

How Far Have We Developed Antibody–Drug Conjugate for the Treatment of Cancer?
 , , ,
, , ,
Abstract
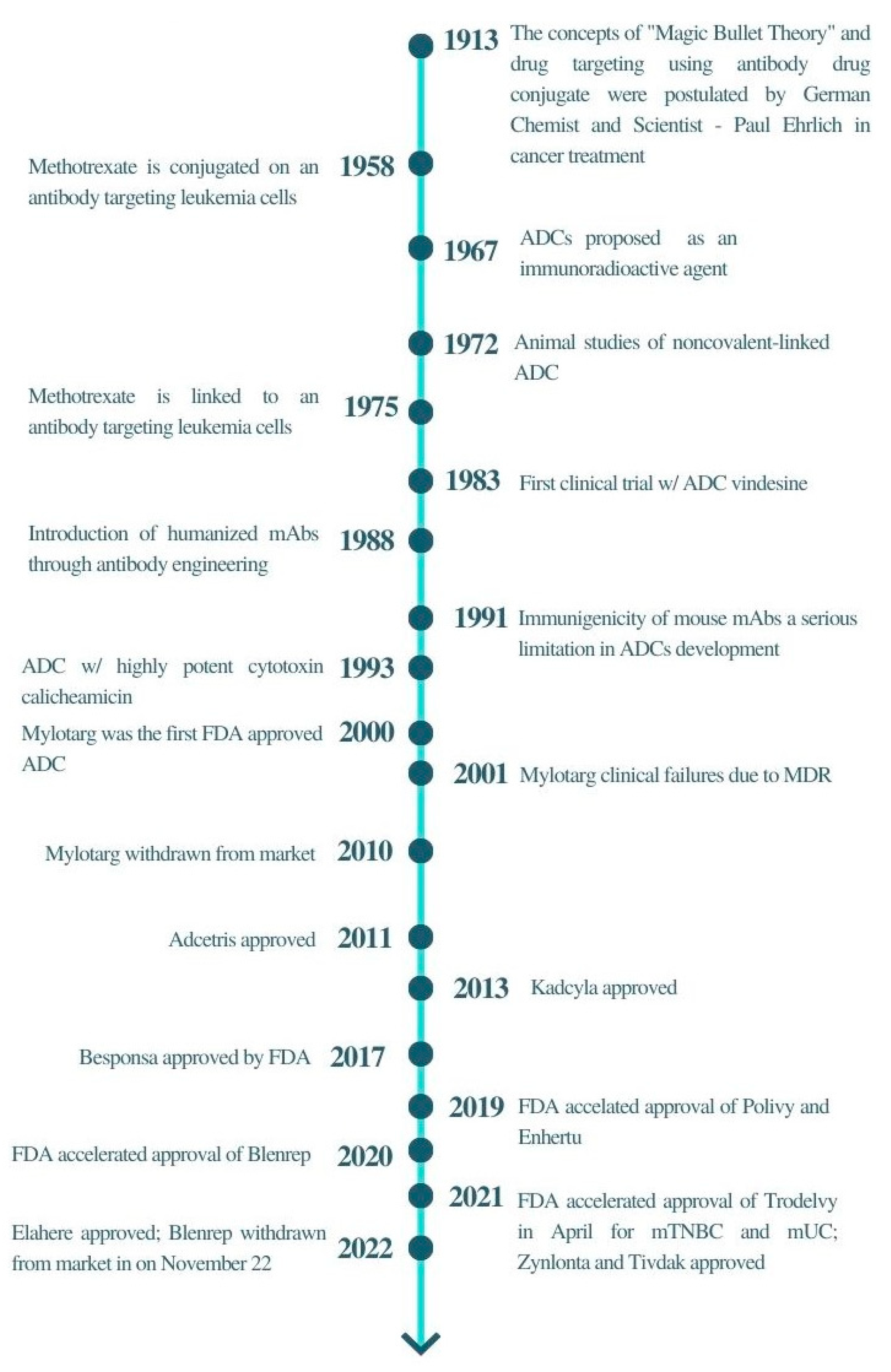
:1. Introduction
2. Principle of Antibody–Drug Conjugate
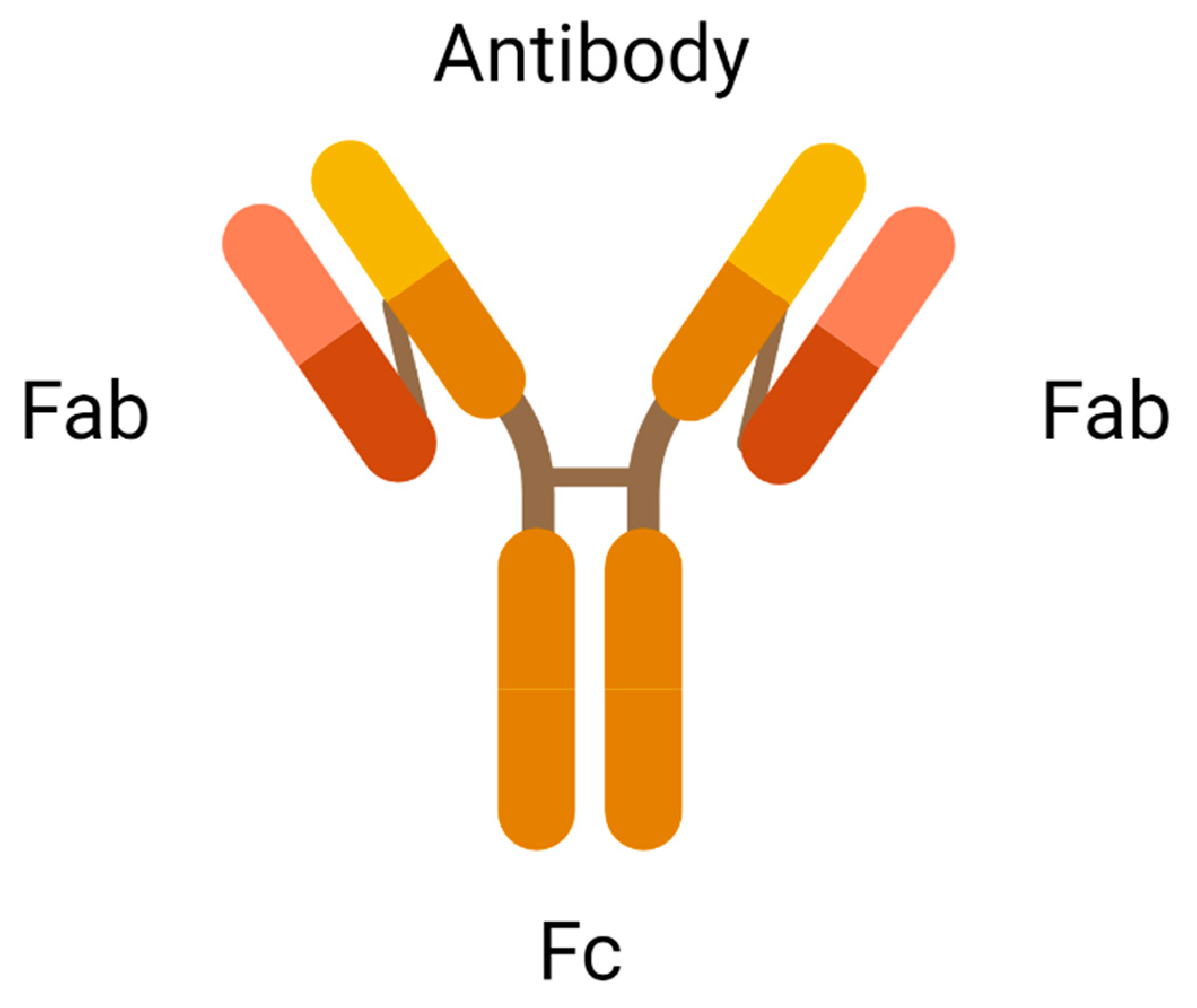
2.1. Antibody
2.2. Cytotoxic Payload
2.3. Linker
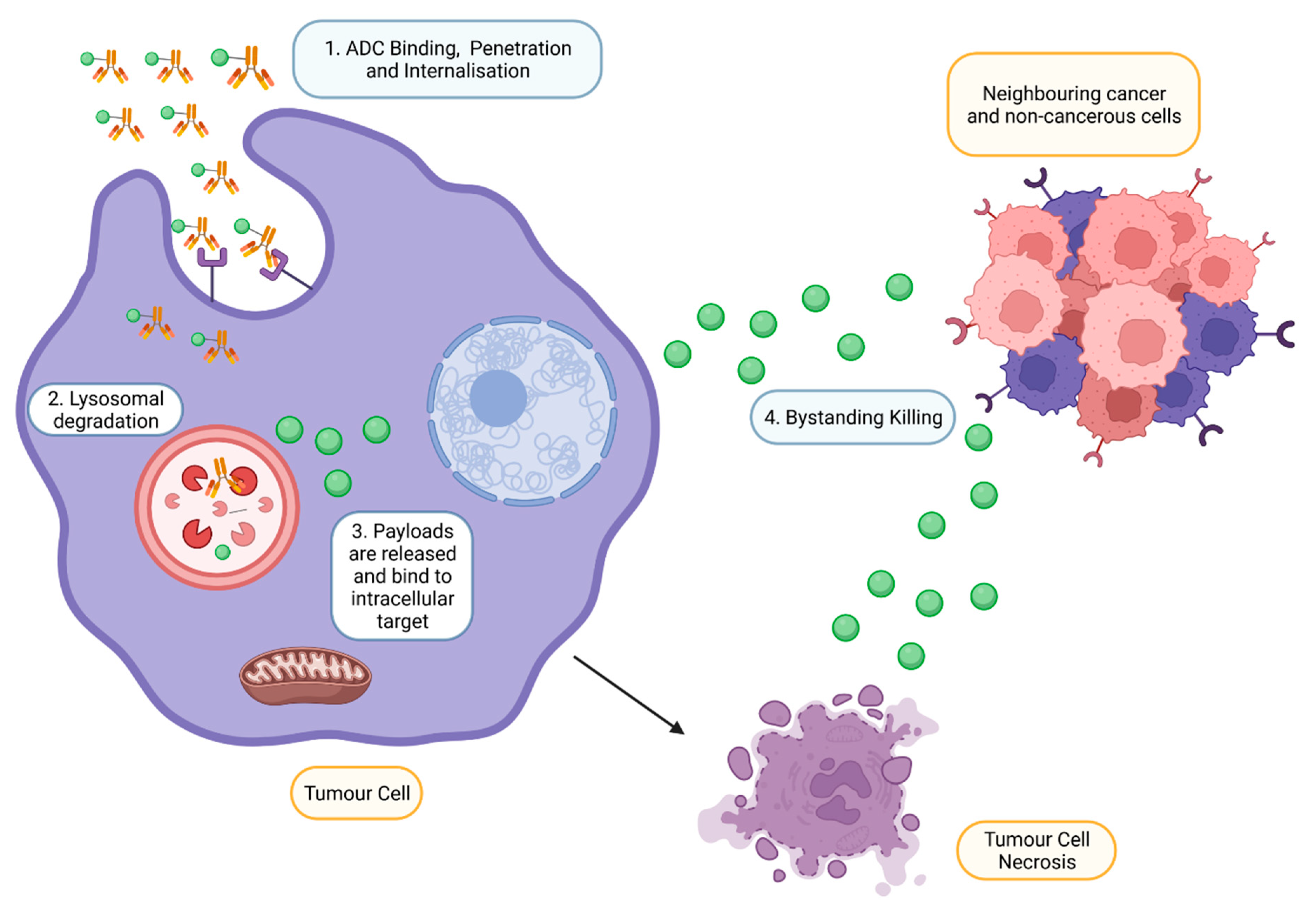
2.4. Mechanism of Action of ADC
3. Antibody–Drug Conjugate Approved by FDA
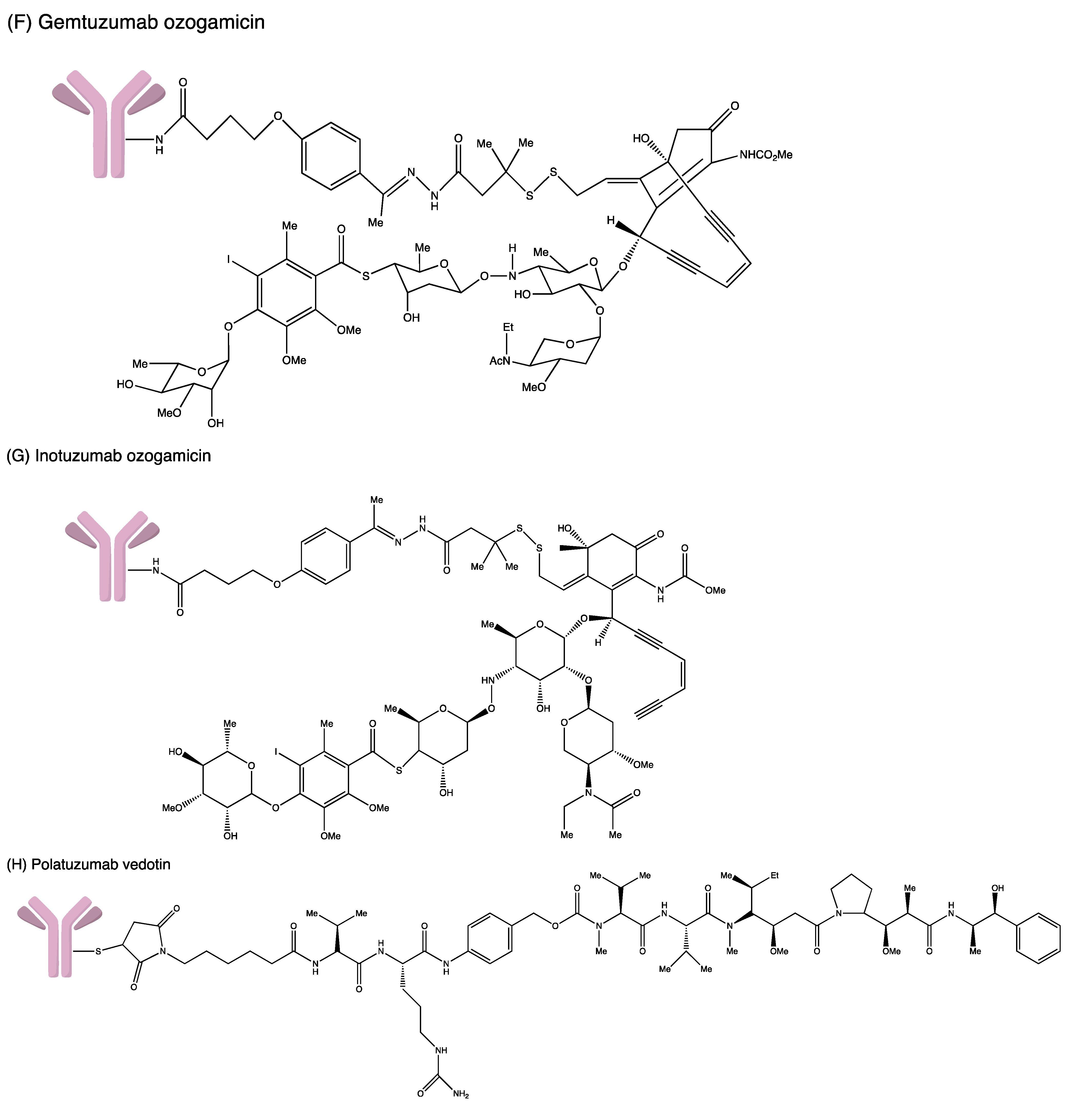
3.1. Gemtuzumab Ozogamicin
3.2. Brentuximab Vedotin
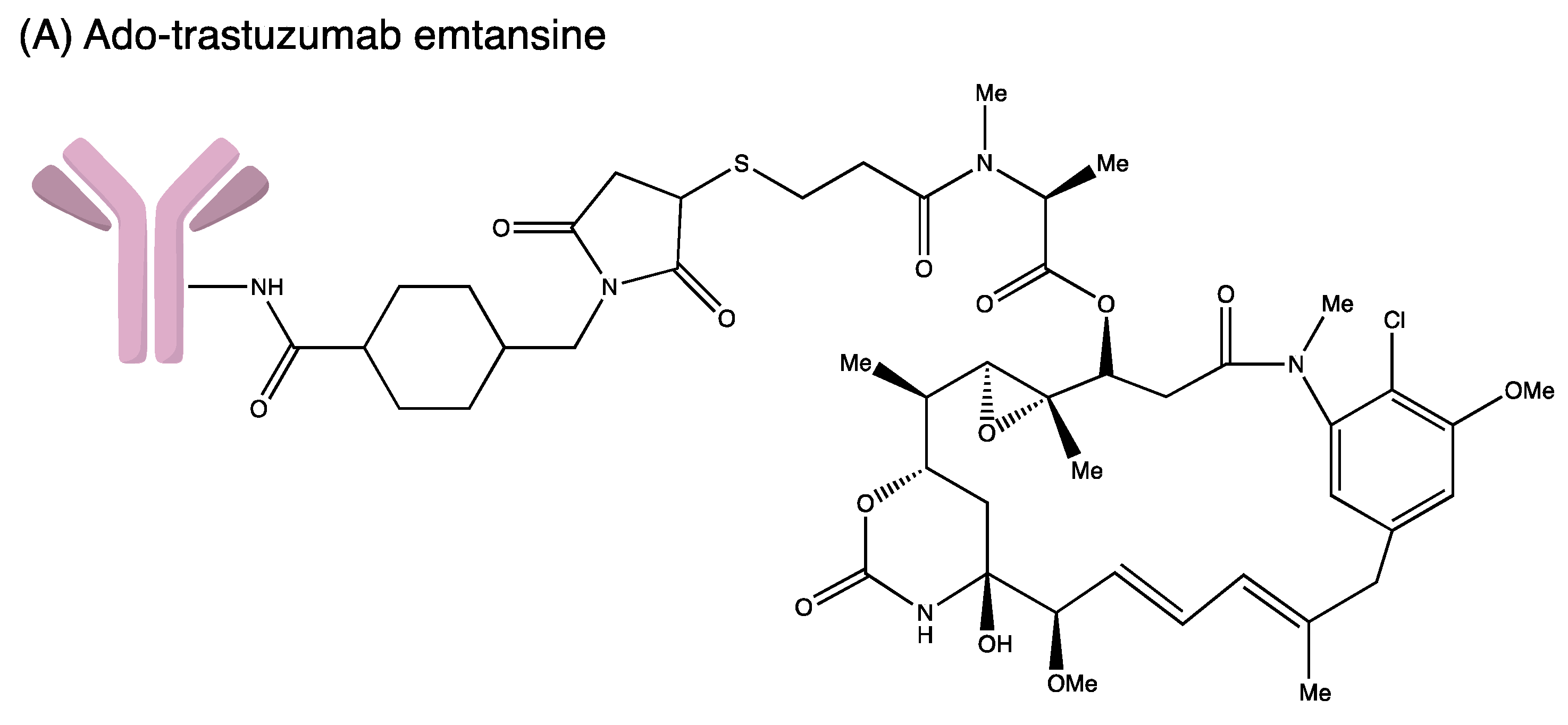
3.3. Ado-Trastuzumab Emtansine
3.4. Inotuzumab Ozogamicin
3.5. Polatuzumab Vedotin-Piiq
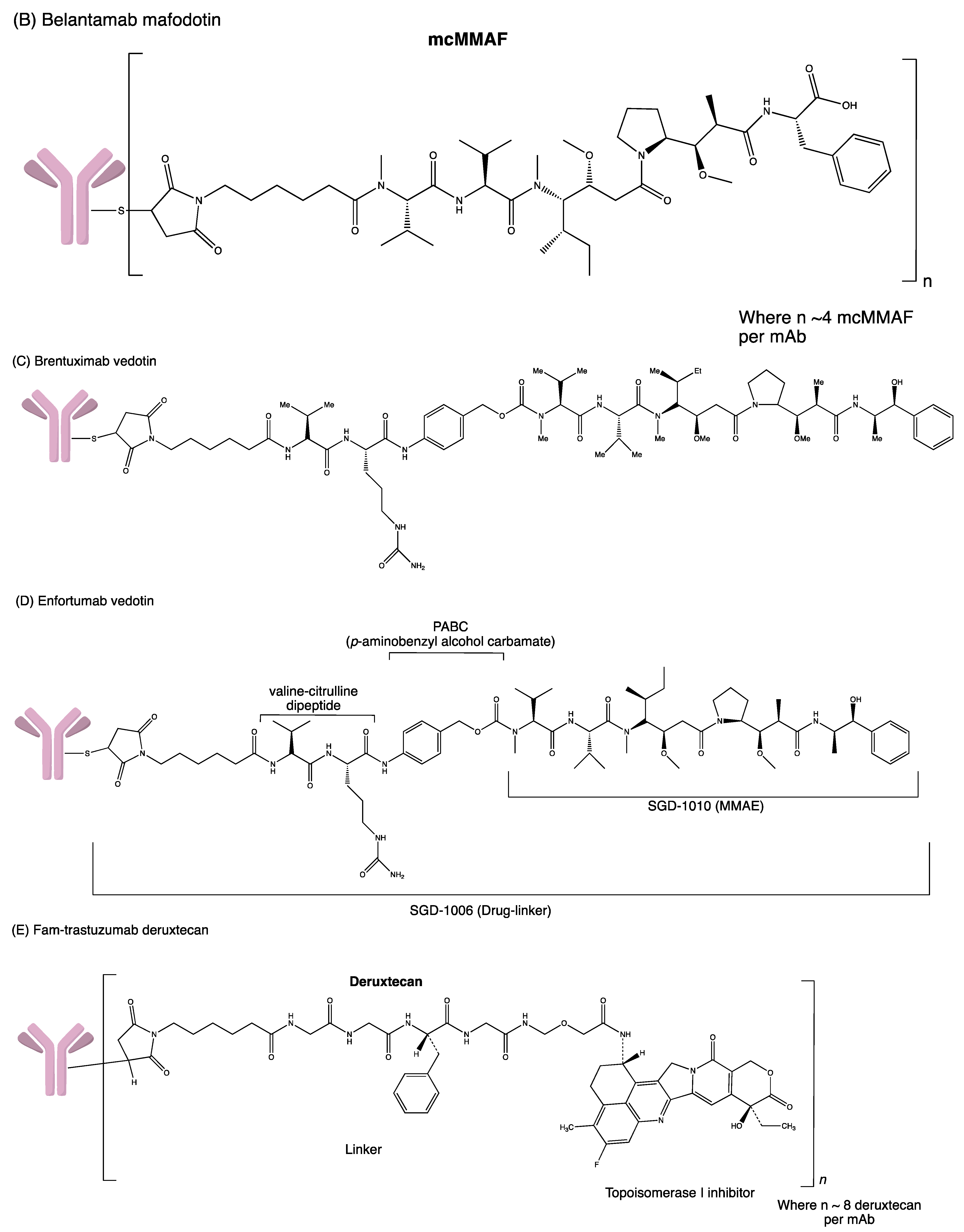
3.6. Enfortumab Vedotin
3.7. Fam-Trastuzumab Deruxtecan
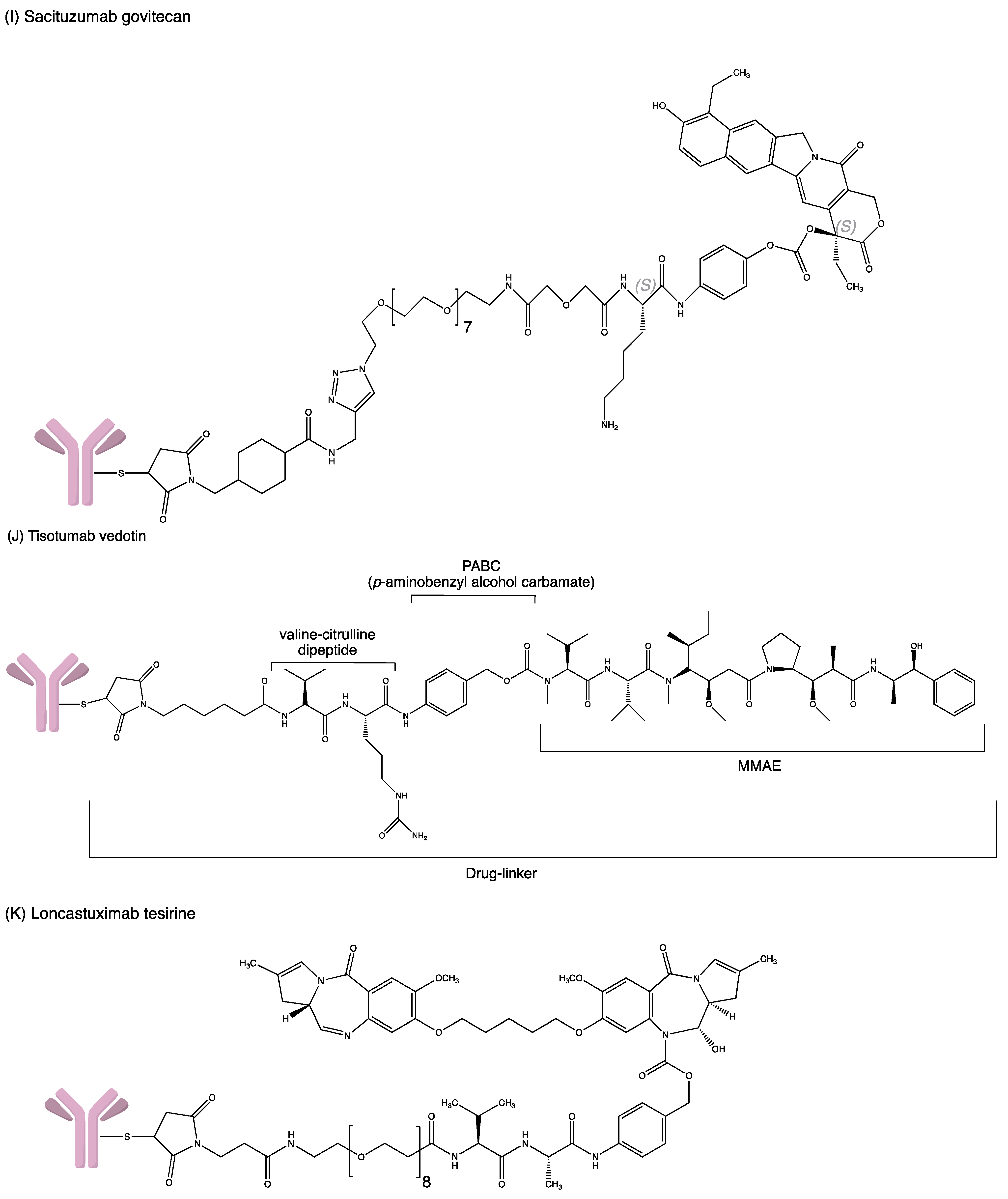
3.8. Sacituzumab Govitecan
3.9. Belantamab Mafodotin-Blmf
3.10. Loncastuximab Tesirine-Lpyl
3.11. Tisotumab Vedotin
3.12. Moxetumomab Pasudotox
4. Antibody–Drug Conjugates under Development
4.1. Potential ADCs to Be Approved
4.1.1. AGS67E
4.1.2. Denintuzumab Mafodotin (SGN-CD19A)
4.1.3. MEDI4276
4.1.4. Mirvetuximab Soravtansine (IMGN853)
4.1.5. Patritumab Deruxtecan (U3-1402)
4.1.6. Telisotuzumab Vedotin (ABBV-399)
4.2. Discontinued/Terminated Clinical Trials
4.2.1. AGS-16C3F
4.2.2. Coltuximab Ravtansine (SAR3419)
4.2.3. Glembatumumab Vedotin (CDX-011)
4.2.4. Lorvotuzumab Mertansine (IMGN-901)
4.2.5. Rovalpituzumab Tesirine (Rova-T)
5. Challenges of the Use of Antibody–Drug Conjugate
5.1. Toxicity of ADC
5.2. Antibody & Antigen Specificity
5.3. Immunogenicity of Antibody
5.4. Stability of Linkers
5.5. Target Features of Successful ADCs
6. Computational Strategies to Address ADC’s Challenges
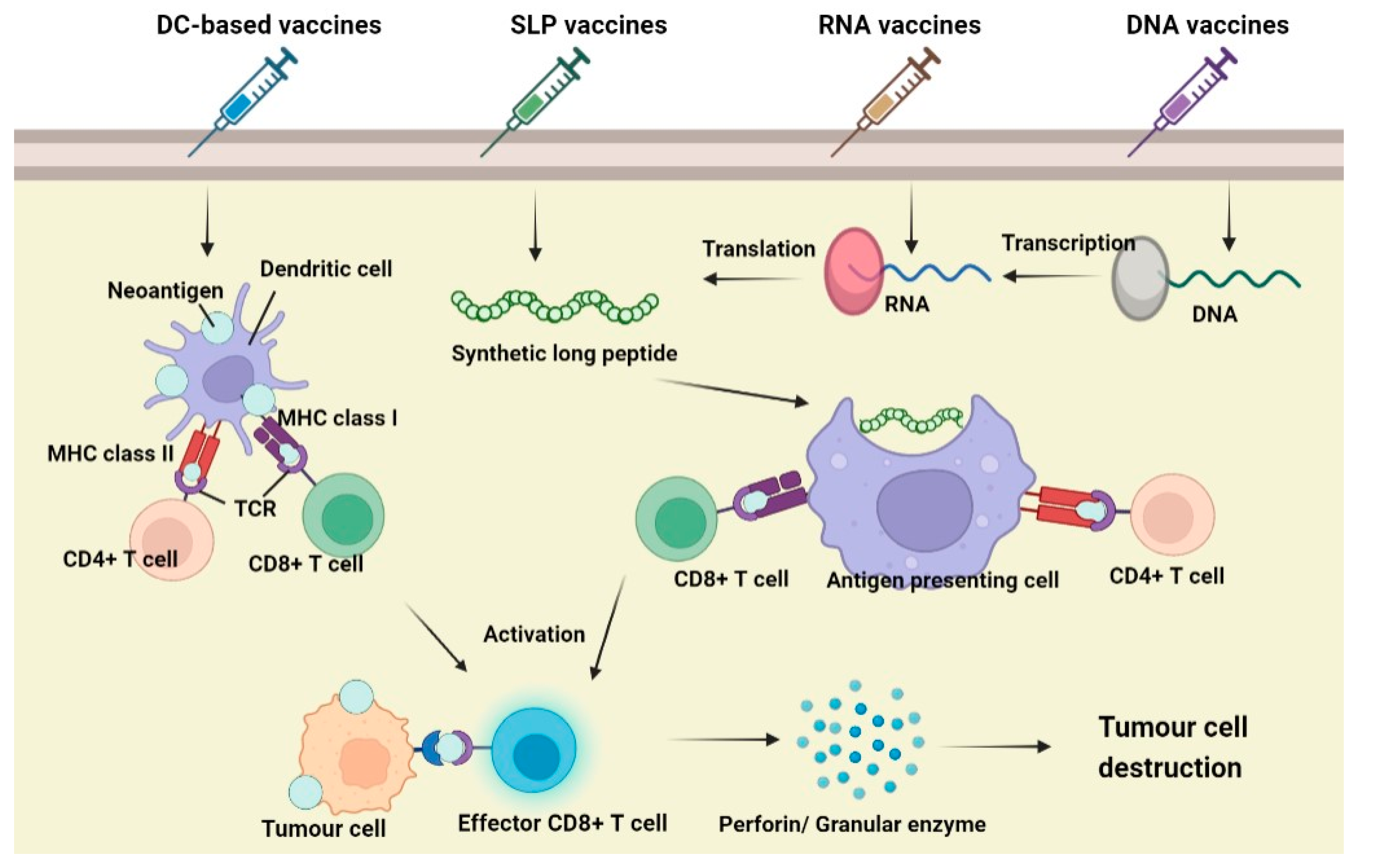
7. Neoantigens Cancer Vaccines
7.1. Neoantigen
7.2. Principle of Neoantigen Vaccines
7.3. Types of Neoantigen Vaccines
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neu, H.C. Paul Ehrlich: Scientist for Life. JAMA 1985, 254, 121. [Google Scholar] [CrossRef]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody–drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the’high-hanging fruit’. Nat. Rev. Drug Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Maturing antibody-drug conjugate pipeline hits 30. Nat. Rev. Drug Discov. 2013, 12, 329–333. [Google Scholar] [CrossRef]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef]
- Tong, J.T.W.; Harris, P.W.R.; Brimble, M.A.; Kavianinia, I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody–Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef]
- Marei, H.E.; Cenciarelli, C.; Hasan, A. Potential of antibody–drug conjugates (ADCs) for cancer therapy. Cancer Cell Int. 2022, 22, 255. [Google Scholar] [CrossRef]
- Birrer, M.J.; Moore, K.N.; Betella, I.; Bates, R. Antibody-drug conjugate-based therapeutics: State of the science. J. Natl. Cancer Inst. 2019, 111, 538–549. [Google Scholar] [CrossRef]
- Hoffmann, R.M.; Coumbe, B.G.; Josephs, D.H.; Mele, S.; Ilieva, K.M.; Cheung, A.; Tutt, A.N.; Spicer, J.F.; Thurston, D.E.; Crescioli, S.; et al. Antibody structure and engineering considerations for the design and function of Antibody Drug Conjugates (ADCs). Oncoimmunology 2018, 7, e1395127. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapymicrotubule-targeting agents for cancer chemotherapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Thorson, J.S.; Sievers, E.L.; Ahlert, J.; Shepard, E.; Whitwam, R.E.; Onwueme, K.C.; Ruppen, M. Understanding and exploiting nature’s chemical arsenal: The past, present and future of calicheamicin research. Curr. Pharm. Des. 2000, 6, 1841–1879. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef]
- Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Linkers having a crucial role in antibody–drug conjugates. Int. J. Mol. Sci. 2016, 17, 561. [Google Scholar] [CrossRef]
- Frigerio, M.; Kyle, A.F. The chemical design and synthesis of linkers used in antibody drug conjugates. Curr. Top. Med. Chem. 2017, 17, 3393–3424. [Google Scholar] [CrossRef]
- Nagayama, A.; Ellisen, L.W.; Chabner, B.; Bardia, A.J.T.O. Antibody–drug conjugates for the treatment of solid tumors: Clinical experience and latest developments. Target. Oncol. 2017, 12, 719–739. [Google Scholar] [CrossRef]
- Gorovits, B.; Krinos-Fiorotti, C. Proposed mechanism of off-target toxicity for antibody–drug conjugates driven by mannose receptor uptake. Cancer Immunol. Immunother. 2013, 62, 217–223. [Google Scholar] [CrossRef]
- Peters, C.; Brown, S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef]
- Li, F.; Emmerton, K.K.; Jonas, M.; Zhang, X.; Miyamoto, J.B.; Setter, J.R.; Nicholas, N.D.; Okeley, N.M.; Lyon, R.P.; Benjamin, D.R.; et al. Intracellular Released Payload Influences Potency and Bystander-Killing Effects of Antibody-Drug Conjugates in Preclinical ModelsReleased Payload Impacts ADC Activity and Bystander Killing. Cancer Res. 2016, 76, 2710–2719. [Google Scholar] [CrossRef]
- Khera, E.; Thurber, G.M. Pharmacokinetic and immunological considerations for expanding the therapeutic window of next-generation antibody–drug conjugates. BioDrugs 2018, 32, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Theocharopoulos, C.; Lialios, P.-P.; Gogas, H.; Ziogas, D.C. An overview of antibody–drug conjugates in oncological practice. Ther. Adv. Med. Oncol. 2020, 12, 1758835920962997. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Estey, E.H.; Appelbaum, F.R.; Lo-Coco, F.; Schiffer, C.A.; Larson, R.A.; Burnett, A.K.; Kantarjian, H.M. Gemtuzumab ozogamicin: Time to resurrect? J. Clin. Oncol. 2012, 30, 3921. [Google Scholar] [CrossRef] [PubMed]
- New York Medical College. Immunochemotherapy and AlloSCT in Patients with High Risk CD33+ AML/MDS; New York Medical College: Valhalla, NY, USA, 2014. [Google Scholar]
- M.D. Anderson Cancer Center. ADCT-602 in Treating Patients with Recurrent or Refractory B-Cell Acute Lymphoblastic Leukemia. NCT03698552; 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03698552 (accessed on 1 October 2022).
- FDA. Highlights of Prescribing Information—Adcetris (Brentuximab Vedotin). 2018; Volume 3, pp. 1–35. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125388s099lbl.pdf (accessed on 2 November 2022).
- Zhao, P.; Zhang, Y.; Li, W.; Jeanty, C.; Xiang, G.; Dong, Y. Recent advances of antibody drug conjugates for clinical applications. Acta Pharm. Sin. B 2020, 10, 1589–1600. [Google Scholar] [CrossRef]
- Van De Donk, N.W.; Dhimolea, E. Brentuximab Vedotin (Adcetris®) for the Treatment of CD30-Positive Hematologic Malignancies. In Handbook of Therapeutic Antibodies; Dübel, S., Reichert, J.M., Eds.; Wiley: New York, NY, USA, 2014; Volume 1, Chapter 49; pp. 1417–1444. [Google Scholar]
- Seagen Inc. A Phase 3 Study of Brentuximab Vedotin (SGN-35) in Patients at High Risk of Residual Hodgkin Lymphoma Following Stem Cell Transplant (The AETHERA Trial). NCT01100502; 2010. Available online: https://clinicaltrials.gov/ct2/show/NCT01100502 (accessed on 1 October 2022).
- M.D. Anderson Cancer Center. Brentuximab Vedotin in Treating Patients with CD30+ Malignant Mesothelioma That Cannot Be Removed by Surgery. NCT03007030; 2016. Available online: https://clinicaltrials.gov/ct2/show/NCT03007030 (accessed on 1 October 2022).
- Roche, H.-L. A Study of Trastuzumab Emtansine Versus Trastuzumab as Adjuvant Therapy in Patients with HER2-Positive Breast Cancer Who Have Residual Tumor in the Breast or Axillary Lymph Nodes Following Preoperative Therapy (KATHERINE). NCT01772472; 2013. Available online: https://clinicaltrials.gov/ct2/show/NCT01772472 (accessed on 1 October 2022).
- Roche, H.-L. A Study of Trastuzumab Emtansine (Kadcyla) Plus Pertuzumab (Perjeta) Following Anthracyclines in Comparison with Trastuzumab (Herceptin) Plus Pertuzumab and a Taxane Following Anthracyclines as Adjuvant Therapy in Participants with Operable HER2-Positive Primary Breast Cancer. NCT01966471; 2013. Available online: https://clinicaltrials.gov/ct2/show/NCT01966471 (accessed on 1 October 2022).
- Roche, H.-L. A Study of Trastuzumab Emtansine versus Capecitabine + Lapatinib in Participants with HER2-Positive Locally Advanced or Metastatic Breast Cancer (EMILIA). NCT00829166; 2009. Available online: https://clinicaltrials.gov/ct2/show/NCT00829166 (accessed on 1 October 2022).
- Daiichi Sankyo Inc. A Study of Trastuzumab Deruxtecan (T-DXd) versus Trastuzumab Emtansine (T-DM1) in High-Risk HER2-Positive Participants with Residual Invasive Breast Cancer Following Neoadjuvant Therapy (DESTINY-Breast05). NCT04622319; 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04622319 (accessed on 1 October 2022).
- Roche, H.-L. A Study of Trastuzumab Emtansine in Combination with Atezolizumab or Placebo as a Treatment for Participants with Human Epidermal Growth Factor 2 (HER2)-Positive and Programmed Death-Ligand 1 (PD-L1)-Positive Locally Advanced (LABC) or Metastatic Breast Cancer (MBC) (KATE3). NCT04740918; 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04740918 (accessed on 1 October 2022).
- Yurkiewicz, I.R.; Muffly, L.; Liedtke, M. Inotuzumab ozogamicin: A CD22 mAb–drug conjugate for adult relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Drug Des. Dev. Ther. 2018, 12, 2293–2300. [Google Scholar] [CrossRef]
- Pfizer. A Study Of Inotuzumab Ozogamicin versus Investigator’s Choice of Chemotherapy in Patients with Relapsed or Refractory Acute Lymphoblastic Leukemia. NCT01564784; 2012. Available online: https://clinicaltrials.gov/ct2/show/NCT01564784 (accessed on 1 October 2022).
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Jain, N.; Maiti, A.; Ravandi, F.; Konopleva, M.; Daver, N.; Kadia, T.; Pemmaraju, N.; Short, N.; Kebriaei, P.; Ning, J.; et al. Inotuzumab ozogamicin with bosutinib for relapsed or refractory Philadelphia chromosome positive acute lymphoblastic leukemia or lymphoid blast phase of chronic myeloid leukemia. Am. J. Hematol. 2021, 96, 1000–1007. [Google Scholar] [CrossRef]
- Deeks, E.D. Polatuzumab vedotin: First global approval. Drugs Future 2019, 79, 1467–1475. [Google Scholar] [CrossRef]
- Sehn, L.H.; Herrera, A.F.; Flowers, C.R.; Kamdar, M.K.; McMillan, A.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab vedotin in relapsed or refractory diffuse large B-cell lymphoma. J. Clin. Oncol. 2020, 38, 155. [Google Scholar] [CrossRef]
- Burke, J.; Morschhauser, F.; Andorsky, D.; Lee, C.; Sharman, J. Antibody–drug conjugates for previously treated aggressive lymphomas: Focus on polatuzumab vedotin. Expert Rev. Clin. Pharmacol. 2020, 13, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab vedotin antibody–drug conjugate targeting nectin-4 is a highly potent therapeutic agent in multiple preclinical cancer modelsADC cancer therapeutic targeting nectin-4. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef] [PubMed]
- Astellas Pharma Inc. A Study of Enfortumab Vedotin for Patients with Locally Advanced or Metastatic Urothelial Bladder Cancer (EV-201). NCT03219333; 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03219333 (accessed on 1 October 2022).
- Powles, T.; Rosenberg, J.E.; Sonpavde, G.P.; Loriot, Y.; Durán, I.; Lee, J.-L.; Matsubara, N.; Vulsteke, C.; Castellano, D.; Wu, C.; et al. Enfortumab vedotin in previously treated advanced urothelial carcinoma. N. Engl. J. Med. 2021, 384, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Astellas Pharma Global Development, I. A Study to Evaluate Enfortumab Vedotin Versus (vs) Chemotherapy in Subjects with Previously Treated Locally Advanced or Metastatic Urothelial Cancer (EV-301). NCT03474107; 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03474107 (accessed on 1 October 2022).
- Daiichi Sankyo Inc. A Study of DS-8201a in Metastatic Breast Cancer Previously Treated with Trastuzumab Emtansine (T-DM1). NCT03248492; 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03248492 (accessed on 1 October 2022).
- AstraZeneca. A Study of T-DXd in Participants with or without Brain Metastasis Who Have Previously Treated Advanced or Metastatic HER2 Positive Breast Cancer (DESTINY-B12). NCT04739761; 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04739761 (accessed on 1 October 2022).
- Goldenberg, D.M.; Sharkey, R.M. Antibody-Drug Conjugates Targeting TROP-2 and Incorporating SN-38: A Case Study of Anti-TROP-2 Sacituzumab Govitecan. MAbs 2019, 11, 987–995. [Google Scholar] [CrossRef]
- GileadSciences. Trial of Sacituzumab Govitecan in Participants with Refractory/Relapsed Metastatic Triple-Negative Breast Cancer (TNBC) (ASCENT). NCT02574455; 2015. Available online: https://clinicaltrials.gov/ct2/show/NCT02574455 (accessed on 1 October 2022).
- GileadSciences. Study of Sacituzumab Govitecan-hziy (IMMU-132) in Adults with Epithelial Cancer. NCT01631552; 2012. Available online: https://clinicaltrials.gov/ct2/show/NCT01631552 (accessed on 1 October 2022).
- Faltas, B.; Goldenberg, D.M.; Ocean, A.J.; Govindan, S.V.; Wilhelm, F.; Sharkey, R.M.; Hajdenberg, J.; Hodes, G.; Nanus, D.M.; Tagawa, S.T. Sacituzumab govitecan, a novel antibody–drug conjugate, in patients with metastatic platinum-resistant urothelial carcinoma. Clin. Genitourin. Cancer 2016, 14, e75–e79. [Google Scholar] [CrossRef]
- GileadSciences. Study of Sacituzumab Govitecan in Participants with Urothelial Cancer That Cannot Be Removed or Has Spread (TROPHY U-01). NCT03547973; 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03547973 (accessed on 1 October 2022).
- GileadSciences. Study of Sacituzumab Govitecan-Hziy versus Treatment of Physician’s Choice in Participants with HR+/HER2- Metastatic Breast Cancer (TROPiCS-02). NCT03901339; 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT03901339 (accessed on 1 October 2022).
- GileadSciences. Study of Sacituzumab Govitecan-hziy in Metastatic Solid Tumors (TROPiCS-03). NCT03964727; 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT03964727 (accessed on 1 October 2022).
- Markham, A. Belantamab mafodotin: First approval. Drugs Future 2020, 80, 1607–1613. [Google Scholar] [CrossRef]
- GlaxoSmithKline. A Study to Investigate the Efficacy and Safety of Two Doses of GSK2857916 in Participants with Multiple Myeloma Who Have Failed Prior Treatment with an Anti-CD38 Antibody. NCT03525678; 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03525678 (accessed on 1 October 2022).
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.-O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- GlaxoSmithKline. Dose Escalation Study to Investigate the Safety, Pharmacokinetics, Pharmacodynamics, Immunogenicity and Clinical Activity of GSK2857916. NCT02064387; 2014. Available online: https://clinicaltrials.gov/ct2/show/NCT02064387 (accessed on 1 October 2022).
- Jain, N.; Stock, W.; Zeidan, A.; Atallah, E.; McCloskey, J.; Heffner, L.; Tomlinson, B.; Bhatnagar, B.; Feingold, J.; Ungar, D.; et al. Loncastuximab tesirine, an anti-CD19 antibody-drug conjugate, in relapsed/refractory B-cell acute lymphoblastic leukemia. Blood Adv. 2020, 4, 449–457. [Google Scholar] [CrossRef]
- Xu, B. Loncastuximab tesirine: An effective therapy for relapsed or refractory diffuse large B-cell lymphoma. Eur. J. Clin. Pharmacol. 2022, 78, 707–719. [Google Scholar] [CrossRef]
- ADC Therapeutics, S.A. Safety and Antitumor Activity Study of Loncastuximab Tesirine and Durvalumab in Diffuse Large B-Cell, Mantle Cell, or Follicular Lymphoma. NCT03685344; 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03685344 (accessed on 1 October 2022).
- ADC Therapeutics, S.A. Study to Evaluate Loncastuximab Tesirine with Rituximab Versus Immunochemotherapy in Participants with Relapsed or Refractory Diffuse Large B-Cell Lymphoma (LOTIS 5). NCT04384484; 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04384484 (accessed on 1 October 2022).
- D’Alessandro, G.; Barra, F.; Boutros, A.; Evangelisti, G.; Desideri, L.F.; Moioli, M.; Ferrero, S. Tisotumab vedotin. Antibody-drug conjugate directed to tissue factor, Tubulin polymerization inhibitor, Treatment of solid tumors. Drugs Future 2020, 45, 717–724. [Google Scholar] [CrossRef]
- Seagen Inc. A Trial of Tisotumab Vedotin in Cervical Cancer. NCT03438396; 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03438396 (accessed on 1 October 2022).
- Coleman, R.L.; Lorusso, D.; Gennigens, C.; González-Martín, A.; Randall, L.; Cibula, D.; Lund, B.; Woelber, L.; Pignata, S.; Forget, F.; et al. Efficacy and safety of tisotumab vedotin in previously treated recurrent or metastatic cervical cancer (innovaTV 204/GOG-3023/ENGOT-cx6): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021, 22, 609–619. [Google Scholar] [CrossRef] [PubMed]
- MedImmune_LLC. Moxetumomab Pasudotox for Advanced Hairy Cell Leukemia. NCT01829711; 2013. Available online: https://clinicaltrials.gov/ct2/show/NCT01829711 (accessed on 1 October 2022).
- Astellas Pharma Global Development, Inc. A Study of AGS-16C3F vs. Axitinib in Metastatic Renal Cell Carcinoma. NCT02639182; 2015. Available online: https://clinicaltrials.gov/ct2/show/NCT02639182 (accessed on 1 October 2022).
- Stokke, J.L.; Bhojwani, D. Antibody-Drug Conjugates for the Treatment of Acute Pediatric Leukemia. J. Clin. Med. 2021, 10, 3556. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Zhou, X.; Wang, X. Antibody-drug conjugates for the treatment of lymphoma: Clinical advances and latest progress. J. Hematol. Oncol. 2021, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- Yale University. Phase II Anetumab Ravtansine in Pre-Treated Mesothelin-Expressing Pancreatic Cancer. NCT03023722; 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03023722 (accessed on 1 October 2022).
- Ambrx, Inc. ARX788 in HER2-Positive, Metastatic Breast Cancer Subjects (ACE-Breast-03) (ACE-Breast03). NCT04829604; 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04829604 (accessed on 1 October 2022).
- Ambrx, Inc. A Dose-escalation, Expansion Study of ARX788, in Advanced Solid Tumors Subjects with HER2 Expression (ACE-Pan Tumor 01). NCT03255070; 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03255070 (accessed on 1 October 2022).
- BioAtla, Inc. Phase 2 CAB-AXL-ADC Safety and Efficacy Study in Adult and Adolescent Patients with Sarcoma. NCT03425279; 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03425279 (accessed on 1 October 2022).
- BioAtla, Inc. CAB-ROR2-ADC Safety and Efficacy Study in Patients with TNBC or Head & Neck Cancer (Ph1) and NSCLC or Melanoma (Ph2). NCT03504488; 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03504488 (accessed on 1 October 2022).
- Clarke, J.; Chu, S.-C.; Siu, L.L.; Machiels, J.-P.; Markman, B.; Heinhuis, K.; Millward, M.; Lolkema, M.; Patel, S.P.; Souza, P.d.; et al. Abstract B057: BMS-986148, an anti-mesothelin antibody-drug conjugate (ADC), alone or in combination with nivolumab demonstrates clinical activity in patients with select advanced solid tumors. Mol. Cancer Ther. 2019, 18 (Suppl. 12), B057. [Google Scholar] [CrossRef]
- Bristol-Myers Squibb. A Study of BMS-986148 in Patients with Select Advanced Solid Tumors. NCT02341625; 2015. Available online: https://clinicaltrials.gov/ct2/show/NCT02341625 (accessed on 1 October 2022).
- ADC Therapeutics, S.A. Study to Evaluate the Efficacy and Safety of Camidanlumab Tesirine (ADCT-301) in Patients with Relapsed or Refractory Hodgkin Lymphoma. NCT04052997; 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT04052997 (accessed on 1 October 2022).
- Sanofi. SAR3419 as Single Agent in Relapsed-Refractory Diffuse Large B-Cell Lymphoma (DLBCL) Patients (STARLYTE). NCT01472887; 2011. Available online: https://clinicaltrials.gov/ct2/show/NCT01472887 (accessed on 1 October 2022).
- Sanofi. SAR3419 in Acute Lymphoblastic Leukemia (MYRALL). NCT01440179; 2011. Available online: https://clinicaltrials.gov/ct2/show/NCT01440179 (accessed on 1 October 2022).
- CytomX Therapeutics. PROCLAIM-CX-2029: A Trial to Find Safe and Active Doses of an Investigational Drug CX-2029 for Patients with Solid Tumors or DLBCL. NCT03543813; 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03543813 (accessed on 1 October 2022).
- Daiichi Sankyo Co., Ltd. First-in-Human Study of DS-1062a for Advanced Solid Tumors (TROPION-PanTumor01). NCT03401385; 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03401385 (accessed on 1 October 2022).
- Seagen, Inc. A Safety Study of SGN-CD19A for B-Cell Lymphoma. NCT01786135; 2013. Available online: https://clinicaltrials.gov/ct2/show/NCT01786135 (accessed on 1 October 2022).
- Seagen, Inc. A Safety Study of SGN-CD19A for Leukemia and Lymphoma. NCT01786096; 2013. Available online: https://clinicaltrials.gov/ct2/show/NCT01786096 (accessed on 1 October 2022).
- Lassman, A.B.; van den Bent, M.J.; Gan, H.K.; Reardon, D.A.; Kumthekar, P.; Butowski, N.; Lwin, Z.; Mikkelsen, T.; Nabors, L.B.; Papadopoulos, K.P.; et al. Safety and efficacy of depatuxizumab mafodotin + temozolomide in patients with EGFR-amplified, recurrent glioblastoma: Results from an international phase I multicenter trial. Neuro-Oncology 2019, 21, 106–114. [Google Scholar] [CrossRef]
- AbbVie. Evaluating the Safety and Pharmacokinetics of ABT-414 for Subjects with Glioblastoma Multiforme. NCT01800695; 2013. Available online: https://clinicaltrials.gov/ct2/show/NCT01800695 (accessed on 1 October 2022).
- AbbVie. Adult Study: ABT-414 Alone or ABT-414 Plus Temozolomide vs. Lomustine or Temozolomide for Recurrent Glioblastoma Pediatric Study: Evaluation of ABT-414 in Children with High Grade Gliomas (INTELLANCE-2). NCT02343406; 2015. Available online: https://clinicaltrials.gov/ct2/show/NCT02343406 (accessed on 1 October 2022).
- AbbVie. A Study of ABT-414 in Participants with Newly Diagnosed Glioblastoma (GBM) with Epidermal Growth Factor Receptor (EGFR) Amplification (Intellance1). NCT02573324; 2015. Available online: https://clinicaltrials.gov/ct2/show/NCT02573324 (accessed on 1 October 2022).
- RemeGen Co., Ltd. Study of RC48-ADC Administered Intravenously to Subjects with HER2-Positive in Advanced Malignant Solid Tumors. NCT02881190; 2016. Available online: https://clinicaltrials.gov/ct2/show/NCT02881190 (accessed on 1 October 2022).
- Daiichi Sankyo Co., Ltd. Study of DS-7300a in Participants with Advanced Solid Malignant Tumors. NCT04145622; 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT04145622 (accessed on 1 October 2022).
- Ramalingam, S.; Lopez, J.; Mau-Sorensen, M.; Thistlethwaite, F.C.; Piha-Paul, S.; Gadgeel, S.; Drew, Y.; Janne, P.; Mansfield, A.; Chen, G.; et al. First-In-human phase 1/2 trial of anti-AXL antibody-drug conjugate (ADC) enapotamab vedotin (EnaV) in advanced NSCLC. J. Thorac. Oncol. 2019, 14, S209. [Google Scholar] [CrossRef]
- Genmab. Enapotamab Vedotin (HuMax-AXL-ADC) Safety Study in Patients with Solid Tumors. NCT02988817; 2016. Available online: https://clinicaltrials.gov/ct2/show/NCT02988817 (accessed on 1 October 2022).
- Celldex Therapeutics. A Study of Glembatumumab Vedotin as Monotherapy or in Combination with Immunotherapies in Patients with Advanced Melanoma. NCT02302339; 2014. Available online: https://clinicaltrials.gov/ct2/show/NCT02302339 (accessed on 1 October 2022).
- Herrera, A.F.; Patel, M.R.; Burke, J.M.; Advani, R.; Cheson, B.D.; Sharman, J.P.; Penuel, E.; Polson, A.G.; Liao, C.D.; Li, C.; et al. Anti-CD79B Antibody-Drug Conjugate DCDS0780A in Patients with B-Cell Non-Hodgkin Lymphoma: Phase 1 Dose-Escalation Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 1294–1301. [Google Scholar] [CrossRef]
- Hoffmann-La_Roche. A Study of Escalating Doses of DCDS0780A in Participants with Relapsed or Refractory B-Cell Non-Hodgkin’s Lymphoma. NCT02453087; 2015. Available online: https://clinicaltrials.gov/ct2/show/NCT02453087 (accessed on 1 October 2022).
- Seagen, Inc. Safety and Efficacy of SGN-LIV1A Plus Pembrolizumab for Patients with Locally-Advanced or Metastatic Triple-Negative Breast Cancer. NCT03310957; 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03310957 (accessed on 1 October 2022).
- Gerber, D.E.; Infante, J.R.; Gordon, M.S.; Goldberg, S.B.; Martín, M.; Felip, E.; Martinez Garcia, M.; Schiller, J.H.; Spigel, D.R.; Cordova, J.; et al. Phase Ia Study of Anti-NaPi2b Antibody-Drug Conjugate Lifastuzumab Vedotin DNIB0600A in Patients with Non-Small Cell Lung Cancer and Platinum-Resistant Ovarian Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 364–372. [Google Scholar] [CrossRef]
- ImmunoGen, Inc. A Study of IMGN901 for Patients with Advanced Solid Tumors and Extensive Stage Small Cell Lung Cancer. NCT01237678; 2010. Available online: https://clinicaltrials.gov/ct2/show/NCT01237678 (accessed on 1 October 2022).
- MedImmune LLC. MEDI2228 in Subjects with Relapsed/Refractory Multiple Myeloma (MEDI2228). NCT03489525; 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03489525 (accessed on 1 October 2022).
- Kunte, S.; Abraham, J.; Montero, A.J. Novel HER2–targeted therapies for HER2–positive metastatic breast cancer. Cancer Cell Int. 2020, 126, 4278–4288. [Google Scholar] [CrossRef]
- MedImmune LLC. A Multiple Ascending Dose Study of MEDI7247 in Participants with Selected Relapsed/Refractory Hematological Malignancies. NCT03106428; 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03106428 (accessed on 1 October 2022).
- ImmunoGen, Inc. A Study of Mirvetuximab Soravtansine vs. Investigator’s Choice of Chemotherapy in Women with Folate Receptor (FR) Alpha Positive Advanced Epithelial Ovarian Cancer (EOC), Primary Peritoneal or Fallopian Tube Cancer (FORWARD I). NCT02631876; 2015. Available online: https://clinicaltrials.gov/ct2/show/NCT02631876 (accessed on 1 October 2022).
- Eisai Inc. A Study of MORAb-202 in Participants with Solid Tumors. NCT03386942; 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03386942 (accessed on 1 October 2022).
- Shimizu, T.; Fujiwara, Y.; Yonemori, K.; Koyama, T.; Sato, J.; Tamura, K.; Shimomura, A.; Ikezawa, H.; Nomoto, M.; Furuuchi, K.; et al. First-in-Human Phase 1 Study of MORAb-202, an Antibody-Drug Conjugate Comprising Farletuzumab Linked to Eribulin Mesylate, in Patients with Folate Receptor-α-Positive Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 3905–3915. [Google Scholar] [CrossRef]
- Stathis, A.; Flinn, I.W.; Madan, S.; Maddocks, K.; Freedman, A.; Weitman, S.; Zucca, E.; Munteanu, M.C.; Lia Palomba, M. Safety, tolerability, and preliminary activity of IMGN529, a CD37-targeted antibody-drug conjugate, in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: A dose-escalation, phase I study. Investig. New Drugs 2018, 36, 869–876. [Google Scholar] [CrossRef] [PubMed]
- ImmunoGen, Inc. IMGN529 in Treating Patients with Relapsed or Refractory Non-Hodgkin’s Lymphoma and Chronic Lymphocytic Leukemia. NCT01534715; 2012. Available online: https://clinicaltrials.gov/ct2/show/NCT01534715 (accessed on 1 October 2022).
- OBI Pharma, I. Phase 1/2 Study of OBI-999 in Patients with Advanced Solid Tumors. NCT04084366; 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT04084366 (accessed on 1 October 2022).
- Daiichi Sankyo, I. U3-1402 in Metastatic or Unresectable Non-Small Cell Lung Cancer. NCT03260491; 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03260491 (accessed on 1 October 2022).
- Genentech, Inc. A Study of Pinatuzumab Vedotin (DCDT2980S) Combined with Rituximab or Polatuzumab Vedotin (DCDS4501A) Combined with Rituximab or Obinutuzumab in Participants with Relapsed or Refractory B-Cell Non-Hodgkin’s Lymphoma (NHL) (ROMULUS). NCT01691898; 2012. Available online: https://clinicaltrials.gov/ct2/show/NCT01691898 (accessed on 1 October 2022).
- Progenics Pharmaceuticals, Inc. A Study of PSMA ADC in Subjects with Metastatic Castration-Resistant Prostate Cancer (mCRPC). NCT01695044; 2012. Available online: https://clinicaltrials.gov/ct2/show/NCT01695044 (accessed on 1 October 2022).
- AbbVie. A Study of Rovalpituzumab Tesirine as Maintenance Therapy Following First- Line Platinum-Based Chemotherapy in Participants With Extensive Stage Small Cell Lung Cancer (MERU) (MERU). NCT03033511; 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03033511 (accessed on 1 October 2022).
- Gomez-Roca, C.A.; Boni, V.; Moreno, V.; Morris, J.C.; Delord, J.-P.; Calvo, E.; Papadopoulos, K.P.; Rixe, O.; Cohen, P.; Tellier, A.; et al. A phase I study of SAR566658, an anti CA6-antibody drug conjugate (ADC), in patients (Pts) with CA6-positive advanced solid tumors (STs)(NCT01156870). J. Clin. Oncol. 2016, 34, 2511. [Google Scholar] [CrossRef]
- Sanofi. First in Man Study of SAR566658 Administered in Patients with CA6-Positive and Refractory Solid Tumor. NCT01156870; 2010. Available online: https://clinicaltrials.gov/ct2/show/NCT01156870 (accessed on 1 October 2022).
- Sanofi. Study Evaluating Efficacy and Safety of SAR566658 Treatment in Patients with CA6 Positive Metastatic Triple Negative Breast Cancer. NCT02984683; 2016. Available online: https://clinicaltrials.gov/ct2/show/NCT02984683 (accessed on 1 October 2022).
- Klus Pharma Inc. Phase I-II, FIH, TROP2 ADC, Advanced Unresectable/Metastatic Solid Tumors, Refractory to Standard Therapies (A264). NCT04152499; 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT04152499 (accessed on 1 October 2022).
- Sutro Biopharma, Inc. Study of STRO-001, an Anti-CD74 Antibody Drug Conjugate, in Patients with Advanced B-Cell Malignancies. NCT03424603; 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03424603 (accessed on 1 October 2022).
- Byondis, B.V. A First-in-Human Dose-Escalation and Expansion Study with the Antibody-Drug Conjugate SYD1875. NCT04202705; 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT04202705 (accessed on 1 October 2022).
- AbbVie. A Study Evaluating the Safety, Pharmacokinetics (PK), and Preliminary Efficacy of ABBV-399 in Participants with Advanced Solid Tumors. NCT02099058; 2014. Available online: https://clinicaltrials.gov/ct2/show/NCT02099058 (accessed on 1 October 2022).
- Daiichi Sankyo Inc. Trastuzumab Deruxtecan (DS-8201a) Versus Investigator’s Choice for HER2-Low Breast Cancer That Has Spread or Cannot Be Surgically Removed [DESTINY-Breast04]. NCT03734029; 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03734029 (accessed on 1 October 2022).
- Byondis, B.V.; SYD985 vs. Physician’s Choice in Participants with HER2-positive Locally Advanced or Metastatic Breast Cancer (TULIP). NCT03262935; 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03262935 (accessed on 1 October 2022).
- Triphase Research and Development III Corp. Study of TRPH-222 in Patients with Relapsed and/or Refractory B-Cell Lymphoma. NCT03682796; 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03682796 (accessed on 1 October 2022).
- Mersana Therapeutics. First-in-Human Study of XMT-1536 in Cancers Likely to Express NaPi2b. NCT03319628; 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03319628 (accessed on 1 October 2022).
- Pierre Fabre Medicament. A Study of a New Investigational Medicinal Product to Treat Patients with Advanced or Metastatic Solid Tumors (Ulysse). NCT03316638; 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03316638 (accessed on 1 October 2022).
- PubChem. Compound Summary of Denintuzumab Mafodotin. 2014. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Denintuzumab-mafodotin (accessed on 1 November 2022).
- Fathi, A.T.; Borate, U.; DeAngelo, D.J.; O’Brien, M.M.; Trippett, T.; Shah, B.D.; Hale, G.A.; Foran, J.M.; Silverman, L.B.; Tibes, R.; et al. A phase 1 study of denintuzumab mafodotin (SGN-CD19A) in adults with relapsed or refractory B-lineage acute leukemia (B-ALL) and highly aggressive lymphoma. Blood 2015, 126, 1328. [Google Scholar] [CrossRef]
- Rinnerthaler, G.; Gampenrieder, S.P.; Greil, R. HER2 Directed Antibody-Drug-Conjugates beyond T-DM1 in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 1115. [Google Scholar] [CrossRef] [PubMed]
- PubChem. Compound Summary for CID 91810695, Mirvetuximab Soravtansine. 2015. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Mirvetuximab-soravtansine (accessed on 1 November 2022).
- Moore, K.N.; Vergote, I.; Oaknin, A.; Colombo, N.; Banerjee, S.; Oza, A.; Pautier, P.; Malek, K.; Birrer, M.J. FORWARD I: A Phase III study of mirvetuximab soravtansine versus chemotherapy in platinum-resistant ovarian cancer. Future Oncol. 2018, 14, 1669–1678. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Birrer, M.J.; O’Malley, D.M.; Moore, K.N.; Konner, J.; Gilbert, L.; Martin, L.P.; Bauer, T.M.; Oza, A.M.; Malek, K.; et al. Evaluation of Prophylactic Corticosteroid Eye Drop Use in the Management of Corneal Abnormalities Induced by the Antibody–Drug Conjugate Mirvetuximab SoravtansinePathogenesis and Management of ADC-induced Ocular Toxicity. Clin. Cancer Res. 2019, 25, 1727–1736. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Koyama, K.; Kamai, Y.; Hirotani, K.; Ogitani, Y.; Zembutsu, A.; Abe, M.; Kaneda, Y.; Maeda, N.; Shiose, Y.; et al. A Novel HER3-Targeting Antibody–Drug Conjugate, U3-1402, Exhibits Potent Therapeutic Efficacy through the Delivery of Cytotoxic Payload by Efficient InternalizationPreclinical Evaluation of U3-1402, a HER3-Targeting ADC. Clin. Cancer Res. 2019, 25, 7151–7161. [Google Scholar] [CrossRef]
- Jänne, P.A.; Baik, C.; Su, W.-C.; Johnson, M.L.; Hayashi, H.; Nishio, M.; Kim, D.-W.; Koczywas, M.; Gold, K.A.; Steuer, C.E.; et al. Efficacy and Safety of Patritumab Deruxtecan (HER3-DXd) in EGFR Inhibitor-Resistant, EGFR-Mutated Non-Small Cell Lung Cancer. Cancer Discov. 2021, 12, 74–89. [Google Scholar] [CrossRef]
- Daiichi Sankyo Inc. HERTHENA-Lung01: Patritumab Deruxtecan in Subjects with Metastatic or Locally Advanced EGFR-Mutated Non-Small Cell Lung Cancer. NCT04619004; 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04619004 (accessed on 1 October 2022).
- Angevin, E.; Strickler, J.H.; Weekes, C.D.; Heist, R.S.; Morgensztern, D.; Nemunaitis, J.J.; Fan, X.; Beaulieu, J.; Motwani, M.; Afar, D.E.; et al. Phase I study of ABBV-399, a c-Met antibody-drug conjugate (ADC), as monotherapy and in combination with erlotinib in patients (pts) with non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2017, 35, 2509. [Google Scholar] [CrossRef]
- Kollmannsberger, C.; Choueiri, T.K.; Heng, D.Y.; George, S.; Jie, F.; Croitoru, R.; Poondru, S.; Thompson, J.A. A Randomized Phase II Study of AGS-16C3F Versus Axitinib in Previously Treated Patients with Metastatic Renal Cell Carcinoma. Oncologist 2021, 26, 182-e361. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Lioure, B.; Kim, S.K.; Atallah, E.; Leguay, T.; Kelly, K.; Marolleau, J.-P.; Escoffre-Barbe, M.; Thomas, X.G.; Cortes, J.; et al. A phase II study of coltuximab ravtansine (SAR3419) monotherapy in patients with relapsed or refractory acute lymphoblastic leukemia. Clin. Lymphoma Myeloma Leuk. 2016, 16, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Trnĕný, M.; Verhoef, G.; Dyer, M.J.; Yehuda, D.B.; Patti, C.; Canales, M.; Lopez, A.; Awan, F.T.; Montgomery, P.G.; Janikova, A.; et al. A phase II multicenter study of the anti-CD19 antibody drug conjugate coltuximab ravtansine (SAR3419) in patients with relapsed or refractory diffuse large B-cell lymphoma previously treated with rituximab-based immunotherapy. Haematologica 2018, 103, 1351. [Google Scholar] [CrossRef]
- Ott, P.A.; Pavlick, A.C.; Johnson, D.B.; Hart, L.L.; Infante, J.R.; Luke, J.J.; Lutzky, J.; Rothschild, N.E.; Spitler, L.E.; Cowey, C.L.; et al. A phase 2 study of glembatumumab vedotin, an antibody-drug conjugate targeting glycoprotein NMB, in patients with advanced melanoma. Cancer Cell Int. 2019, 125, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Kaye, F.J.; Spigel, D.R.; Kudrik, F.J.; Ponce, S.; Ellis, P.M.; Majem, M.; Lorigan, P.; Gandhi, L.; Gutierrez, M.E.; et al. Phase 1/2 study of the CD56-targeting antibody-drug conjugate lorvotuzumab mertansine (IMGN901) in combination with carboplatin/etoposide in small-cell lung cancer patients with extensive-stage disease. Clin. Lung Cancer 2017, 18, 68–76. [Google Scholar] [CrossRef] [PubMed]
- PubChem. Compound Summary for CID 131954443, Rovalpituzumab Tesirine. 2018. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/131954443 (accessed on 2 November 2022).
- AbbVie. AbbVie Discontinues Rovalpituzumab Tesirine (Rova-T) Research and Development Program. 2019. Available online: https://news.abbvie.com/news/press-releases/abbvie-discontinues-rovalpituzumab-tesirine-rova-t-research-and-development-program.htm (accessed on 1 November 2022).
- Nejadmoghaddam, M.-R.; Minai-Tehrani, A.; Ghahremanzadeh, R.; Mahmoudi, M.; Dinarvand, R.; Zarnani, A.-H. Antibody-drug conjugates: Possibilities and challenges. Avicenna J. Med. Biotechnol. 2019, 11, 3. [Google Scholar]
- Plenderleith, I.H. Treating the treatment: Toxicity of cancer chemotherapy. Can. Fam. Physician 1990, 36, 1827. [Google Scholar] [PubMed]
- Elder, D.; Treichler, S. Antibody-drug conjugates: Challenges, solutions and future potential. Eur. Pharm. Rev. 2021, 26, 14–18. [Google Scholar]
- Hafeez, U.; Parakh, S.; Gan, H.K.; Scott, A.M. Antibody–drug conjugates for cancer therapy. Molecules 2020, 25, 4764. [Google Scholar] [CrossRef]
- Staudacher, A.H.; Brown, M.P. Antibody drug conjugates and bystander killing: Is antigen-dependent internalisation required? Br. J. Cancer 2017, 117, 1736–1742. [Google Scholar] [CrossRef]
- Yurkovetskiy, A.V.; Bodyak, N.D.; Yin, M.; Thomas, J.D.; Clardy, S.M.; Conlon, P.R.; Stevenson, C.A.; Uttard, A.; Qin, L.; Gumerov, D.R. Dolaflexin: A Novel Antibody–Drug Conjugate Platform Featuring High Drug Loading and a Controlled Bystander EffectDolaflexin: A Novel Auristatin-based ADC Platform. Mol. Cancer Ther. 2021, 20, 885–895. [Google Scholar] [CrossRef]
- Farràs, M.; Miret, J.; Camps, M.; Román, R.; Martínez, Ó.; Pujol, X.; Erb, S.; Ehkirch, A.; Cianferani, S.; Casablancas, A. Homogeneous antibody-drug conjugates: DAR 2 anti-HER2 obtained by conjugation on isolated light chain followed by mAb assembly. MAbs 2020, 12, 1702262. [Google Scholar] [CrossRef] [PubMed]
- Szot, C.; Saha, S.; Zhang, X.M.; Zhu, Z.; Hilton, M.B.; Morris, K.; Seaman, S.; Dunleavey, J.M.; Hsu, K.-S.; Yu, G.-J.; et al. Tumor stroma–targeted antibody-drug conjugate triggers localized anticancer drug release. J. Clin. Investig. 2018, 128, 2927–2943. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Chari, R.V. Antibody conjugate therapeutics: Challenges and potential. Clin. Cancer Res. 2011, 17, 6389–6397. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive ReviewAntibody–Drug Conjugates in Cancer Immunotherapy. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef]
- Su, D.; Zhang, D. Linker design impacts antibody-drug conjugate pharmacokinetics and efficacy via modulating the stability and payload release efficiency. Front. Pharmacol. 2021, 12, 687926. [Google Scholar] [CrossRef]
- Melo, R.; Lemos, A.; Preto, A.; de Almeida, J.G.; Correia, J.; Sensoy, O.; Moreira, I. Computational Approaches in Antibody-Drug Conjugate Optimization for Targeted Cancer Therapy. Curr. Top. Med. Chem. 2018, 18, 1091–1109. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R. Computational Studies on Antibody Drug Conjugates (ADCs) for Precision Oncology. ChemistrySelect 2022, 7, e202202259. [Google Scholar] [CrossRef]
- Zhang, Z.; Lu, M.; Qin, Y.; Gao, W.; Tao, L.; Su, W.; Zhong, J. Neoantigen: A New Breakthrough in Tumor Immunotherapy. Front. Immunol. 2021, 12, 672356. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.R.; Tran, S.; Jain, M.; Narendran, A. Neoantigen Cancer Vaccines: Generation, Optimization, and Therapeutic Targeting Strategies. J. Clin. Oncol. 2022, 10, 196. [Google Scholar] [CrossRef]
- Butterfield, L.H. Cancer vaccines. BMJ 2015, 350, h988. [Google Scholar] [CrossRef]
- Xu, P.; Luo, H.; Kong, Y.; Lai, W.-F.; Cui, L.; Zhu, X. Cancer neoantigen: Boosting immunotherapy. Biomed. Pharmacother. 2020, 131, 110640. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.-Y. Therapeutic cancer vaccines: Past, present, and future. Adv. Cancer Res. 2013, 119, 421–475. [Google Scholar] [PubMed]
- Li, L.; Goedegebuure, S.; Gillanders, W.E. Preclinical and clinical development of neoantigen vaccines. Ann. Oncol. 2017, 28, xii11–xii17. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components of ADC | ||
|---|---|---|
| Antibody | Subunit | Function |
| Antigen-binding Fragment (Fab) | Mediate antigen recognition expressed on tumour cells. | |
| Constant Fragment (Fc) | Facilitate binding of Fab to immune cells. | |
| Linker | Type | Mechanism of Drug Release |
| Cleavable | Cleavage depends on the physiological environment.
| |
| Non-cleavable | It depends on lysosomal proteolytic degradation and requires optimal trafficking to lysosome. Thioether linker is cleaved by proteases in the cytosolic milieu, e.g., Trastuzumab Emtansine (T–DM1). | |
| Cytotoxic Payload | Target Site | Examples of Payload with Mechanism of Action |
| DNA |
| |
| Tubulin |
| |
| ADC | Antibody | Target Antigen | Linker | Cytotoxic Payload | Indication | Year of Approval |
|---|---|---|---|---|---|---|
| Gemtuzumab ozogamicin | Humanised IgG4 | CD33 | Acid–labile hydrazone | Calicheamicin | Relapse or refractory AML and newly diagnosed CD33+ AML | Approved in 2000, withdrawn in 2010 but then reapproved in 2017 for relapsed/refractory malignancies FDA approved for newly diagnosed CD33+ AML in ≥1-month paediatric patient |
| Brentuximab vedotin | Chimeric IgG1 | CD30 | Protease-cleavable dipeptide | MMAE | HL, ALCL and different subtypes of T-cell lymphomas | FDA accelerated approval in 2011 In 2018, FDA approved the treatment of previously untreated stage III-IV HL and previously untreated ALCL and other CD30+ peripheral T-cell lymphomas. |
| Ado-trastuzumab emtansine | Humanised IgG1 | HER2 | SMCC | DM1 | Metastatic HER2+ breast cancer, previously treated with trastuzumab and a taxane and as adjuvant treatment for HER2+ early breast cancer with the residual invasive disease after neoadjuvant taxane and trastuzumab | FDA approved in 2013 FDA approved for adjuvant treatment in 2019 |
| Inotuzumab Ozogamicin | Humanised IgG4 | CD22 | Acid–labile hydrazone | Calicheamicin | Relapsed or refractory B-cell precursor ALL | FDA approved in February 2017 |
| Monotherapy treatment of relapsed or refractory CD22-positive B-cell precursor | EMA approved in June 2017 | |||||
| Polatuzumab vedotin | Humanised IgG1 | CD79b | Cleavable dipeptide | MMAE | Relapsed or refractory diffuse large B-cell lymphoma | FDA accelerated approval in 2019 |
| Enfortumab vedotin | Fully human IgG1 | Nectin 4 | Protease-cleavable dipeptide (Val–Cit) linker | MMAE | Locally advanced or metastatic urothelial cancer in adult patients who received prior treatment with a PD-1/L1 inhibitor and platinum-based chemotherapy in neoadjuvant/adjuvant setting | FDA accelerated approved in 2019 |
| Trastuzumab deruxtecan | Humanised IgG1 | HER2/ERB2 | Protease-cleavable tetra-peptide (Gly–Gly–Phe–Gly) linker | DXd | Unresectable locally advanced or metastatic HER2+ breast cancer, previously treated with trastuzumab and a taxane, adjuvant treatment for HER2+ early breast cancer with the residual invasive disease after neoadjuvant taxane and trastuzumab | FDA accelerated approved in 2019 |
| Sacituzumab govitecan | Humanised IgG1 | TROP-2 | Acid–labile ester (CL2 linker) | SN-38 | Triple-negative breast cancer, urothelial and other cancers | FDA accelerated approval in April 2020 for mTNBC |
| FDA regular approval for in April 2021 TNBC | ||||||
| FDA accelerated approval in April 2021 for mUC | ||||||
| Belantamab mafodotin | Humanised IgG1 | BCMA | Protease- resistant maleimidohexanoic linker | MMAF | Relapsed or refractory multiple myeloma in adults who have received at least four prior therapies | FDA accelerated approval in 2020 |
| Loncastuximab tesirine- lpyl | Humanised IgG1 | CD19 | Valine–alanine dipeptide | PDB dimer | Relapsed or refractory large B-cell lymphoma | FDA accelerated approval in April 2021 |
| Tisotumab vedotin | IgG1 | Tissue factor (TF) | mc–val–cit–PABC | MMAE | Recurrent or metastatic cervical cancer in patients with disease progression during or following chemotherapy | FDA accelerated approval in September 2021 |
| Moxetumomab Pasudotox | - | CD22 | Recombinant covalently fused | Pseudotox | Relapsed or refractory HCL who received at least two prior systemic therapies | FDA approval in September 2018 |
| Compound Name | Target Antigen (A), Linker (L), Cytotoxin (C) | Indication | Phase | Efficacy | Side Effect | Reference |
|---|---|---|---|---|---|---|
| AGS-16C3F | A: ENPP3 L: Non-cleavable maleimidocaproyl linker C: MMAF | Renal cell carcinoma | II | Overall Survival 13.1 months Progression-Free Survival 2.9 months | Fatigue Nausea Blurred vision | NCT02639182 Status: Completed [68] |
| AGS62P1 | A: FLT3 L: Non-cleavable linker C: AGL-0182-30 | Acute Myeloid Leukemia | I | N/A Last Update Posted: 18 June 2019 | NCT02864290 Status: Terminated (due to lack of efficacy) [69] | |
| AGS67E | A: CD37 L: Protease cleavable linker C: MMAE | Refractory or Relapsed Lymphoid Malignancies | I | Overall Response Rate 22% Complete Response Rate 14% | Peripheral neuropathy Neutropenia | NCT02175433 Status: Completed [70] |
| Anetumab Ravtansine (Bay 94-9343) | A: Mesothelin L: Reducible SPDB C: DM4 | Mesothelin-expressing Pancreatic Cancer | II | Response Rate Progressive disease: 85.7% Stable disease: 14.3% Time to Progression Median: 63.5 days | Anaemia Cardiac disorders Eye disorders | NCT03023722 Status: Completed [71] |
| ARX788 | A: HER2 L: pAcF C: MMAF | HER2 Positive Metastatic Breast Cancer | II | N/A (Ongoing) Last Update Posted: 11 February 2022 Estimated Study Completion Date: February 2025 | NCT04829604 Status: Recruiting [72] | |
| Breast Neoplasms, Gastric Neoplasm and Solid tumours | I | N/A (Ongoing) Last Update Posted: 2 September 2021 Estimated Study Completion Date: March 2023 | NCT03255070 Status: Recruiting [73] | |||
| BA3011 (CAB-AXL-ADC) | A: AXL L: Cleavable linker C: MMAE | Solid tumours | I/II | N/A (Ongoing) Last Update Posted: 13 September 2021 Estimated Study Completion Date: January 2022 | NCT03425279 Status: Recruiting [74] | |
| BA3021 (CAB-ROR2-ADC) | A: Ror2 L: N/A C: N/A | Solid tumours | I/II | N/A (Ongoing) Last Update Posted: 13 September 2021 Estimated Study Completion Date: 30 June 2023 | NCT03504488 Status: Recruiting [75] | |
| BMS-986148 | A: Mesothelin L: N/A C: Tubulysin | Advance solid tumours: mesothelioma, non-small cell lung cancer, ovarian cancer, pancreatic cancer, and gastric cancer. | I/II | Objective Response Rate Monotherapy: 6% Combo-therapy: 20% [76] | AST increased ALT increased Fatigue Nausea [76] | NCT02341625 Status: Terminated (due to business reasons not related to safety) [77] |
| Camidanlumab Tesirine (ADCT-301) | A: CD25 L: Cleavable valine–alanine C: PBD dimer | Relapsed or refractory Hodgkin’s Lymphoma | II | N/A (Ongoing) Last Update Posted: 5 February 2021 Estimated Study Completion Date: 15 May 2024 | NCT04052997 Status: Active, not-recruiting [78] | |
| Coltuximab ravtansine (SAR3419) | A: CD19 L: Cleavable disulfide, C: DM4 | Diffuse large B-cell lymphoma | II | Overall Response Rate 43.9% Complete Responses: 14.6% Partial Responses: 29.3% | AstheniaFatigue Nausea Diarrhoea Cough AST increased Hematologic disorders | NCT01472887 Status: Completed [79] |
| Acute Lymphoblastic Leukaemia | Objective Response Rate 25.47% Duration of Response 1.94 months | Pyrexia Diarrhoea Nausea Haematologic disorders | NCT01440179 Status: Terminated (due to very modest activity compared to competitors) [80] | |||
| CX-2029 | A: CD71 L: Cleavable protease C: MMAE | Solid tumours, diffuse large B-cell lymphoma | I/II | N/A (Ongoing) Last Update Posted: 15 December 2021 Estimated Study Completion Date: December 2022 | NCT03543813 Status: Active, not-recruiting [81] | |
| Datopotamab deruxtecan DS-1062a | A: TACSTD L: Tetrapeptide C: Topoisomerase I inhibitor | Advanced Solid Tumours | I | N/A (Ongoing) Last Update Posted: 26 January 2022 Estimated Study Completion Date: 1 January 2024 | NCT03401385 Status: Recruiting [82] | |
| Denintuzumab mafodotin (SGN-CD19A) | A: CD19 L: Non-cleavable maleimidocaproxyl-valine-citrulline C: MMAF | B-cell lymphoma | I | Complete Remission Q3wk dosing relapsed pts: 7 Partial Remission Q3wk dosing relapsed pts: 4 Complete Remission Rate Q3wk dosing relapsed pts: 32% Q3wk dosing refractory pts: 10% | Blurred vision Dry eye Fatigue Keratopathy Constipation Photophobia Nausea | NCT01786135 Status: Completed [83] |
| Leukaemia and Lymphoma | Complete Remission Weekly dosing: 6 Q3wk dosing: 3 Partial Remission Weekly dosing: 1 Complete Remission Rate Weekly dosing: 19% Q3wk dosing: 35% | Pyrexia Nausea Fatigue Headache Chills Vomiting Blurred vision Anaemia | NCT01786096 Status: Completed [84] | |||
| Depatuxizumab mafodotin (ABT-414) | A: EGFR L: Non-cleavable maleimidocaproyl linker C: MMAF | Glioblastoma Multiforme | I | Objective Response Rate 14.3% 6-month Progression-Free Survival Rate 25.2% 6-month Overall Survival Rate 69.1% [85] | Blurred vision Fatigue Photophobia [85] | NCT01800695 Status: Completed [86] |
| Glioblastoma | II | Overall Survival 15.5 months Progression-Free Survival 3.5 months | Keratitis Fatigue Dry eye Headache | NCT02343406 Status: Completed [87] | ||
| Glioblastoma and Gliosarcoma | II/III | N/A (Ongoing) Last Update Posted: 11 January 2022 Estimated Study Completion Date: 31 December 2021 | NCT02573324 Status: Active, not recruiting [88] | |||
| Disitamab vedotin (RC-48) | A: HER2/neu L: Cleavable linker C: MMAE | HER2-positive advanced malignant solid tumours | I | Overall Response Rate: 21% Disease Control Rate 49.1% | Neutropenia Hypoesthesia Increased conjugation of blood bilirubin | NCT02881190 Status: Completed [89] |
| DS-7300a (B7-H3 ADC) | A: B7-H3 (CD276) L: Cleavable peptide linker C: DXd (exatecan derivative) | Advanced and malignant solid tumours | I/II | N/A (Ongoing) Last Update Posted: 14 December 2021 Estimated Study Completion Date: 1 October 2023 | NCT04145622 Status: Recruiting [90] | |
| Enapotamab vedotin (HuMax-AXL-ADC) | A: AXL L: Cleavable mc–val–cit–PABC C: MMAE | Non-small cell lung cancer | I/II | Objective Response Rate 19% Disease Control Rate 50% [91] | Constipation Nausea Decreased appetite [91] | NCT02988817 Status: Completed [92] |
| Epratuzumab tesirine (ADCT-602) | A: CD22 L: Cleavable valine–alanine peptide C: PBD dimer | Recurrent or refractory B-cell acute lymphoblastic leukaemia | I/II | N/A (Ongoing) Last Update Posted: 26 January 2022 Estimated Study Completion Date: 31 December 2022 | NCT03698552 Status: Recruiting [25] | |
| Glembatumumab vedotin (CDX-011) | A: gpNMB, L: Cleavable Dipeptide C: MMAE | Melanoma | II | Overall Survival 8.8 months Progression-Free Survival 4.4 months | Alopecia Peripheral sensory neuropathy Fatigue Nausea Decreased appetite | NCT02302339 Status: Terminated (due to development of glembatumumab vedotin was discontinued) [93] |
| Iladatuzumab vedotin (DCDS0780A) | A: CD79B L: Cleavable linker C: MMAE | Non-Hodgkin’s Lymphoma | I | Response rate 47% Complete responses: 28% Partial responses: 18% [94] | Blurred vision Fatigue Corneal deposits Neutropenia Nausea Peripheral neuropathy [94] | NCT02453087 Status: Completed [95] |
| Ladiratuzumab vedotin (SGN-LIV1A) | A: Zinc transporter LIV-1 L: Protease-cleavable linker C: MMAE | Locally advanced, Metastatic triple-negative breast cancer | I/II | N/A (Ongoing) Last Update Posted: 11 January 2022 Estimated Study Completion Date: 31 December 2024 | NCT03310957 Status: Recruiting [96] | |
| Lifastuzumab vedotin (DNIB0600A) | A: Phosphate-sodium cotransporter SLC34A2 L: Cleavable maleimidocaproyl-valyl-citrullinyl-p-aminobenzyloxycarbonyl C: MMAE | Non-Small Cell Lung Cancer (NSCLC) and Platinum Resistant Ovarian Cancer | I | Duration of Response NSCLC: 5 months PROC: 11.4 months | Fatigue Nausea Decreased appetite Vomiting Peripheral sensory neuropathy | NCT01363947 Status: Completed [97] |
| Lorvotuzumab mertansine (IMGN901) | A: CD56 L: Cleavable disulfide C: DM1 | Small Cell Lung Cancer | II | Overall Survival 10.1 months Progression-Free Survival 6.2 months | Peripheral Sensory Neuropathy Anaemia Neutropenia Fatigue | NCT01237678 Status: Terminated(Study was stopped earlydue to lack of efficacy signal and safety concerns) [98] |
| MEDI2228 | A: BCMA L: Cleavable linker C: PBD dimer | Relapsed/Refractory Multiple Myeloma | I | N/A (Ongoing) Last Update Posted: 11 November 2021 Estimated Study Completion Date: 30 June 2022 | NCT03489525 Status: Active, not recruiting [99] | |
| MEDI4276 | A: HER2; ERBB2 L: Maleimidocaproyl linker C: MMETA (AZ13599185) | HER2-expressing Advanced Solid Tumours | I | N/A Last Update Posted: 18 June 2019 | Nausea Fatigue Transaminitis | NCT02576548 Status: Completed [100] |
| MEDI7247 | A: ASCT2 L: Cleavable linker C: PBD dimer | Haematological Malignancies | I | Progression-Free Survival AML pts: 0.8 months DLBCL pts: 2.8 months Overall Survival AML pts: 1.3 months DLBCL pts: 5.4 months [101] | Thrombocytopenia Hypophosphataemia Anaemia Neutropenia [101] | NCT03106428 Status: Completed [101] |
| Mirvetuximab soravtansine (IMGN853) | A: FOLR1 L: Cleavable disulfide C: DM4 | Ovarian, Endometrial, Non-small cell lung cancer | III | Cancer Antigen-125 Response 51% Objective Response Rate 22% | Eye disorder GI disorder Neuropathy disorder | NCT02631876 Status: Completed [102] |
| MORAb-202 | A: FOLR1 L: Cathepsin-cleavable linker C: Eribulin mesylate | FRα- positive solid tumours | I | Objective Response Rate Complete responses: 5% Partial responses: 41% Stable Disease 36% [103] | Leukopenia Neutropenia Interstitial lung disease [104] | NCT03386942 Status: Active, not recruiting [103] |
| Naratuximab emtansine (IMGN529) | A: CD37 L: Non-reducible thioether bond C: DM1 | Non-Hodgkin’s Lymphoma and Chronic Lymphocytic Leukaemia | I | Overall Response Rate: 13% Complete Responses: 1 Partial Responses: 4 [105] | Fatigue Neutropenia Pyrexia Thrombocytopenia Febrile neutropenia Peripheral neuropathy [105] | NCT01534715 Status: Completed [106] |
| OBI-999 (Anti-Globo H ADC) | A: Globo H L: Site-specific ThioBridge C: MMAE | Locally advanced solid tumours | I/II | N/A (Ongoing) Last Update Posted: 16 December 2021 Estimated Study Completion Date: 9 December 2023 | NCT04084366 Status: Recruiting [107] | |
| Patritumab deruxtecan (U3-1402, HER3-DXd) | A: HER3 L: Cleavable peptide C: Deruxtecan | EGFR inhibitor-resistant and EGFR-mutated non-small cell lung cancer | I | Objective Response Rate Confirmed: 39% (2% complete responses, 37% partial responses) Time to Response 2.6 months | Thrombocytopenia Neutropenia Fatigue | NCT03260491 Status: Active, not recruiting [108] |
| Pinatuzumab Vedotin (DCDT2980S) | A: CD22 L: Cleavable mc–val–cit–PABC C: MMAE | Diffuse Large B-Cell Lymphoma | II | Overall Survival 16.493 months Progression-Free Survival 5.388 months | Fatigue Peripheral neuropathy Diarrhoea Neutropenia | NCT01691898 Status: Completed [109] |
| PSMA ADC (Prostate-Specific Membrane Antigen Antibody Drug Conjugate) | A: PSMA L: Cleavable Dipeptide C: MMAE | Prostate Cancer | II | PSA Response (>50% Decrease) Chemotherapy-experienced:11% Chemotherapy-naive: 21% Overall Radiologic Response (Stable Disease) Chemotherapy-experienced: 61% Chemotherapy-naive: 69% | Anaemia GI disorders Fatigue Decreased appetite. Peripheral neuropathy | NCT01695044 Status: Completed [110] |
| Rovalpituzumab tesirine (Rova-T) | A: DLL3 L: Cleavable dipeptide C: PBD dimer | Small-cell lung cancer | III | Overall Survival Rova-T/Dexamethasone: 8.8 months Placebo 9.89 months | Thrombocytopenia Pericardial effusion Abdominal pain Pneumonia | NCT03033511 Status: Terminated [111] |
| SAR566658 | A: CA6 L: SPDB C: DM4 | Neoplasm Malignant | I | Stable Disease 39% [112] | Fatigue Peripheral neuropathy GI disorders Neutropenia [112] | NCT01156870 Status: Completed [113] |
| Triple-Negative Breast Cancer | II | - | Keratitis Asthenia Keratopathy | NCT02984683 Status: Terminated (Limited clinical benefit combined serious ophthalmological event) [114] | ||
| SKB264 | A: TROP2 L: N/A C: Belotecan derivatives | Locally advanced unresectable/metastatic solid tumours | I/II | N/A (Ongoing) Last Update Posted: 11 January 2022 Estimated Study Completion Date: December 2022 | NCT04152499 Status: Recruiting [115] | |
| STRO-001 | A: CD74 L: Non-cleavable dibenzocyclooctyne (DBCO) linker C: Maytansoid | B-Cell Malignancies | I | N/A (Ongoing) Last Update Posted: 19 October 2020 Estimated Study Completion Date: November 2023 | NCT03424603 Status: Recruiting [116] | |
| SYD1875 | A: 5T4 L: Cleavable valine-citrulline-seco C: Duocarmycin analogues | Solid tumour | I | N/A (Ongoing) Last Update Posted: 13 January 2022 Estimated Study Completion Date: January 2024 | NCT04202705 Status: Active, not recruiting [117] | |
| Telisotuzumab vedotin (ABBV-399) | A: ABT-700 L: Cleavable dipeptide C: MMAE | Advanced solid tumours cancer and non-small cell lung cancer | I | N/A (Ongoing) Last Update Posted: 1 February 2022 Estimated Study Completion Date: 26 December 2024 | NCT02099058 Status: Recruiting [118] | |
| Trastuzumab Deruxtecan (DS-8201a) | A: ERBB2 L: Cleavable tetrapeptide linker C: Topoisomerase I inhibitor | HER2-Low Advanced Breast Cancer | III | Progression-Free Survival 9.9 months Overall Survival 23.4 months | Nausea Alopecia Neutropenia Anaemia Fatigue | NCT03734029 Status: Active, not recruiting [119] |
| Trastuzumab duocarmazine (SYD985) | A: HER2 L: Cleavable linker C: Duocarmycin | Metastatic Breast Cancer | III | N/A (Ongoing) Last Update Posted: 12 January 2022 Estimated Study Completion Date: May 2022 | NCT03262935 Status: Active, not recruiting [120] | |
| TRPH-222 | A: CD22 L: 4AP non-cleavable linker C: Maytansine | Relapsed and/or Refractory B-Cell Lymphoma | I | N/A (Ongoing) Last Update Posted: 1 April 2021 Estimated Study Completion Date: 30 August 2022 | NCT03682796 Status: Recruiting [121] | |
| Upifitamab rilsodotin (XMT-1536) | A: NaPi2b L: Cleavable linker C: Auristatin F | Ovarian cancer and non-small cell lung cancer | I | N/A (Ongoing) Last Update Posted: 24 September 2021 Estimated Study Completion Date: 31 December 2022 | NCT03319628 Status: Recruiting [122] | |
| W0101 | A: IGF-1R L: Non-cleavable maleimidocaproyl linker C: Auristatin derivative | Advanced or metastatic solid tumours | I/II | N/A (Ongoing) Last Update Posted: 12 August 2021 Estimated Study Completion Date: 30 December 2024 | NCT03316638 Status: Recruiting [123] | |
| Zilovertamab Vedotin (MK-2140) | A: ROR1 L: Cleavable mc–val-cit–PABC linker C: MMAE | Relapsed or Refractory Diffuse Large B-Cell Lymphoma | II | N/A (Ongoing) Last Update Posted: 26 January 2023 Estimated Study Completion Date: 10 June 2025 | NCT05144841 Status: Recruiting | |
| Main Topics | Summary Points |
|---|---|
| Toxicity of ADC |
|
| Antibody & Antigen Specificity |
|
| Immunogenicity of Antibody |
|
| Stability of linkers |
|
| Advantages | Disadvantages |
|---|---|
| ADC has increased the tumour specificity and selectivity and enhances the delivery of the cytotoxic payload to the cancer cell. | ADC is unable to carry a large amount of cytotoxic payload which in turn required a higher dose to achieve the therapeutic effect leading to an increased risk of toxicity. |
| ADC can carry a very toxic payload that was previously discarded due to safety issues. | ADC bystander killing effect. |
| With a stable linker, the therapeutic window of ADC will be expanded even if higher dosages of toxin-carrying ADC be administered to patients. | The nonspecific binding of inappropriate selection of antibodies might lead to undesirable toxicity. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, Y.J.; Clarissa Lau, P.S.; Low, S.X.; Ng, S.L.; Ong, M.Y.; Pang, H.M.; Lee, Z.Y.; Yow, H.Y.; Hamzah, S.B.; Sellappans, R.; et al. How Far Have We Developed Antibody–Drug Conjugate for the Treatment of Cancer? Drugs Drug Candidates 2023, 2, 377-421. https://doi.org/10.3390/ddc2020020
Lim YJ, Clarissa Lau PS, Low SX, Ng SL, Ong MY, Pang HM, Lee ZY, Yow HY, Hamzah SB, Sellappans R, et al. How Far Have We Developed Antibody–Drug Conjugate for the Treatment of Cancer? Drugs and Drug Candidates. 2023; 2(2):377-421. https://doi.org/10.3390/ddc2020020
Chicago/Turabian StyleLim, Yu Jun, Pei Sze Clarissa Lau, Shi Xuan Low, Shong Li Ng, Min Yee Ong, Huey Ming Pang, Zheng Yang Lee, Hui Yin Yow, Sharina Binti Hamzah, Renukha Sellappans, and et al. 2023. "How Far Have We Developed Antibody–Drug Conjugate for the Treatment of Cancer?" Drugs and Drug Candidates 2, no. 2: 377-421. https://doi.org/10.3390/ddc2020020