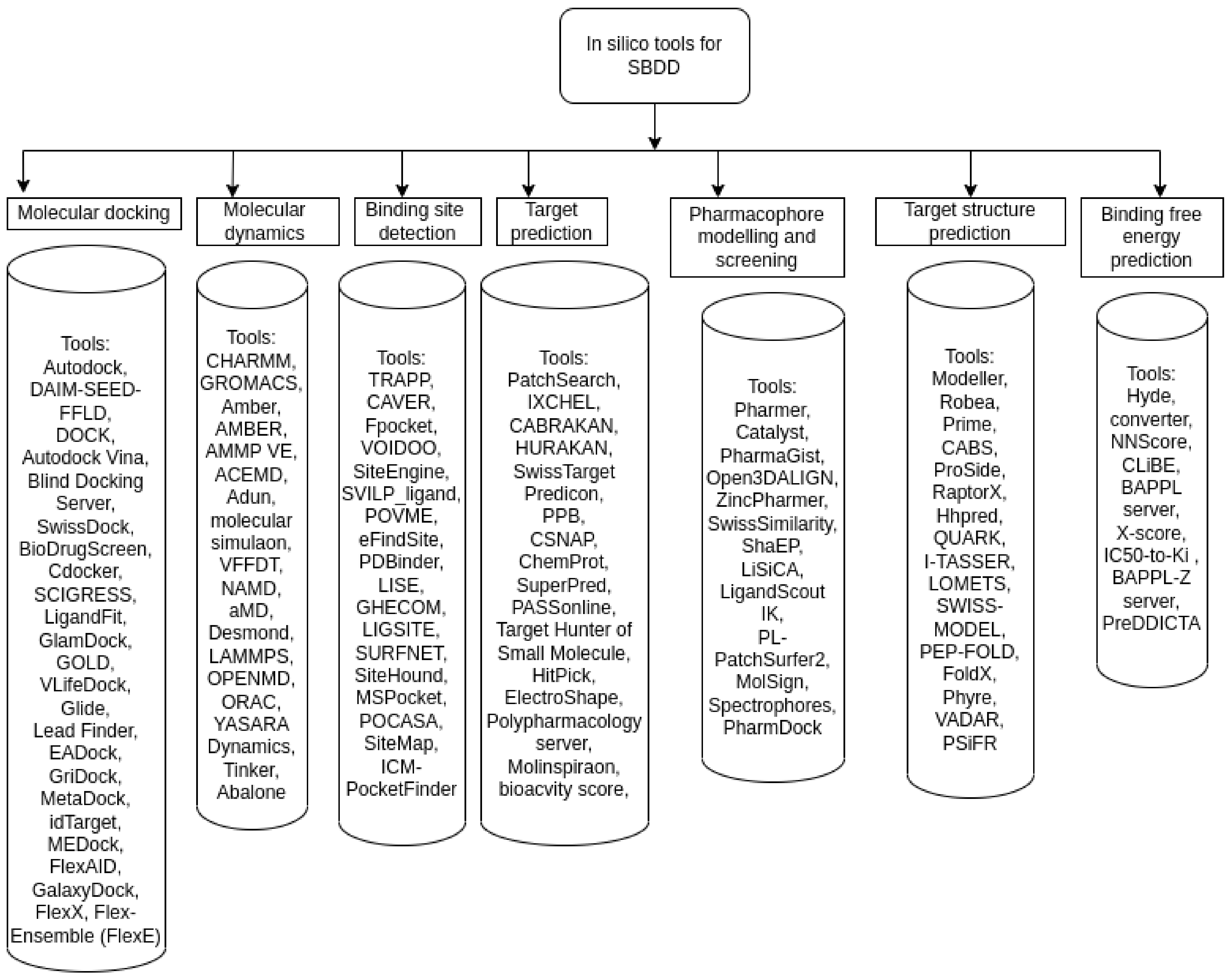
6.1. Structure-Based Drug Discovery
6.2. Ligand-Based Drug Discovery
6.2.1. QSAR of Ligand-Based Drug Discovery
- (1)
- Curated chemical dataset.
- (2)
- Creation of molecular descriptor.
- (3)
- Split dataset into training and testing datasets. Build QSAR model.
- (4)
- Validation of QSAR model and virtual design of ligand.
- (5)
- Predict the best ligand and test the QSAR model’s accuracy.
- (6)
6.2.2. Pharmacophore Modeling
- (1)
- Selection of a set of active ligands: A set of active ligands known to bind to the target of interest is selected. These ligands may come from experimental data or from virtual screening studies.
- (2)
- Structural alignment of ligands: The ligands in the set are structurally aligned based on common features such as functional groups or rings.
- (3)
- Identification of pharmacophoric features: The aligned ligands are analyzed to identify common pharmacophoric features, usually functional groups or other chemical properties important for ligand binding to the target. Examples of pharmacophoric features include hydrogen bond acceptors, hydrogen bond donors, aromatic rings, and hydrophobic regions.
- (4)
- Generation of a pharmacophore model: The pharmacophoric features identified in step 3 are used to generate a pharmacophore model, which is a three-dimensional representation of the common features required for ligand binding to the target. The model may be visualized using software programs that allow for manipulation and refinement of the model.
- (5)
- Validation of the pharmacophore model: The pharmacophore model is validated using techniques such as molecular docking or virtual screening to test whether the model can accurately predict the binding affinity of new ligands to the target.
6.3. System-Based Drug Discovery
7. Drug Molecule Design
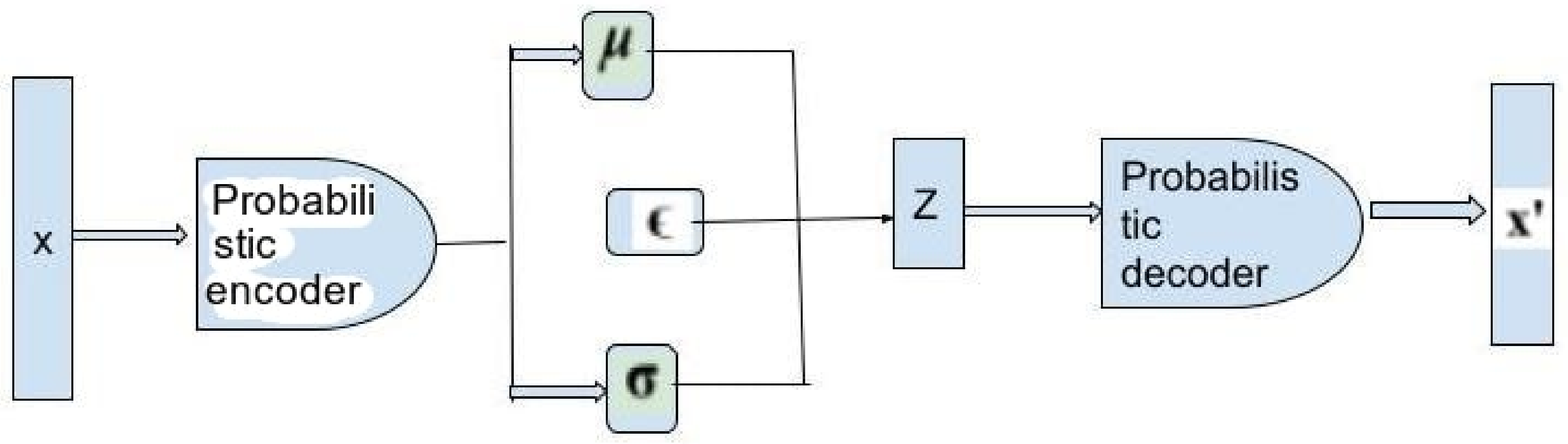
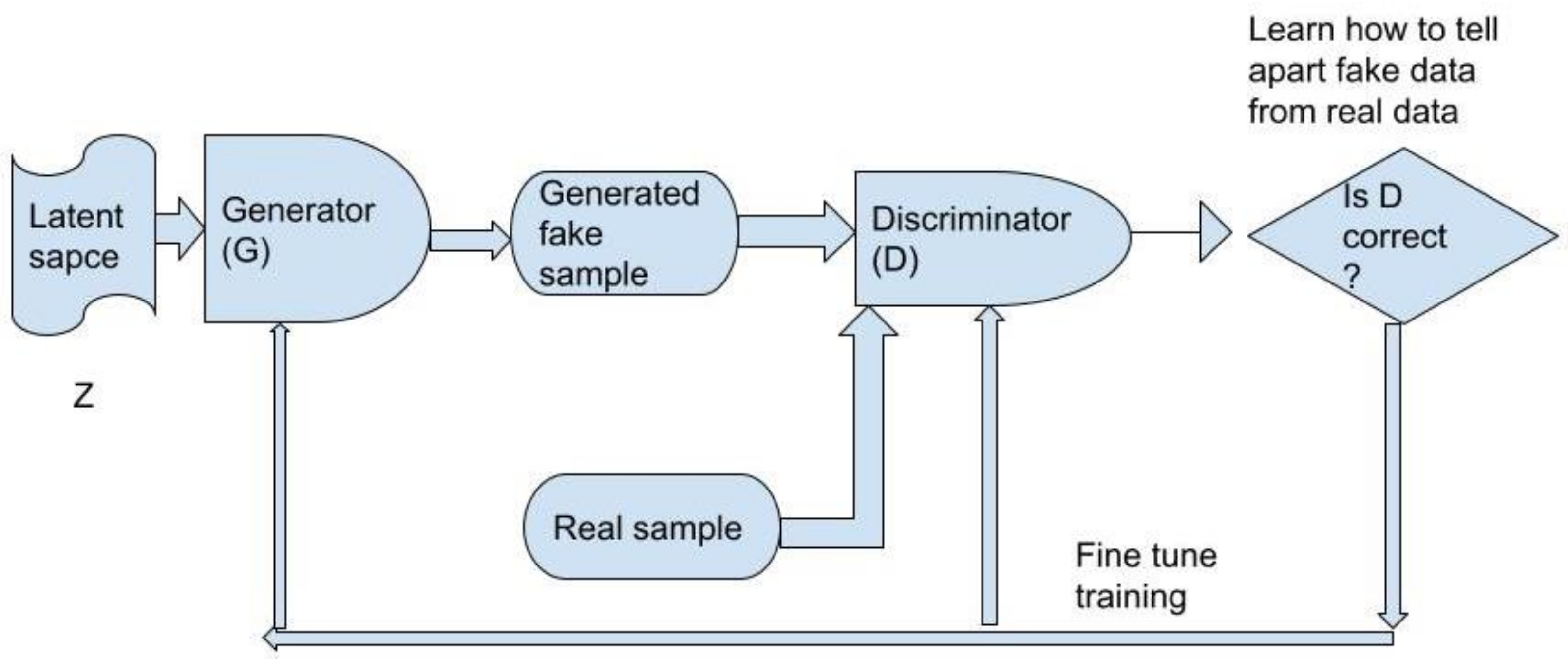
Methods for Deep Generative Model
8. Design of Kinase Inhibitors Using CADD
9. Evaluation Methods for Different Machine Learning and Deep Learning Generative Techniques for Drug Design
9.1. Simple Numeric Methods
- Validity [101] is the ratio of valid molecules to the total number of molecules in the generated molecules dataset. A valid molecule is one where all atoms’ corresponding bonds match their valency and validity estimates the model’s ability to learn the valency of atoms.
- Novelty [101] is the ratio of molecules that do not appear in the training set to the total number of molecules in the generated molecules dataset. It estimates the ability of the model to tap into the unknown chemical space.
- Uniqueness [101] is the ratio of unique molecules to the total number of molecules in the generated molecules dataset. It estimates the generative repetitiveness of a model and a high unique score is ideal.
- Diversity [102] is classified into two categories: internal diversity (IntDiv) and external diversity (ExtDiv). IntDiv is the measure of similarity between molecules in the generated molecules dataset. ExtDiv is the measure of similarity between molecules in the generated molecules dataset and the training dataset. It uses the power (p) mean of the pairwise Tanimoto similarity (S) between the generated (G) dataset and the training (T) dataset.
9.2. Probabilistic Distribution Methods
- Kullback–Leibler Divergence (KL-Divergence) [103] is a measure of the statistical distance between two probability distributions of various physicochemical descriptors from the training and generated molecules datasets. A low KLD for any descriptor implies the model has successfully learned its distribution. The formula for KLD for a descriptor (D) between the generated (G) and training (T) distribution is shown:
- Frechet ChemNet Distance (FCD) [104] uses the means () and covariances (C) of the features of the training (T) and generated (G) datasets from the penultimate layer of ChemNet. Lower values are better as they imply the distributions are closer.
9.3. Optimization Evaluation Methods
- 1.
- Synthetic accessibility score (SAS) [105] is a value used to estimate the ease of synthesis of a molecule. A low score implies ease in the synthesis of the drug-like molecule. Its range is from 0 to 10.
- 2.
- Quantitative Estimate of Drug-likeness (QED) [106] is used to calculate the drug-likeness of a molecule using descriptors from various drugs in the market, and is calculated by taking the geometric mean of all the desirable functions, each corresponding to different descriptors. Its range is from 0 to 1.
- 3.
- Octanol–water partition coefficient (LogP) [107] is used to calculate how hydrophobic/hydrophilic a molecule is. Its range is on average from −3 to 7.
- 4.
- Topological polar surface area (TPSA) [108] calculates the molecular polar surface area of the polar atoms, which provides insight into the transport properties of drugs.
- 5.
- GuacaMol [103] is a benchmarking suite for drug-like molecules that uses 5 distribution-learning benchmarks (novelty, validity, uniqueness, KLD, and FCD) and 20 goal-directed benchmarks (e.g., Scaffold Hop, Valsartan SMARTS, Celecoxib rediscovery, Albuterol similarity, Median molecules, Osimertinib MPO).
- 6.
- Vina [109] is a scoring function that measures the protein–ligand binding affinity by summing the important energy factors in protein–ligand binding.
- 7.
- Celecoxib rediscovery [110] is a rediscovery method that attempts to rediscover the target molecule when removed from the training dataset. Its range lies from 0 to 1.
9.4. 3D Similarity Methods
- Root-mean-squared deviation [111] calculates the 3D alignment similarity between two molecule conformations from training set and generated molecule . is found by rotating and translating the original conformation to obtain RMSD(R,R’).
- SHApeFeaTure Similarity (SHAFTS) [112] uses a hybrid similarity method using molecular shape and chemical groups appended by pharmacophore features for 3D similarity calculation. The hybrid similarity has two parts: shape-density overlap (ShapeScore) is the intersection between two molecules A and B, which is the sum of the overlap integrals of single atomic shape-densities for which a Gaussian function was used. is the interatomic distance.
- FeatureScore is the sum of overlap between the feature points in A and B of the same type. is the distance between the features of A and B and is the overlap tolerance.
- Finally, the hybrid score is defined as a weighted sum of the ShapeScore and FeatureScore scaled to [0, 2].
- Rapid overlay of chemical structures (ROCS) [113] uses unweighted sums to aggregate many features of similarity, resulting in parameter-free models. It measures the chemical and shape similarity of two molecules by calculating the Tanimoto coefficients of the aligned overlap volumes:
10. Drug Development Database
11. Discussion
- Expand the chemical space that is medically relevant.
- Design and screen extremely large chemical libraries rationally.
- Extract lead compounds and unknown hits from screening libraries.
- Improve multi-target drug design.
- Identify responsible region in genome.
- Improve targeting protein–protein interaction module.
- Try to reduce off-target binding during clinical trials.
- For multi-target drug design, reduce toxicity.
- Compound and library enumeration.
- Improve medically relevant 3D drug molecule design.
- In molecule generation methods using deep learning we face many challenges, such as out-of-distribution generation, lack of interoperability, lack of unified evaluation protocol, generation in low-data regime, etc.
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of Open Access Journals |
| TLA | Three-letter acronym |
| CADD | Computer-aided drug design |
| SBDD | Structure-based drug design |
| LBDD | Ligand-based drug design |
| MD | Molecular dynamics |
| ADMET | Adsorption, distribution, metabolism, excretion and toxicity |
References
- Parvu, L. QSAR—A piece of drug design. J. Cell Mol. Med. 2003, 7, 333–335. [Google Scholar] [CrossRef] [PubMed]
- Duong-Ly, K.C.; Peterson, J.R. The human kinome and kinase inhibition. Curr. Protoc. Pharmacol. 2013, 60, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, B.; Abed, S.N.; Al-Attraqchi, O.; Kuche, K.; Tekade, R.K. Chapter 21—Computer-Aided Prediction of Pharmacokinetic (ADMET) Properties. In Dosage Form Design Parameters; Academic Press: Cambridge, MA, USA, 2018; pp. 731–755. [Google Scholar]
- Lengauer, T.; Rarey, M. Computational methods for biomolecular docking. Curr. Opin. Struct. Biol. 1996, 6, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Fefpia, M.; Marshall, S.; Burghaus, R.; Cosson, V.; Cheung, S.; Chenel, M.; Dellapasqua, O.; Frey, N.; Hamrén, B.; Harnisch L., T. Good Practices in Model-Informed Drug Discovery and Development Practice Application and Documentation. CPT Pharm. Syst. Pharmacol. 2016, 5, 93–122. [Google Scholar]
- Ghosh, B.; Choudhuri, S.T. Drug Design for Malaria with Artificial Intelligence (AI). In Plasmodium Species and Drug Resistance; IntechOpen: London, UK, 2021. [Google Scholar]
- Mullard Asher, T. Biotech R&D spend jumps by more than 15%. Nat. Rev. Drug Discov. 2016, 15, 447–448. [Google Scholar]
- Choudhuri, S.; Mallik, S.; Ghosh, B.; Si, T.; Bhadra, T.; Maulik, U.; Li, A. A Review of Computational Learning and IoT Applications to High-Throughput Array-Based Sequencing and Medical Imaging Data in Drug Discovery and Other Health Care Systems. In Applied Smart Health Care Informatics: A Computational Intelligence Perspective; John Wiley & Sons: New York, NY, USA, 2022. [Google Scholar]
- Barabási, A.L.; Gulbahce, N.; Loscalzo, J. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Laurie, A.T.; Jackson, R.M. Q-sitefinder An energy-based method for the prediction of protein-ligand binding sites. Bioinformatics 2005, 21, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- Ewing, T.J.; Makino, S.; Skillman, A.G.; Kuntz, I.D. search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001, 15, 411–428. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Olson, A.J. Automated docking of substrates to proteins by simulated annealing. Proteins 1990, 8, 195–202. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Cournia, Z.; Chipot, C.; Roux, B.; York, D.M.; Sherman, W. Free Energy Methods in Drug Discovery—Introduction. ACS Symp. Ser. 2021, 1397, 1–38. [Google Scholar]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/ GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2010, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Hansson, T.; Marelius, J.; Åqvist, J. Ligand binding affinity prediction by linear interaction energy methods. J. Comput. Aided Mol. Des. 1998, 12, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Kandel, J.; Tayara, H.; Chong, K.T. PUResNet prediction of protein-ligand binding sites using deep residual neural network. J. Cheminform. 2021, 13, 65. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Mam, B.; Sowdhamini, R. A Deep Learning Approach to Predict Protein-Ligand Binding Affinity. Bioinform. Biol. Insights 2021, 15, 11779322211030364. [Google Scholar] [CrossRef]
- Eramian, M.Y.S.; Pieper, U.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 2006, 5, 6. [Google Scholar]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL an automated protein homology-modeling server. Nucleic Acids Res 2003, 31, 3381–3385. [Google Scholar] [CrossRef] [PubMed]
- Bernard, D.; Coop, A.; MacKerell, A.D., Jr. Conformationally sampled pharmacophore for peptidic delta. J. Med. Chem. 2005, 48, 73–80. [Google Scholar] [CrossRef]
- Duchowicz, P.R.; Castro, E.A.; Fernández, F.M.; Gonzalez, M.P. A new search algorithm of QSPR/QSAR theories Normal boiling points of some organic molecules. Chem. Phys. Lett. 2005, 412, 376–380. [Google Scholar] [CrossRef]
- Wade, R.C.; Henrich, S.; Wang, T.T. Using 3D protein structures to derive 3D-QSARs. Drug Discov. Today Technol. 2004, 1, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Acharya, C.; Coop, A.; Polli, J.E.; Mackerell, A.D., Jr. Recent advances in ligand-based drug design relevance and utility of the conformationally sampled pharmacophore approach. Curr. Comput. Aided Drug Des. 2011, 7, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Bohl, C.E.; Chang, C.; Mohler, M.L.; Chen, J.; Miller, D.D.; Swaan, P.W.; Dalton, J.T. A ligand-based approach to identify quantitative structure-activity relationships for the androgen receptor. J. Med. Chem. 2004, 47, 3765–3776. [Google Scholar] [CrossRef]
- Winkler David Alan, T. Overview of Quantitative Structure—Activity Relationships (QSAR). In Molecular Analysis and Genome Discovery; John Wiley & Sons: New York, NY, USA, 2004; pp. 347–367. [Google Scholar]
- Topliss, J.G.; Edwards, R.P.T. Chance factors in studies of quantitative structure-activity relationships. J. Med. Chem 1979, 22, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, D.M.; Basak, S.C.; Shi, X. QSAR with few compounds and many features. J. Chem. Inform. Comput. Sci. 2001, 41, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.P.; Modi, S.; Bender, A.; Robinson, R.L.M.; Kirchmair, J.; Promkat, T. The challenges involved in modeling toxicity data in silico a review. Curr. Pharm. Des. 2012, 18, 1266–1291. [Google Scholar] [CrossRef] [PubMed]
- Pujol, A.; Mosca, R.; Farrés, J.; Aloy, P. Unveiling the role of network and systems biology in drug discovery. Trends Pharmacol. Sci. 2010, 31, 115–123. [Google Scholar] [CrossRef]
- Butcher, E.C.; Berg, E.L.; Kunkel, E.J. Systems biology in drug discovery. Nat. Biotechnol. 2004, 22, 1253–1259. [Google Scholar] [CrossRef]
- Barabási, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine a network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Ryu, S.; Kim, J.W.; Kim, W.Y. Molecular generative model based on conditional variational autoencoder for de novo molecular design. J. Cheminform. 2018, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Cho, K. Conditional molecular design with deep generative models. J. Chem. Inf. Model. 2018, 59, 43–52. [Google Scholar] [CrossRef]
- Harel, S.; Radinsky, K. Prototype-based compound discovery using deep generative models. Mol. Pharm. 2018, 15, 4406–4416. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Blaschke, T.; Olivecrona, M.; Engkvist, O.; Bajorath, J.; Chen, H. Application of generative autoencoder in de novo molecular design. Mol. Inform. 2017, 37, 1700123. [Google Scholar] [CrossRef] [PubMed]
- Sattarov, B.; Baskin, I.I.; Horvath, D.; Marcou, G.; Bjerrum, E.J.; Varnek, A. De novo molecular design by combining deep autoencoder recurrent neural networks with generative topographic mapping. J. Chem. Inf. Model. 2019, 59, 1182–1196. [Google Scholar] [CrossRef]
- Kusner, M.J.; Paige, B.; Hernández-Lobato, J.M. Grammar Variational Autoencoder. arXiv 2017, arXiv:1703.01925. [Google Scholar]
- Jørgensen, P.B.; Mesta, M.; Shil, S.; García Lastra, J.M.; Jacobsen, K.W.; Thygesen, K.S.; Schmidt, M.N. Machine learning-based screening of complex molecules for polymer solar cells. J. Chem. Phys. 2018, 148, 241735. [Google Scholar] [CrossRef]
- Jørgensen, P.B.; Schmidt, M.N.; Winther, O. Deep generative models for molecular science. Mol. Inform. 2018, 37, 1700133. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Tian, Y.; Dai, B.; Skiena, S.; Song, L. Syntax-directed variational autoencoder for structured data. arXiv 2018, arXiv:1802.08786. [Google Scholar]
- Liu, Q.; Allamanis, M.; Brockschmidt, M.; Gaunt, A. Constrained graph variational autoencoders for molecule design. arXiv 2018, arXiv:1805.09076. [Google Scholar]
- Samanta, B.; De, A.; Ganguly, N.; Gomez-Rodriguez, M. Designing random graph models using variational autoencoders with applications to chemical design. arXiv 2018, arXiv:1802.05283. [Google Scholar]
- Winter, R.; Montanari, F.; Noé, F.; Clevert, D.A. Learning Continuous and Data-Driven Molecular Descriptors by Translating Equivalent Chemical Representations. Chem. Sci. 2019, 10, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Kajino, H. Molecular Hypergraph Grammar with its Application to Molecular Optimization. arXiv 2018, arXiv:1803.03324. [Google Scholar]
- Jin, W.; Barzilay, R.; Jaakkola, T. Junction tree variational autoencoder for molecular graph generation. In Proceedings of the International Conference on Learning Representations, Stockholm, Sweden, 10–15 July 2018. [Google Scholar]
- Jin, W.; Yang, K.; Barzilay, R.; Jaakkola, T. Learning Multimodal Graph-to-Graph Translation for Molecular Optimization. In Proceedings of the International Conference on Learning Representations, New Orleans, LA, USA, 6–9 May 2019. [Google Scholar]
- Samanta, B.; De, A.; Jana, G.; Gómez, V.; Chattaraj, P.K.; Ganguly, N.; Gomez-Rodriguez, M. NeVAE A Deep Generative Model for Molecular Graphs. arXiv 2019, arXiv:1802.05283. [Google Scholar] [CrossRef]
- Simonovsky, M.; Komodakis, N. Graphvae Towards generation of small graphs using variational autoencoders. arXiv 2018, arXiv:1802.03480. [Google Scholar]
- Ma, T.; Chen, J.; Xiao, C. Constrained Generation of Semantically Valid Graphs via Regularizing Variational Autoencoders. In Proceedings of the Advances in Neural Information Processing Systems 32: Annual Conference on Neural Information Processing Systems 2019, NeurIPS 2019, Vancouver, BC, Canada, 8–14 December 2019. [Google Scholar]
- Skalic, M.; Jiménez, J.; Sabbadin, D.; De Fabritiis, G. Shape-based generative modeling for de-novo drug design. J. Chem. Inf. Model. 2019, 59, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Kuzminykh, D.; Polykovskiy, D.; Kadurin, A.; Zhebrak, A.; Baskov, I.; Nikolenko, S.; Shayakhmetov, R.; Zhavoronkov, A. 3D molecular representations based on the wave transform for convolutional neural networks. Mol. Pharm. 2018, 15, 4378–4385. [Google Scholar] [CrossRef] [PubMed]
- Steven, M. Kearnes Li Li and Patrick Riley, T. Decoding molecular graph embeddings with reinforcement learning. arXiv 2019, arXiv:1904.08915. [Google Scholar]
- Guimaraes, G.L.; Sanchez-Lengeling, B.; Outeiral, C.; Farias, P.L.C.; Aspuru-Guzik, A. Objective-Reinforced Generative Adversarial Networks (ORGAN) for Sequence Generation Models. arXiv 2017, arXiv:1705.10843. [Google Scholar]
- Putin, E.; Asadulaev, A.; Ivanenkov, Y.; Aladinskiy, V.; Sanchez-Lengeling, B.; Aspuru-Guzik, A.; Zhavoronkov, A. Reinforced adversarial neural computer for de novo molecular design. J. Chem. Inf. Model. 2018, 58, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Putin, E.; Asadulaev, A.; Vanhaelen, Q.; Ivanenkov, Y.; Aladinskaya, A.V.; Aliper, A.; Zhavoronkov, A. Adversarial threshold neural computer for molecular de novo design. Mol. Pharm. 2018, 15, 4386–4397. [Google Scholar] [CrossRef] [PubMed]
- Kadurin, A.; Nikolenko, S.; Khrabrov, K.; Aliper, A.; Zhavoronkov, A. druGAN An advanced generative adversarial autoencoder model for de novo generation of new molecules with desired molecular properties in silico. Mol. Pharm. 2017, 14, 3098–3104. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Lucio, O.; Baillif, B.; Clevert, D.A.; Rouquié, D.; Wichard, J. De novo generation of hit-like molecules from gene expression signatures using artificial intelligence. Nat. Commun. 2020, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- De Cao, N.; Kipf, T. MolGAN An implicit generative model for small molecular graphs. In Proceedings of the ICML 2018 Workshop on Theoretical Foundations and Applications of Deep Generative Models, Stockholm, Sweden, 14–15 July 2018. [Google Scholar]
- De Cao, N.; Kipf, T. MolGAN An implicit generative model for small molecular graphs. arXiv 2018, arXiv:1805.11973. [Google Scholar]
- Maziarka, Ł.; Pocha, A.; Kaczmarczyk, J.; Rataj, K.; Danel, T.; Warchoł, M. Mol-cyclegan—A generative model for molecular optimization. J. Cheminform. 2020, 12, 1758–2946. [Google Scholar] [CrossRef]
- Merk, D.; Friedrich, L.; Grisoni, F.; Schneider, G. De novo design of bioactive small molecules by artificial intelligence. Mol. Inform. 2018, 37, 1700153. [Google Scholar] [CrossRef]
- Merk, D.; Grisoni, F.; Friedrich, L.; Schneider, G. Tuning artificial intelligence on the de novo design of natural-product-inspired retinoid X receptor modulators. Commun. Chem. 2018, 1, 68. [Google Scholar] [CrossRef]
- Ertl, P.; Lewis, R.; Martin, E.; Polyakov, V. In silico generation of novel drug-like chemical matter using the LSTM neural network. arXiv 2017, arXiv:1712.07449. [Google Scholar]
- Neil, D.; Segler, M.; Guasch, L.; Ahmed, M.; Plumbley, D.; Sellwood, M.; Brown, N. Exploring deep recurrent models with reinforcement learning for molecule design. In Proceedings of the International Conference on Learning Representations, Vancouver, BC, Canada, 30 April–3 May 2018. [Google Scholar]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Müller, A.T.; Huisman, B.J.; Fuchs, J.A.; Schneider, P.; Schneider, G. Generative recurrent networks for de novo drug design. Mol. Inform. 2017, 37, 1700111. [Google Scholar] [CrossRef] [PubMed]
- Segler, M.H.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 2017, 4, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Vinyals, O.; Dyer, C.; Pascanu, R.; Battaglia, P. Learning Deep Generative Models of Graphs. arXiv 2018, arXiv:1803.03324. [Google Scholar]
- Pogány, P.; Arad, N.; Genway, S.; Pickett, S.D. De novo molecule design by translating from reduced graphs to SMILES. J. Chem. Inf. Model. 2018, 59, 1136–1146. [Google Scholar] [CrossRef]
- Bjerrum, E.J.; Threlfall, R. Molecular Generation with Recurrent Neural Networks (RNNs). arXiv 2017, arXiv:1705.04612. [Google Scholar]
- Yang, X.; Zhang, J.; Yoshizoe, K.; Terayama, K.; Tsuda, K. ChemTS an efficient python library for de novo molecular generation. Sci. Technol. Adv. Mater. 2017, 18, 972–976. [Google Scholar] [CrossRef] [PubMed]
- Cherti, M.; Kégl, B.; Kazakçi, A.O. De novo drug design with deep generative models an empirical study. In Proceedings of the International Conference on Learning Representations Work-Shop Track, Toulon, France, 24–26 April 2017. [Google Scholar]
- Zheng, S.; Yan, X.; Gu, Q.; Yang, Y.; Du, Y.; Lu, Y.; Xu, J. QBMG quasi-biogenic molecule generator with deep recurrent neural network. J. Cheminform. 2019, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. Molecular de-novo design through deep re-inforcement learning. J. Cheminform. 2017, 9, 1–4. [Google Scholar] [CrossRef]
- Sumita, M.; Yang, X.; Ishihara, S.; Tamura, R.; Tsuda, K. Hunting for organic molecules with artificial intelligence Molecules optimized for desired excitation energies. ACS Cent. Sci. 2018, 4, 1126–1133. [Google Scholar] [CrossRef]
- Arús-Pous, J.; Blaschke, T.; Ulander, S.; Reymond, J.L.; Chen, H.; Engkvist, O. Exploring the GDB-13 Chemical Space Using Deep Generative Models. J. Cheminform. 2018, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kadurin, A.; Aliper, A.; Kazennov, A.; Mamoshina, P.; Vanhaelen, Q.; Khrabrov, K.; Zhavoronkov, A. The cornucopia of meaningful leads Applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 2016, 8, 10883. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lengeling, B.; Outeiral, C.; Guimaraes, G.L.; Aspuru-Guzik, A. Optimizing distributions over molecular space. An Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry (ORGANIC). ChemRxiv 2017. [Google Scholar] [CrossRef]
- You, J.; Liu, B.; Ying, Z.; Pande, V.; Leskovec, J. Graph Convolutional Policy Network for Goal-Directed Molecular Graph Generation. arXiv 2018, arXiv:1806.02473. [Google Scholar]
- Grattarola, D.; Livi, L.; Alippi, C. Ad-versarial autoencoders with constant-curvature latent manifolds. arXiv 2018, arXiv:1812.04314. [Google Scholar]
- Ikebata, H.; Hongo, K.; Isomura, T.; Maezono, R.; Yoshida, R. Bayesian molecular design with a chem20 ical language model. J. -Comput.-Aided Mol. Des. 2017, 31, 379–391. [Google Scholar] [CrossRef]
- Goodfellow, I.; Bengio, Y.; Courville, T. Deep Learning; MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Chollet, F. Deep learning with Python; Manning Publications Co: Shelter Island, NY, USA, 2018. [Google Scholar]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative adversarial nets. NIPS 2014, 63, 2672–2680. [Google Scholar]
- Nowozin, S.; Cseke, B.; Tomioka, R. f-GAN Training generative neural samplers using variational divergence minimization. Adv. Neural Inf. Process. Syst. 2016, 29, 271–279. [Google Scholar]
- Arjovsky, M.; Chintala, S.; Bottou, L. Wasserstein generative adversarial networks. In Proceedings of the International Conference on Machine Learning, Sydney, Australia, 6–11 August 2017; pp. 214–223. [Google Scholar]
- Yuan, H.; Zhuang, J.; Hu, S.; Li, H.; Xu, J.; Hu, Y.; Xiong, X.; Chen, Y.; Lu, T. Molecular Modeling of Exquisitely Selective c-Met Inhibitors through 3D-QSAR and Molecular Dynamics Simulations. J. Chem. Inf. Model. 2014, 54, 2544–2554. [Google Scholar] [CrossRef]
- Kilchmann, F.; Marcaida, M.J.; Kotak, S.; Schick, T.; Boss, S.D.; Awale, M.; Gönczy, P.; Reymond, J.-L. Discovery of a Selective Aurora A Kinase Inhibitor by Virtual Screening. J. Med. Chem. 2016, 59, 7188–7211. [Google Scholar] [CrossRef]
- Jones, R.L.; Judson, I.R. The development and application of imatinib. Expert Opin. Drug Saf. 2005, 4, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Radford, I.R. The development and application of imatinib. Curr. Opin. Investig. Drugs 2002, 3, 492–499. [Google Scholar] [PubMed]
- Sanachai, K.; Mahalapbutr, P.; Hengphasatporn, K.; Shigeta, Y.; Seetaha, S.; Tabtimmai, L.; Wolschann, P.; Kittikool, T.; Yotphan, S.; Choowongkomon, K.; et al. Pharmacophore-Based Virtual Screening and Experimental Validation of Pyrazolone-Derived Inhibitors toward Janus Kinases. ACS Omega 2022, 7, 33548–33559. [Google Scholar] [CrossRef] [PubMed]
- Asiedu, S.O.; Kwofie, S.K.; Broni, E.; Wilson, M.D. Computational Identification of Potential Anti-Inflammatory Natural Compounds Targeting the p38 Mitogen-Activated Protein Kinase (MAPK): Implications for COVID-19-Induced Cytokine Storm. Biomolecules 2021, 29, 653. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Jiang, S.; Ye, W.; Ren, X.; Wang, X.; Zhang, Y.; Yin, M.; Wang, K.; Tao, Y.; Yang, J.; et al. Combined treatment of mitoxantrone sensitizes breast cancer cells to rapalogs through blocking eEF-2K-mediated activation of Akt and autophagy. Cell. Death Dis. 2020, 11, 948. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G. The Development of CK2 Inhibitors: From Traditional Pharmacology to in Silico Rational Drug Design. Pharmaceuticals 2017, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Makhouri, F.R.; Ghasemi, J.B. High-throughput Docking and Molecular Dynamics Simulations towards the Identification of Novel Peptidomimetic Inhibitors against CDC7. Mol. Inform. 2018, 37, 653. [Google Scholar] [CrossRef] [PubMed]
- Pereira, T. Diversity oriented Deep Reinforcement Learning for targeted molecule generation. J. Cheminform. 2021, 13, 9–10. [Google Scholar] [CrossRef]
- Mostapha Benhenda, T. ChemGAN challenge for drug discovery can AI reproduce natural chemical diversity? arXiv 2017, 3, 2–3. [Google Scholar]
- Brown, N.; Fiscato, M.; Segler, M.H.; Vaucher, A.C. GuacaMolbenchmarking models for de novo molecular design. J. Cheminform. 2019, 59, 1096–1108. [Google Scholar]
- Preuer, K.; Renz, P.; Unterthiner, T.; Hochreiter, S.; Klambauer, G. Fréchet ChemNet Distance A Metric for Generative Models for Molecules in Drug Discovery. J. Chem. Inf. Model. 2018, 58, 1736–1741. [Google Scholar] [CrossRef]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 3–5. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Jacek, K.; Hanna, P.; Anna, M.; Beata, D. The log P Parameter as a Molecular Descriptor in the Computer-aided Drug Design—An Overview. Comput. Methods Sci. Technol. 2012, 18, 81–88. [Google Scholar]
- Prasanna, S.; Doerksen, R.J. Topological polar surface area a useful descriptor in 2D-QSAR. Curr. Med. Chem. 2009, 16, 21–41. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina improving the speed and accuracy of docking with a new scoring function efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 2–4. [Google Scholar] [CrossRef]
- Leguy, J.; Cauchy, T.; Glavatskikh, M.; Duval, B.; Da Mota, B. EvoMol a flexible and interpretable evolutionary algorithm for unbiased de novo molecular generation. J. Cheminform. 2020, 12, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar]
- Liu, X.; Jiang, H.; Li, H. SHAFTS A Hybrid Approach for 3D Molecular Similarity Calculation. 1. Method and Assessment of Virtual Screening. J. Chem. Inf. Model. 2011, 51, 2372–2385. [Google Scholar] [CrossRef] [PubMed]
- Rush, T.S.; Grant, J.A.; Mosyak, L.; Nicholls, A. A Shape-Based 3-D Scaffold Hopping Method and Its Application to a Bacterial Protein—Protein Interaction. J. Med. Chem. 2005, 48, 1489–1495. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Dral, P.O.; Rupp, M.; Von Lilienfeld, O.A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data 2014, 1, 1–7. [Google Scholar] [CrossRef]
- Ruddigkeit, L.; Van Deursen, R.; Blum, L.C.; Reymond, J.L. Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J. Chem. Inf. Model. 2012, 52, 2864–2875. [Google Scholar] [CrossRef] [PubMed]
- TSterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Polykovskiy, D.; Zhebrak, A.; Sanchez-Lengeling, B.; Golovanov, S.; Tatanov, O.; Belyaev, S.; Kurbanov, R.; Artamonov, A.; Aladinskiy, V. Molecular sets (MOSES) a benchmarking platform for molecular generation models. Front. Pharmacol. 2020, 11, 565644. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; AlLazikani, B.; et al. ChEMBL a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- Blum, L.C.; Reymond, J.L. 970 million druglike small molecules for virtual screening in the chemical universe database gdb-13. J. Am. Chem. Soc. 2009, 131, 8732–8733. [Google Scholar] [CrossRef]
- Axelrod, S.; Gomez-Bombarelli, R. GEOM Energy-annotated molecular conformations for property prediction and molecular generation. arXiv 2020, arXiv:2006.05531. [Google Scholar] [CrossRef]
- Schütt, K.T.; Arbabzadah, F.; Chmiela, S.; Müller, K.R.; Tkatchenko, A. Quantum-chemical insights from deep tensor neural networks. Nat. Commun. 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Xu, Z.; Luo, Y.; Zhang, X.; Xu, X.; Xie, Y.; Liu, M.; Dickerson, k.; Deng, C.; Nakata, M.; Ji, S. Molecule3D A benchmark for predicting 3d geometries from molecular graphs. arXiv 2021, arXiv:2110.01717. [Google Scholar]
- Francoeur, P.G.; Masuda, T.; Sunseri, J.; Jia, A.; Iovanisci, R.B.; Snyder, I.; Koes, D.R. Three-dimensional convolutional neural networks and a cross-docked data set for structure-based drug design. J. Chem. Inf. Model. 2020, 60, 4200–4215. [Google Scholar] [CrossRef]
- Desaphy, J.; Bret, G.; Rognan, D.; Kellenberger, E. sc-PDB a 3d-database of ligandable binding sites—10 years on. Nucleic Acids Res. 2015, 43, D399–D404. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys enhanced (dud-e) better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Borah, K.; Bora, K.; Mallik, S.; Zhao, Z. Potential Therapeutic Agents on Alzheimer’s Disease through Molecular Docking and Molecular Dynamics Simulation Study of Plant-Based Compounds. Comput. Methods Appl. Biol. Chem. Sci. 2022, 20, e202200684. [Google Scholar]
- Bora, K.; Mahanta, L.B.; Borah, K.; Chyrmang, G.; Barua, B.; Mallik, S.; Das, H.S.; Zhao, Z. Machine Learning Based Approach for Automated Cervical Dysplasia Detection Using Multi-Resolution Transform Domain Features. Mathematics 2022, 21, 4126. [Google Scholar] [CrossRef]
- Khandelwal, M.; Kumar Rout, R.; Umer, S.; Mallik, S.; Li, A. Multifactorial feature extraction and site prognosis model for protein methylation data. Briefings Funct. Genom. 2023, 22, 20–30. [Google Scholar] [CrossRef]
- Ghosh, A.; Jana, N.D.; Mallik, S.; Zhao, Z. Designing optimal convolutional neural network architecture using differential evolution algorithm. Patterns 2022, 3, 100567. [Google Scholar] [CrossRef] [PubMed]
- Dhar, R.; Mallik, S.; Devi, A. Exosomal microRNAs (exoMIRs): Micromolecules with macro impact in oral cancer. Biotech 2022, 12, 155. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Architecture | Representation | Dataset | References |
|---|---|---|---|
| VAE | SMILES | ZINC | [36] |
| VAE | SMILES | ZINC | [37] |
| VAE | SMILES | ZINC | [38] |
| VAE | SMILES | ZINC/QM9 | [39] |
| VAE | SMILES | ChEMBL | [40] |
| VAE | SMILES | ChEMBL | [40] |
| VAE | SMILES | ChEMBL23 | [41] |
| GVAE | CFG (SMILES) | ZINC | [42] |
| GVAE | CFG (custom) | PSC | [43,44] |
| SD-VAE | CFG (custom) | ZINC | [45] |
| CVAE | Graph | ZINC/CEPDB | [46] |
| VAE | Graph | ZINC/QM9 | [47] |
| VAE | Graph | ZINC+PubChem | [48] |
| MHG-VAE | Graph (MHG) | ZINC | [49] |
| JT-VAE | Graph (operation) | ZINC | [50] |
| JT-VAE | Graph (operation) | ZINC | [51] |
| VAE | Graph (Tensor) | ZINC | [52] |
| VAE | Graph (Tensor) | ZINC/QM9 | [53] |
| VAE | Graph (Tensor) | ZINC | [54] |
| CVAE | 3D density | ZINC | [55] |
| VAE | 3D wave transform | ZINC | [56] |
| VAE+RL | MPNN+graph ops | ZINC | [57] |
| GAN | SMILES | GBD-17 | [58] |
| GAN (ANC) | SMILES | ZINC/CHEMDIV | [59] |
| GAN (ATNC) | SMILES | ZINC/CHEMDIV | [60] |
| GAN | MACCS (166 bit) | MCF-7 | [61] |
| sGAN | MACCS (166 bit) | L1000 | [62] |
| GAN | Graph (tensors) | QM9 | [63,64] |
| CycleGAN | Graph operation | ZINC | [65] |
| RNN | SMILES | ChEMBL | [66] |
| RNN | SMILES | ChEMBL | [67] |
| RNN | SMILES | ChEMBL | [68] |
| RNN | SMILES | ChEMBL | [69] |
| RNN | SMILES | ChEMBL | [70] |
| RNN | SMILES | ChEMBL | [71] |
| RNN | SMILES | ChEMBL | [72] |
| RNN | Graph operations | ChEMBL | [73] |
| RNN | RG+SMILES | ChEMBL | [74] |
| RNN | SMILES | ZINC | [75] |
| RNN | SMILES | ZINC | [76] |
| RNN | SMILES | ZINC | [77] |
| RNN | SMILES | ZINC | [78] |
| RNN | SMILES | DRD2 | [79] |
| RNN | SMILES | PubChemQC | [80] |
| RNN | SMILES | GDB-13 | [81] |
| AAE | MACCS (166 bit) | MCF-7 | [82] |
| AAE | SMILES | HCEP | [83] |
| GCPN | Graph | ZINC | [84] |
| CCM-AAE | Graph (tensors) | QM9 | [85] |
| BMI | SMILES | PubChem | [86] |
| ML Models | Performance Analysis Metric |
|---|---|
| Linear regression | RMSE |
| Logistic regression | RMSE |
| SVM | Accuracy or F-1 score |
| Q-learning | Cumulative reward |
| R-learning | IQM on performance profiles |
| Dataset | Approximate Amount | Description |
|---|---|---|
| QM9 [114,115] | 134,000 | This is a subset of GDB-13 (a database of nearly 1 billion stable and synthetically accessible organic molecules) composed of all molecules of up to 23 atoms including 9 heavy atoms. QM9 provides quantum chemical properties for the chemical space of small organic molecules. |
| ZINC [116] | 250,000 | It comprises over 230 million compounds in ready-to-dock, 3D formats. |
| Molecular Sets (MOSES) [117] | 1,937,000 | The set is based on the ZINC Clean Leads collection. This dataset has been filtered from the ZINC dataset. These are the drug-like molecules. |
| ChEMBL [118] | 2,100,000 | A database of bioactive compounds with drug-like molecules, which is manually curated. |
| GDB13 [119] | 970,000,000 | In this dataset, we have small organic compounds with up to 13 atoms, using chemical stability and synthetic feasibility principles. It is the largest publicly available small organic molecule database. |
| GEOM-QM9 [120] | 450,000; 37,000,000 | The 3D conformer ensembles are annotated by GEOM-QM9 using sophisticated sampling and semiempirical density functional theory. The dataset contains around 133K 3D molecules. |
| GEOM-Drugs [120] | 317,000 | This dataset also uses advanced sampling and semiempirical density functional theory to annotate the 3D conformer ensembles. |
| ISO17 [121] | 200; 431,000 | This dataset contains 197 2D molecules and 430,692 molecule-conformation pairs. |
| Molecule3D [122] | 4 million | This dataset contains almost 4 million molecules and researchers use density functional theory to create exact ground-state geometries for the molecules in the dataset. |
| CrossDock2020 [123] | 22,500,000 | The CrossDocked2020 collection contains 22.5 million docked ligand poses in various binding pockets that are similar across the Protein Data Bank. |
| scPDB [124] | An annotated database of druggable binding sites from the Protein Data Bank. It registers 9283 binding sites from 3678 unique proteins and 5608 unique ligands, with a total of 16,034 entries. | |
| DUD-E [125] | DUD-E contains 102 target-specific affinity scores and 22,886 active molecules. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choudhuri, S.; Yendluri, M.; Poddar, S.; Li, A.; Mallick, K.; Mallik, S.; Ghosh, B. Recent Advancements in Computational Drug Design Algorithms through Machine Learning and Optimization. Kinases Phosphatases 2023, 1, 117-140. https://doi.org/10.3390/kinasesphosphatases1020008
Choudhuri S, Yendluri M, Poddar S, Li A, Mallick K, Mallik S, Ghosh B. Recent Advancements in Computational Drug Design Algorithms through Machine Learning and Optimization. Kinases and Phosphatases. 2023; 1(2):117-140. https://doi.org/10.3390/kinasesphosphatases1020008
Chicago/Turabian StyleChoudhuri, Soham, Manas Yendluri, Sudip Poddar, Aimin Li, Koushik Mallick, Saurav Mallik, and Bhaswar Ghosh. 2023. "Recent Advancements in Computational Drug Design Algorithms through Machine Learning and Optimization" Kinases and Phosphatases 1, no. 2: 117-140. https://doi.org/10.3390/kinasesphosphatases1020008