
Degradative Signaling in ATG7-Deficient Skeletal Muscle Following Cardiotoxin Injury
Abstract
:1. Introduction
2. Results
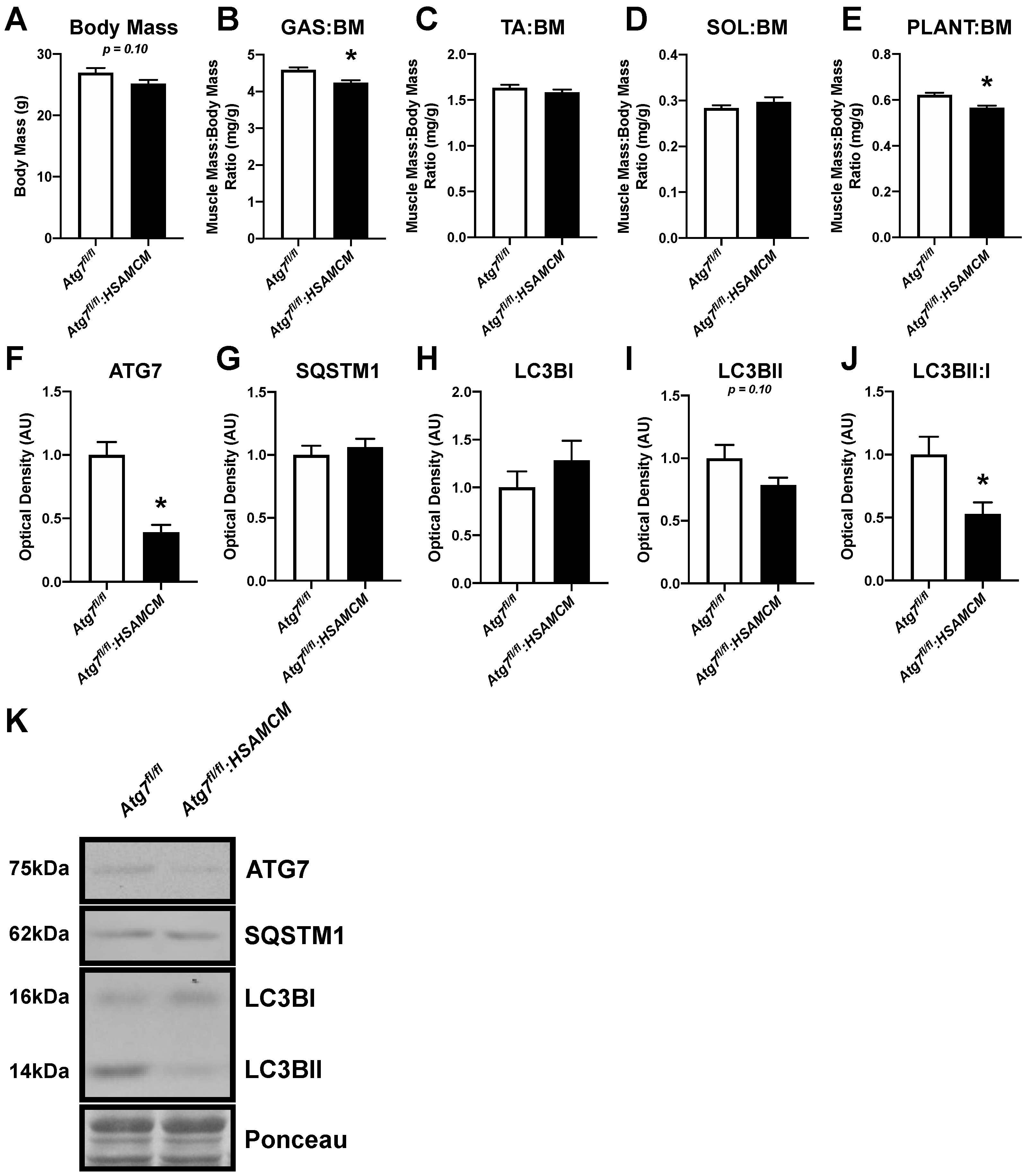
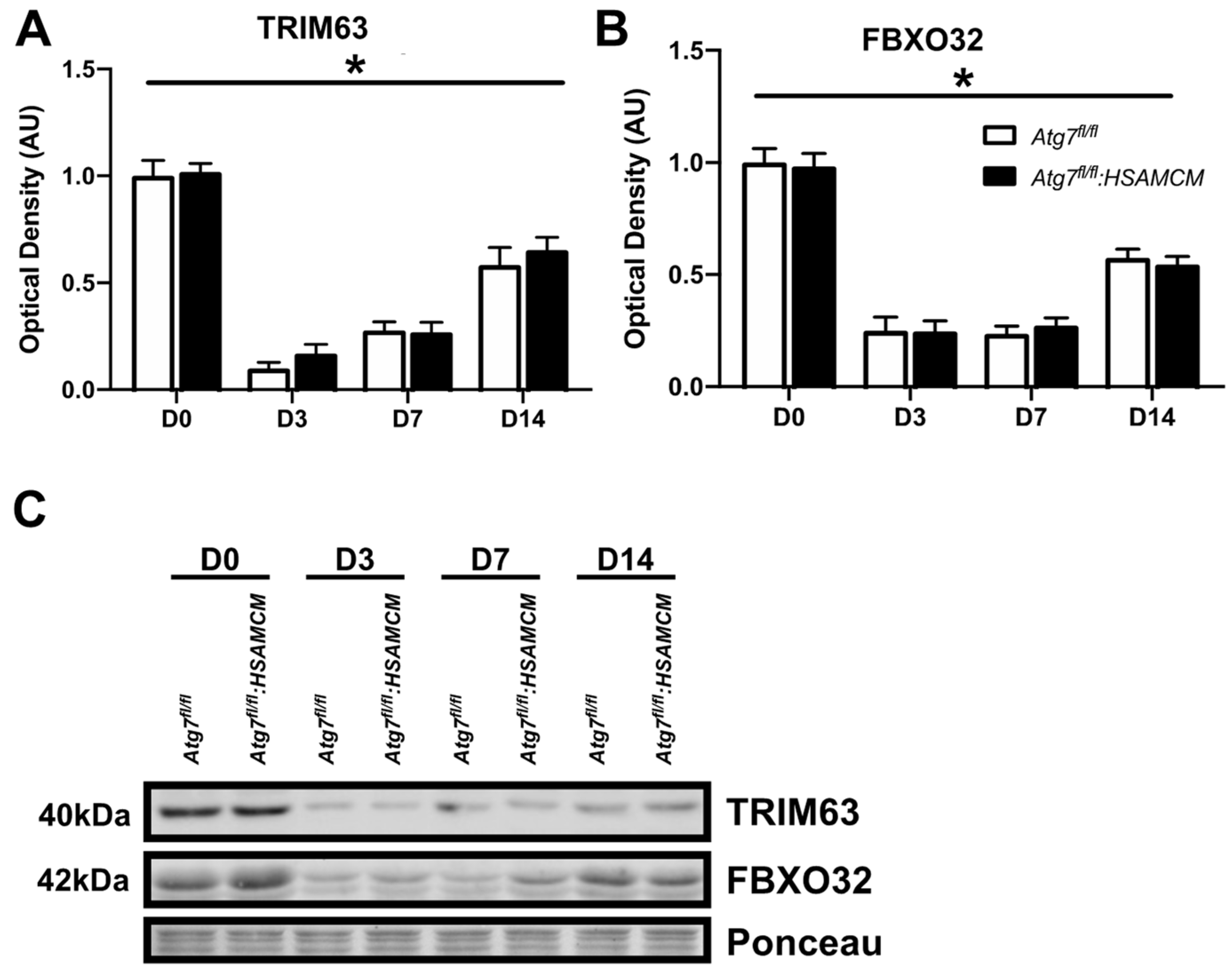
2.1. Atg7 Knockout in Skeletal Muscle Reduces Muscle Mass and Autophagic Markers
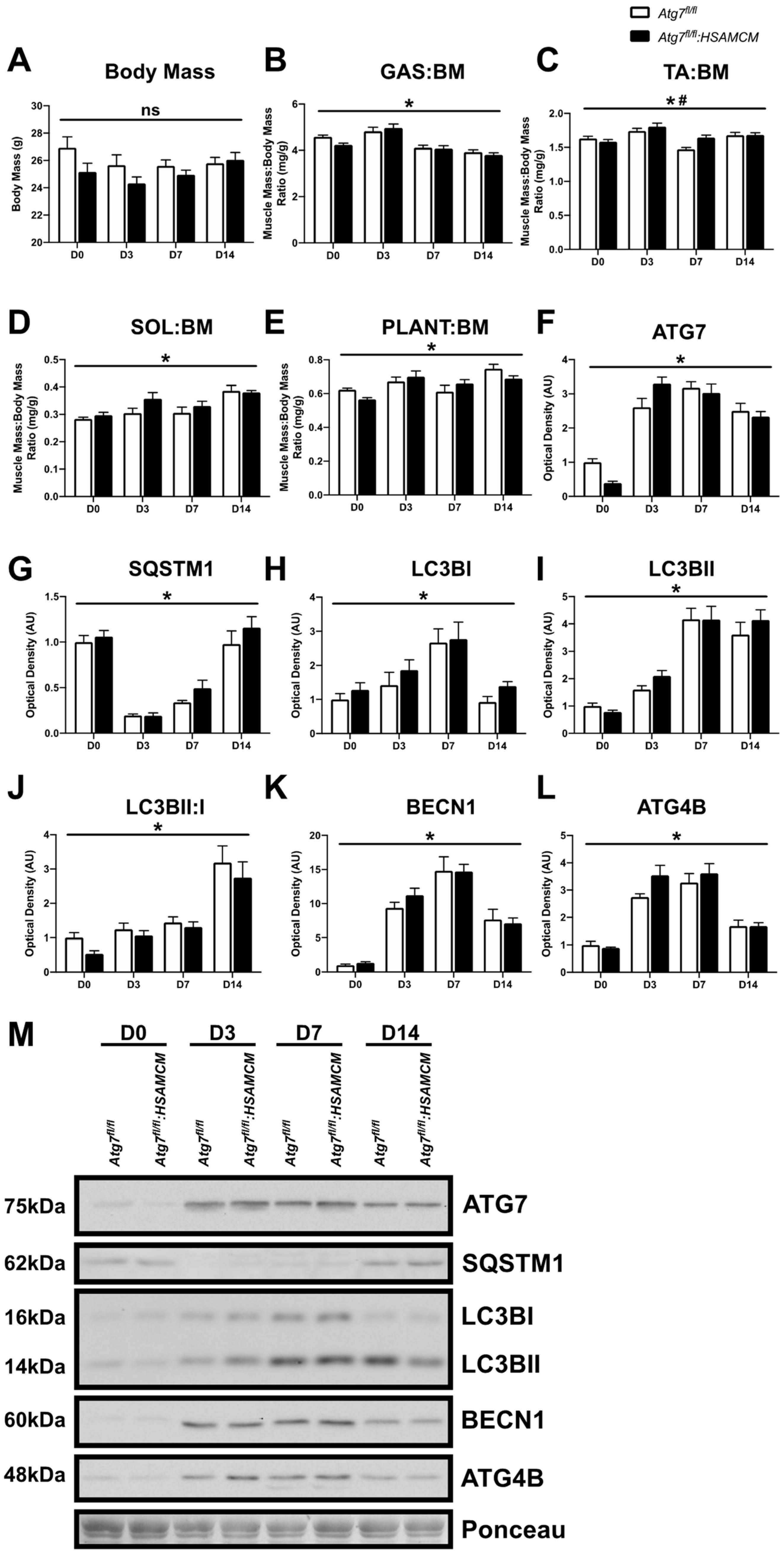
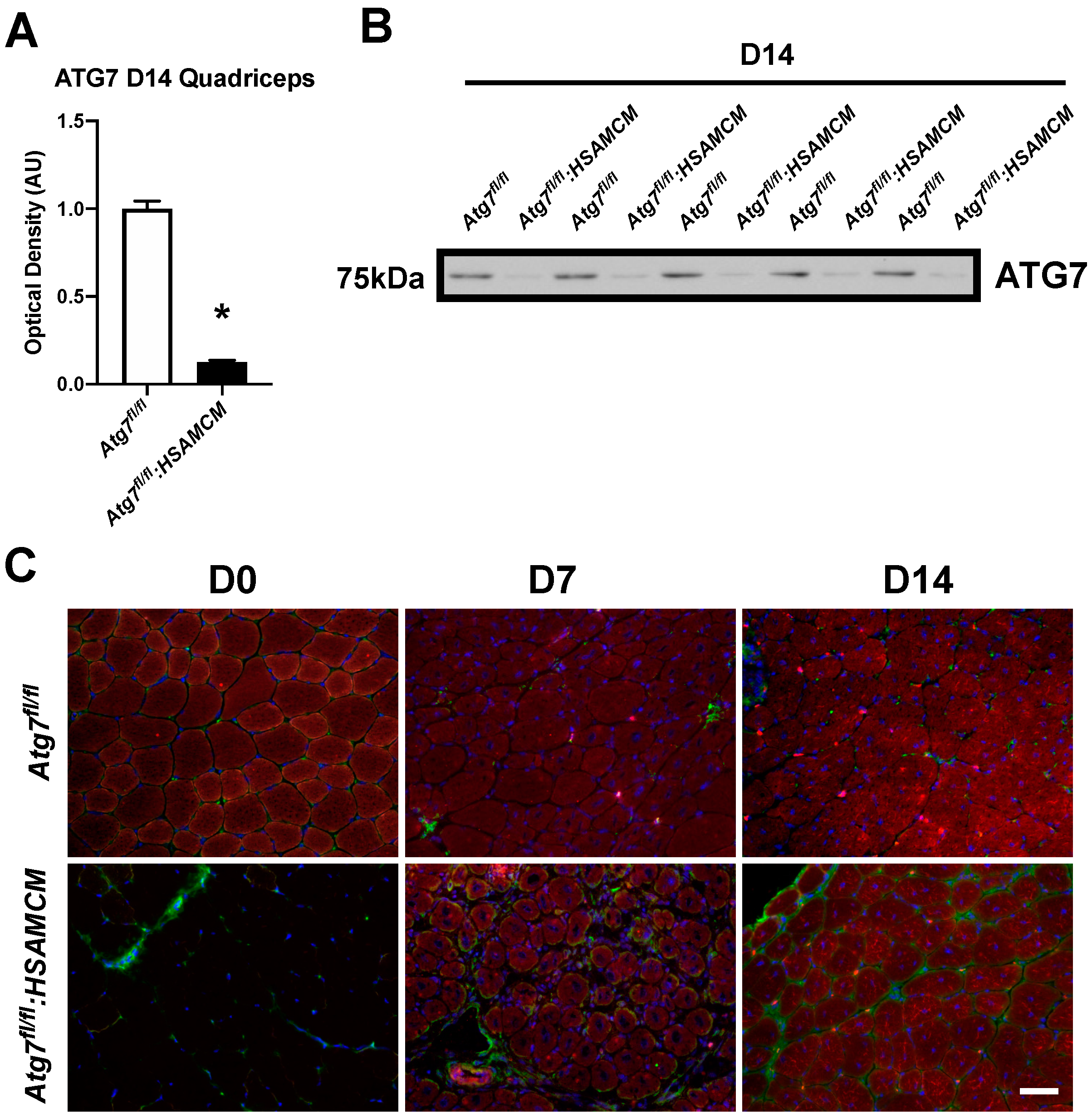
2.2. CTX-Injury Does Not Alter Mass Parameters in Skeletal Muscle of Atg7 Knockout Mice
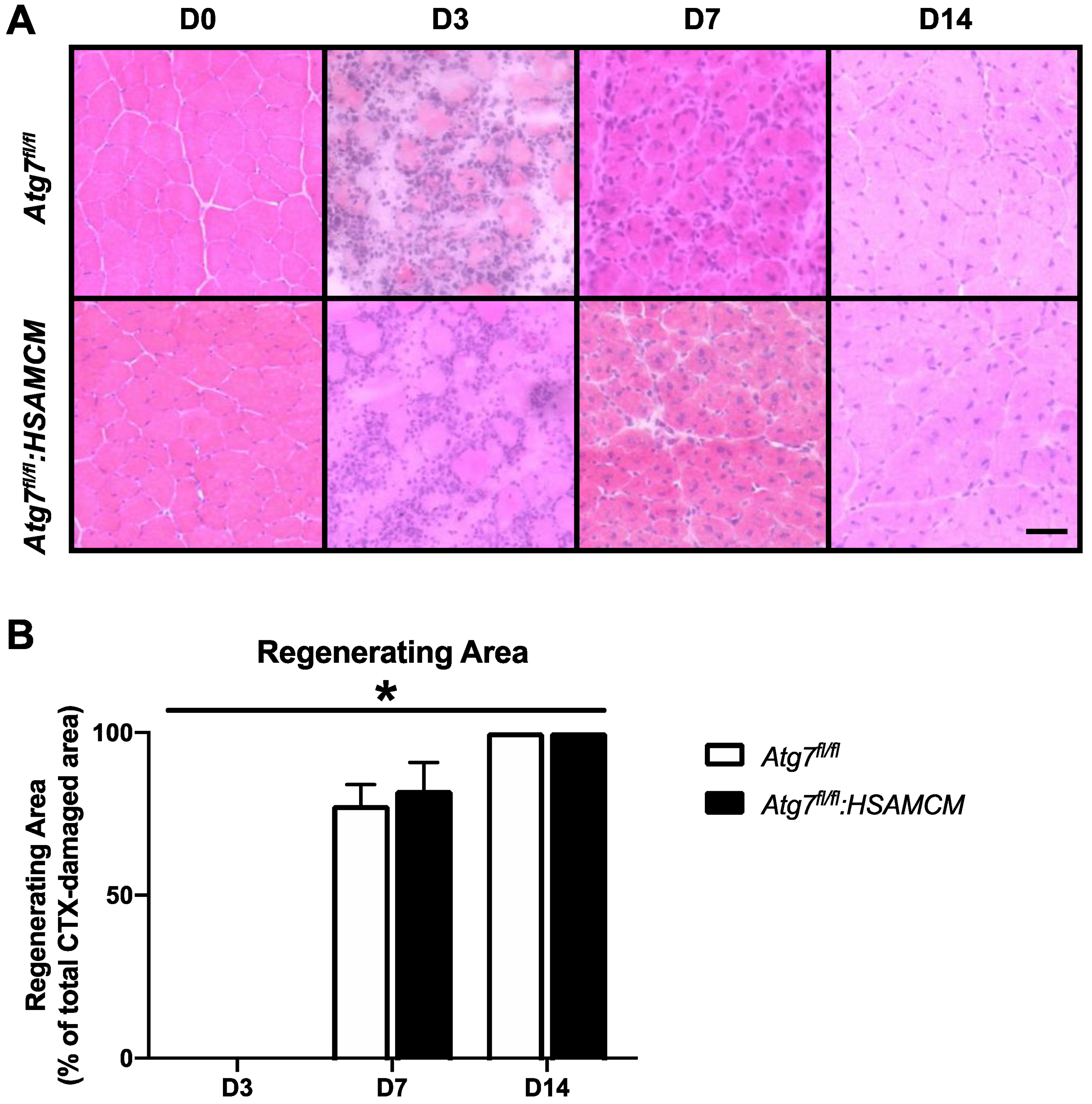
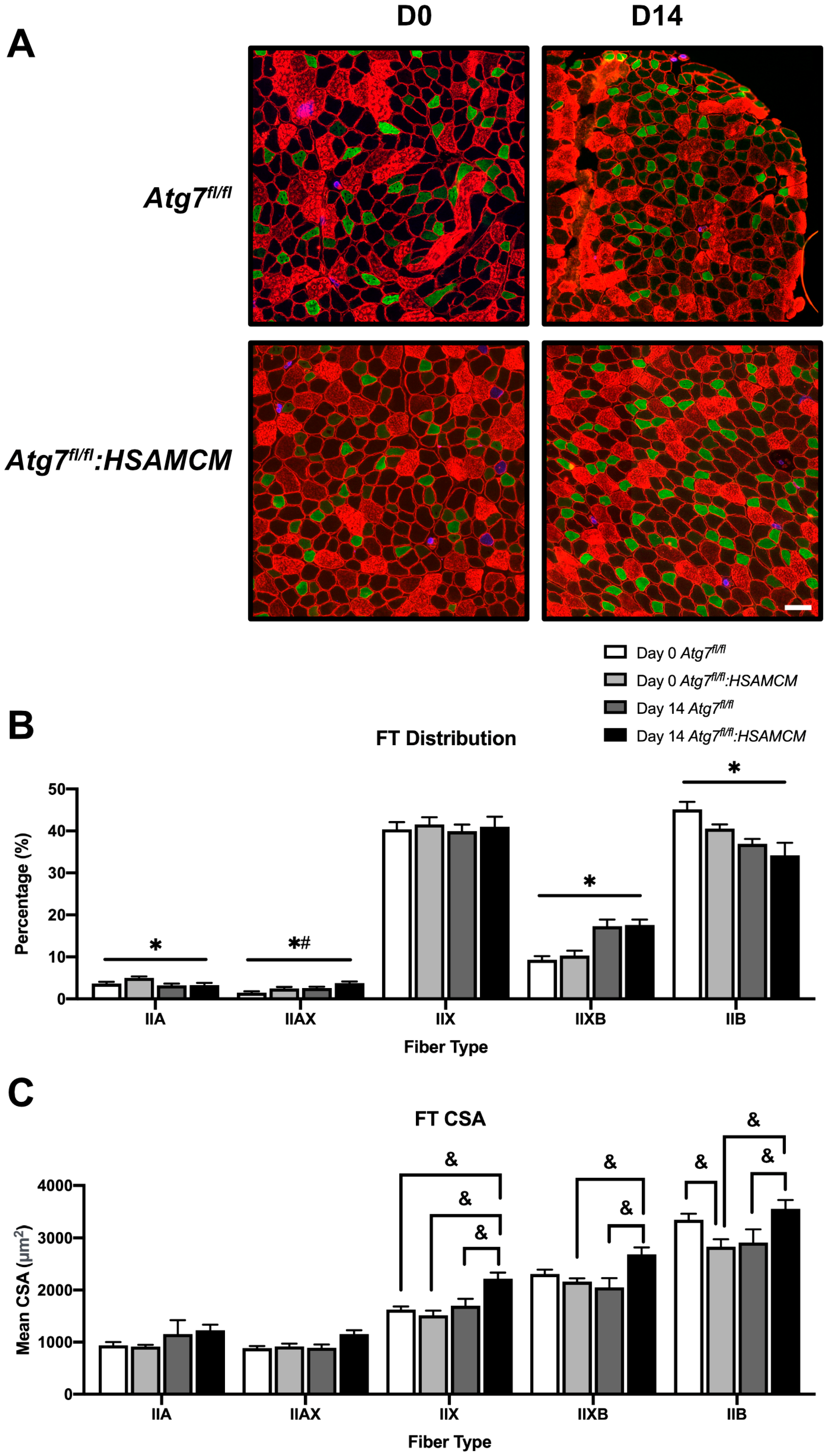
2.3. Fiber-Type Distribution Remains Largely Unchanged but CSA in Glycolytic Fibers Is Slightly Larger in Atg7fl/fl:HSAMCM Animals
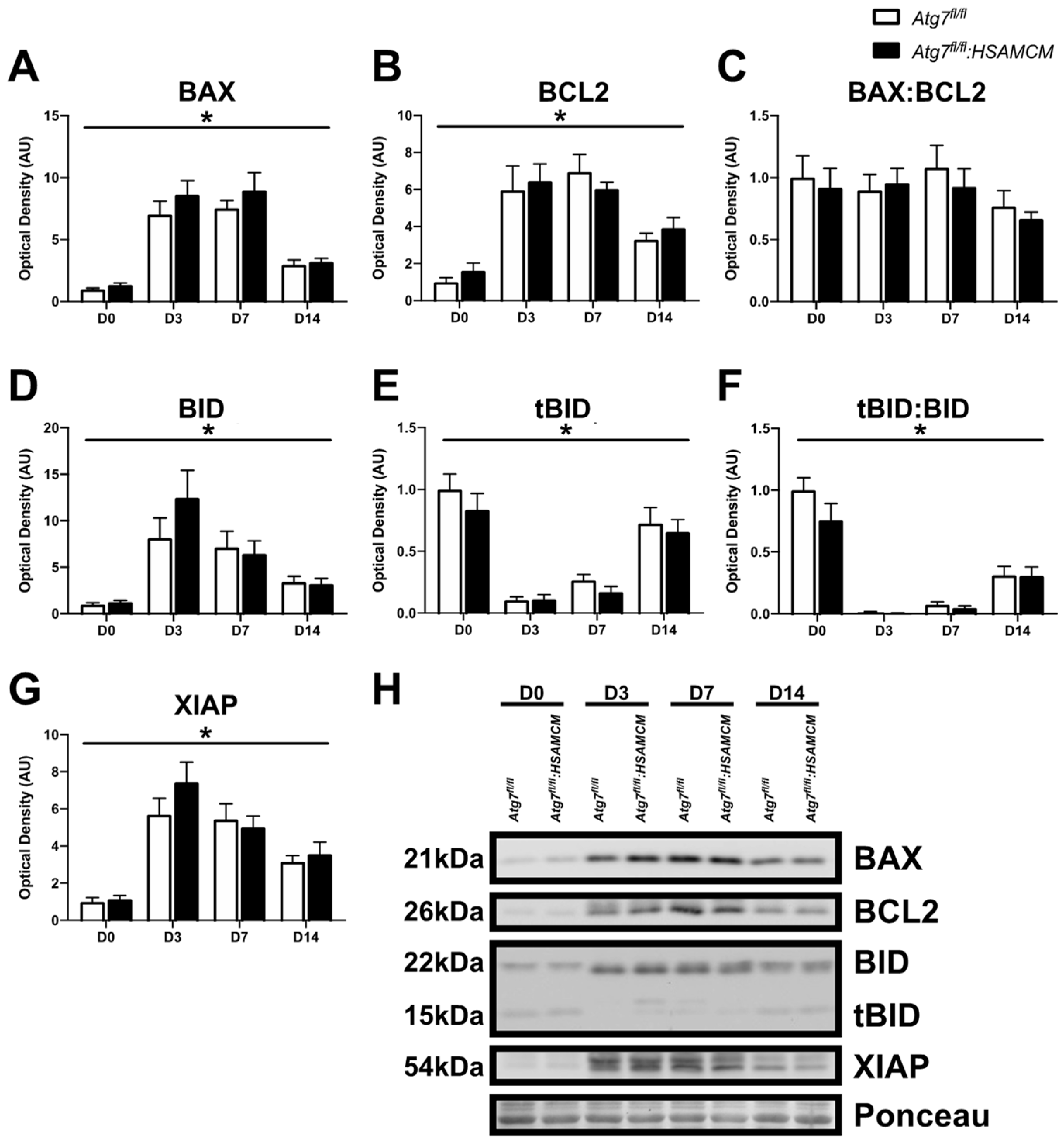
2.4. Atg7 Knockout Does Not Impact Atrophic and Apoptotic Protein Markers in Response to CTX Injury
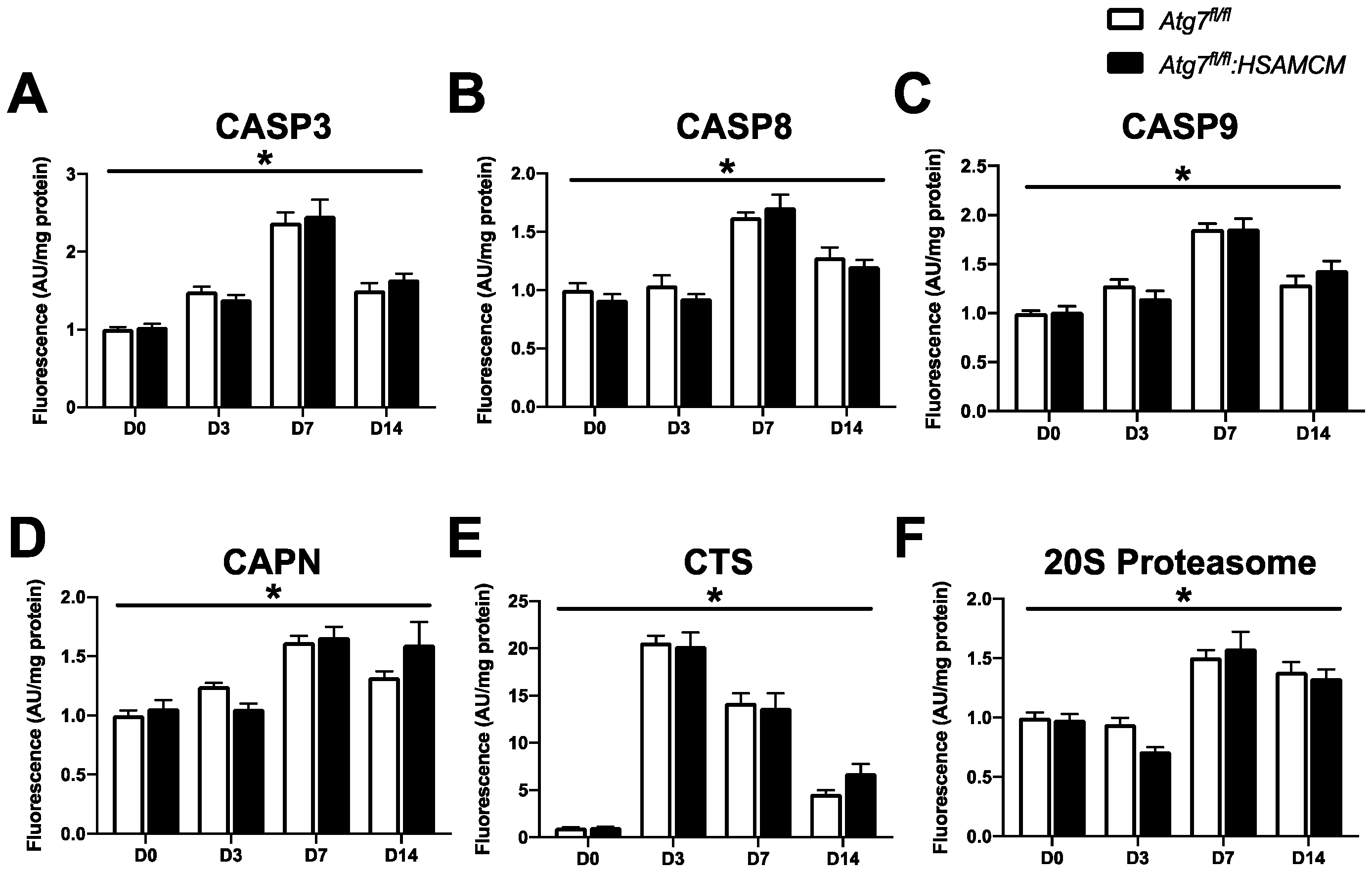
2.5. CTX-Induced Skeletal Muscle Damage Mediates a Transient Elevation in Apoptotic Enzyme Activity
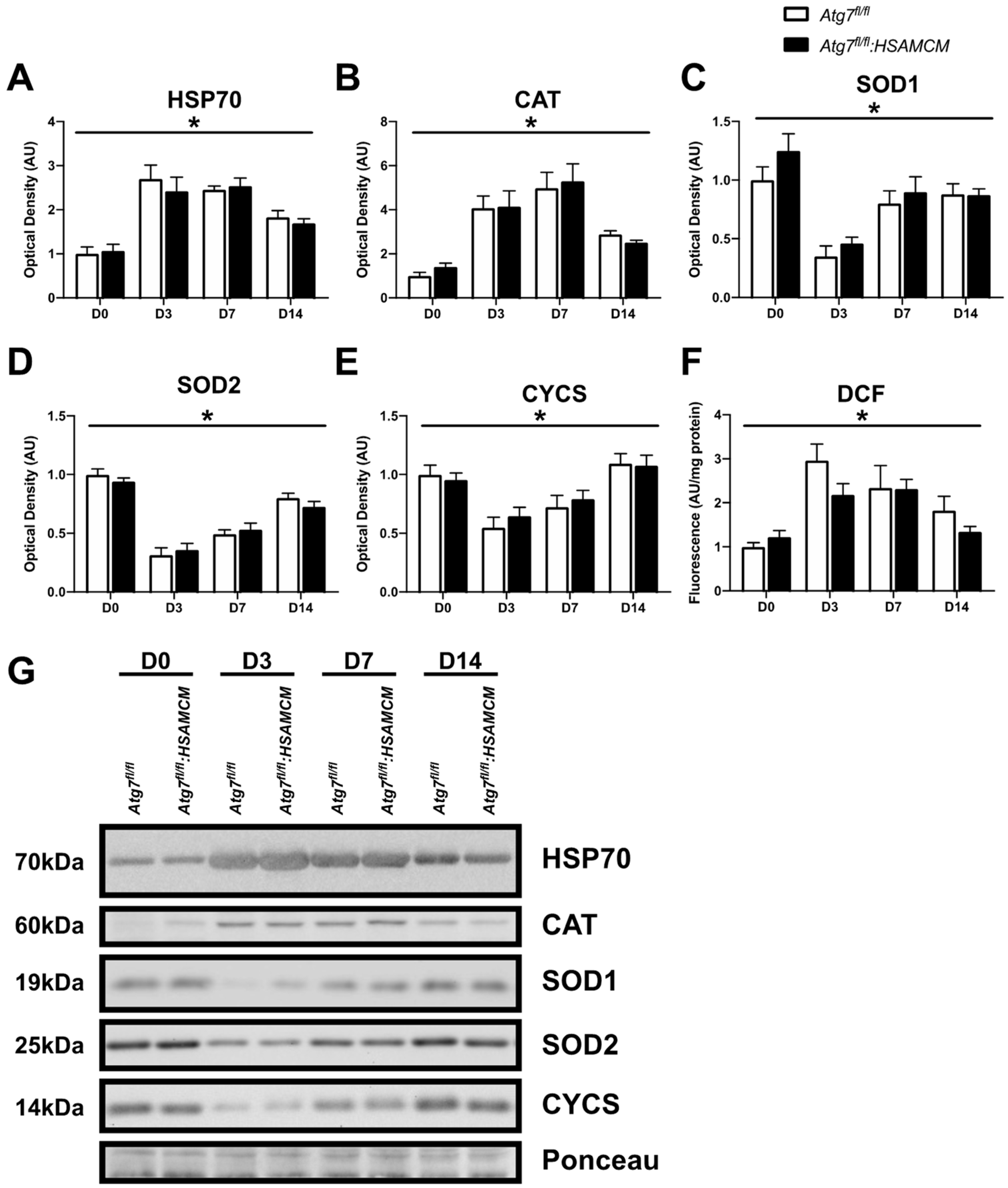
2.6. Regenerating Skeletal Muscle Displays an Early Increase in ROS, Which Declines in Concert with Elevated Antioxidant Enzymes
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Skeletal Muscle Collection
4.3. Immunoblot Analyses
4.4. Enzyme Activity Assays
4.5. Caspase and Calpain Assay
4.6. Cathepsin Assay
4.7. 20S Proteasomal Activity Assay
4.8. Reactive Oxygen Species Generation
4.9. Histological Analyses
4.10. Immunofluorescence
4.11. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McNeil, P.L.; Khakee, R. Disruptions of Muscle Fiber Plasma Membranes. Role in Exercise-Induced Damage. Am. J. Pathol. 1992, 140, 1097–1109. [Google Scholar] [PubMed]
- Sloper, J.C.; Barrett, M.C.; Partridge, T.A. The Muscle Cell. J. Clin. Pathol. 1978, 12, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Ciciliot, S.; Schiaffino, S. Regeneration of Mammalian Skeletal Muscle. Basic Mechanisms and Clinical Implications. Curr. Pharm. Des. 2010, 16, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Hawke, T.J.; Garry, D.J. Myogenic Satellite Cells: Physiology to Molecular Biology. J. Appl. Physiol. 2001, 91, 534–551. [Google Scholar] [CrossRef]
- Rahman, F.A.; Krause, M.P. PAI-1, the Plasminogen System, and Skeletal Muscle. Int. J. Mol. Sci. 2020, 21, 7066. [Google Scholar] [CrossRef]
- McMillan, E.M.; Quadrilatero, J. Autophagy Is Required and Protects against Apoptosis during Myoblast Differentiation. Biochem. J. 2014, 462, 267–277. [Google Scholar] [CrossRef]
- Baechler, B.L.; Bloemberg, D.; Quadrilatero, J. Mitophagy Regulates Mitochondrial Network Signaling, Oxidative Stress, and Apoptosis during Myoblast Differentiation. Autophagy 2019, 15, 1606–1619. [Google Scholar] [CrossRef]
- Bloemberg, D.; Quadrilatero, J. Autophagy, Apoptosis, and Mitochondria: Molecular Integration and Physiological Relevance in Skeletal Muscle. Am. J. Physiol.-Cell Physiol. 2019, 317, C111–C130. [Google Scholar] [CrossRef]
- Paolini, A.; Omairi, S.; Mitchell, R.; Vaughan, D.; Matsakas, A.; Vaiyapuri, S.; Ricketts, T.; Rubinsztein, D.C.; Patel, K. Attenuation of Autophagy Impacts on Muscle Fibre Development, Starvation Induced Stress and Fibre Regeneration Following Acute Injury. Sci. Rep. 2018, 8, 9062. [Google Scholar] [CrossRef]
- Nichenko, A.S.; Southern, W.M.; Atuan, M.; Luan, J.; Peissig, K.B.; Foltz, S.J.; Beedle, A.M.; Warren, G.L.; Call, J.A. Mitochondrial Maintenance via Autophagy Contributes to Functional Skeletal Muscle Regeneration and Remodeling. Am. J. Physiol.-Cell Physiol. 2016, 311, C190–C200. [Google Scholar] [CrossRef]
- Nichenko, A.S.; Southern, W.M.; Tehrani, K.F.; Qualls, A.E.; Flemington, A.B.; Mercer, G.H.; Yin, A.; Mortensen, L.J.; Yin, H.; Call, J.A. Mitochondrial-Specific Autophagy Linked to Mitochondrial Dysfunction Following Traumatic Freeze Injury in Mice. Am. J. Physiol.-Cell Physiol. 2020, 318, C242–C252. [Google Scholar] [CrossRef] [PubMed]
- Nitta, A.; Hori, K.; Tanida, I.; Igarashi, A.; Deyama, Y.; Ueno, T.; Kominami, E.; Sugai, M.; Aoki, K. Blocking LC3 Lipidation and ATG12 Conjugation Reactions by ATG7 Mutant Protein Containing C572S. Biochem. Biophys. Res. Commun. 2019, 508, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy Is Required to Maintain Muscle Mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Paré, M.F.; Baechler, B.L.; Fajardo, V.A.; Earl, E.; Wong, E.; Campbell, T.L.; Tupling, A.R.; Quadrilatero, J. Effect of Acute and Chronic Autophagy Deficiency on Skeletal Muscle Apoptotic Signaling, Morphology, and Function. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Rahman, F.A.; Angus, S.A.; Stokes, K.; Karpowicz, P.; Krause, M.P. Impaired ECM Remodeling and Macrophage Activity Define Necrosis and Regeneration Following Damage in Aged Skeletal Muscle. Int. J. Mol. Sci. 2020, 21, 4575. [Google Scholar] [CrossRef]
- Moriscot, A.S.; Baptista, I.L.; Silva, W.J.; Silvestre, J.G.; Adams, V.; Gasch, A.; Bogomolovas, J.; Labeit, S. MuRF1 and MuRF2 Are Key Players in Skeletal Muscle Regeneration Involving Myogenic Deficit and Deregulation of the Chromatin-Remodeling Complex. JCSM Rapid Commun. 2019, 2, 1–25. [Google Scholar] [CrossRef]
- Ogasawara, S.; Cheng, X.W.; Inoue, A.; Hu, L.; Piao, L.; Yu, C.; Goto, H.; Xu, W.; Zhao, G.; Lei, Y.; et al. Cathepsin K Activity Controls Cardiotoxin-induced Skeletal Muscle Repair in Mice. J. Cachexia Sarcopenia Muscle 2018, 9, 160–175. [Google Scholar] [CrossRef]
- Mukasa, T.; Momoi, T.; Momoi, M.Y. Activation of Caspase-3 Apoptotic Pathways in Skeletal Muscle Fibers in Laminin Alpha2-Deficient Mice. Biochem. Biophys. Res. Commun. 1999, 260, 139–142. [Google Scholar] [CrossRef]
- Sandri, M.; El Meslemani, A.H.; Sandri, C.; Schjerling, P.; Vissing, K.; Andersen, J.L.; Rossini, K.; Carraro, U.; Angelini, C. Caspase 3 Expression Correlates with Skeletal Muscle Apoptosis in Duchenne and Facioscapulo Human Muscular Dystrophy. A Potential Target for Pharmacological Treatment? J. Neuropathol. Exp. Neurol. 2001, 60, 302–312. [Google Scholar] [CrossRef]
- Rahman, F.A.; Quadrilatero, J. Mitochondrial Apoptotic Signaling Involvement in Remodeling During Myogenesis and Skeletal Muscle Atrophy. Semin. Cell Dev. Biol. 2022, 143, 66–74. [Google Scholar] [CrossRef]
- Fernando, P.; Kelly, J.F.; Balazsi, K.; Slack, R.S.; Megeney, L.A. Caspase 3 Activity Is Required for Skeletal Muscle Differentiation. Proc. Natl. Acad. Sci. USA 2002, 99, 11025–11030. [Google Scholar] [CrossRef] [PubMed]
- Dick, S.A.; Chang, N.C.; Dumont, N.A.; Bell, R.A.V.; Putinski, C.; Kawabe, Y.; Litchfield, D.W.; Rudnicki, M.A.; Megeney, L.A. Caspase 3 Cleavage of Pax7 Inhibits Self-Renewal of Satellite Cells. Proc. Natl. Acad. Sci. USA 2015, 112, E5246–E5252. [Google Scholar] [CrossRef] [PubMed]
- Bloemberg, D.; Quadrilatero, J. Mitochondrial Pro-Apoptotic Indices Do Not Precede the Transient Caspase Activation Associated with Myogenesis. Biochim. Biophys. Acta 2014, 1843, 2926–2936. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.; Bentzinger, C.; Sincennes, M.-C.; Rudnicki, M. Satellite Cells and Skeletal Muscle Regeneration. Compr. Physiol. 2015, 5, 1027–1059. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Wang, Y.X.; Rudnicki, M.A. Intrinsic and Extrinsic Mechanisms Regulating Satellite Cell Function. Development 2015, 142, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Rudnicki, M.A. Satellite Cells, the Engines of Muscle Repair. Nat. Rev. Mol. Cell Biol. 2012, 13, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Rong-hua, W.; Man, X.; Pan-feng, Z.; Mei, L. The mRNA Alteration and Correlation of Calpains and FoxOs during Gastrocnemius Muscle Atrophy Induced by Sciatic Nerve Injury and Cardiotoxin Injection. Chin. J. Tissue Eng. Res. 2012, 16, 1173. [Google Scholar] [CrossRef]
- Kitajima, Y.; Suzuki, N.; Nunomiya, A.; Osana, S.; Yoshioka, K.; Tashiro, Y.; Takahashi, R.; Ono, Y.; Aoki, M.; Nagatomi, R. The Ubiquitin-Proteasome System Is Indispensable for the Maintenance of Muscle Stem Cells. Stem Cell Rep. 2018, 11, 1523–1538. [Google Scholar] [CrossRef]
- Hauerslev, S.; Sveen, M.-L.; Duno, M.; Angelini, C.; Vissing, J.; Krag, T.O. Calpain 3 Is Important for Muscle Regeneration: Evidence from Patients with Limb Girdle Muscular Dystrophies. BMC Musculoskelet. Disord. 2012, 13, 43. [Google Scholar] [CrossRef]
- Richard, I.; Broux, O.; Allamand, V.; Fougerousse, F.; Chiannilkulchai, N.; Bourg, N.; Brenguier, L.; Devaud, C.; Pasturaud, P.; Roudaut, C. Mutations in the Proteolytic Enzyme Calpain 3 Cause Limb-Girdle Muscular Dystrophy Type 2A. Cell 1995, 81, 27–40. [Google Scholar] [CrossRef]
- Ojima, K.; Kawabata, Y.; Nakao, H.; Nakao, K.; Doi, N.; Kitamura, F.; Ono, Y.; Hata, S.; Suzuki, H.; Kawahara, H.; et al. Dynamic Distribution of Muscle-Specific Calpain in Mice Has a Key Role in Physical-Stress Adaptation and Is Impaired in Muscular Dystrophy. J. Clin. Investig. 2010, 120, 2672–2683. [Google Scholar] [CrossRef] [PubMed]
- Ermolova, N.; Kudryashova, E.; DiFranco, M.; Vergara, J.; Kramerova, I.; Spencer, M.J. Pathogenity of Some Limb Girdle Muscular Dystrophy Mutations Can Result from Reduced Anchorage to Myofibrils and Altered Stability of Calpain 3. Hum. Mol. Genet. 2011, 20, 3331–3345. [Google Scholar] [CrossRef]
- Hayashi, C.; Ono, Y.; Doi, N.; Kitamura, F.; Tagami, M.; Mineki, R.; Arai, T.; Taguchi, H.; Yanagida, M.; Hirner, S.; et al. Multiple Molecular Interactions Implicate the Connectin/Titin N2A Region as a Modulating Scaffold for P94/Calpain 3 Activity in Skeletal Muscle. J. Biol. Chem. 2008, 283, 14801–14814. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.M.; Lamb, G.D. Endogenous Calpain-3 Activation Is Primarily Governed by Small Increases in Resting Cytoplasmic [Ca2+] and Is Not Dependent on Stretch. J. Biol. Chem. 2009, 284, 7811–7819. [Google Scholar] [CrossRef] [PubMed]
- Cohen, N.; Kudryashova, E.; Kramerova, I.; Anderson, L.V.B.; Beckmann, J.S.; Bushby, K.; Spencer, M.J. Identification of Putative in Vivo Substrates of Calpain 3 by Comparative Proteomics of Overexpressing Transgenic and Nontransgenic Mice. Proteomics 2006, 6, 6075–6084. [Google Scholar] [CrossRef]
- Mellgren, R.L.; Miyake, K.; Kramerova, I.; Spencer, M.J.; Bourg, N.; Bartoli, M.; Richard, I.; Greer, P.A.; McNeil, P.L. Calcium-Dependent Plasma Membrane Repair Requires m- or Mu-Calpain, but Not Calpain-3, the Proteasome, or Caspases. Biochim. Biophys. Acta 2009, 1793, 1886–1893. [Google Scholar] [CrossRef]
- Lek, A.; Evesson, F.J.; Lemckert, F.A.; Redpath, G.M.I.; Lueders, A.-K.; Turnbull, L.; Whitchurch, C.B.; North, K.N.; Cooper, S.T. Calpains, Cleaved Mini-DysferlinC72, and L-Type Channels Underpin Calcium-Dependent Muscle Membrane Repair. J. Neurosci. 2013, 33, 5085–5094. [Google Scholar] [CrossRef]
- Tidball, J.G. Inflammatory Processes in Muscle Injury and Repair. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2005, 288, R345–R353. [Google Scholar] [CrossRef]
- Tidball, J.G. Mechanisms of Muscle Injury, Repair, and Regeneration. Compr. Physiol. 2011, 1, 2029–2062. [Google Scholar] [CrossRef]
- Kozakowska, M.; Pietraszek-Gremplewicz, K.; Jozkowicz, A.; Dulak, J. The Role of Oxidative Stress in Skeletal Muscle Injury and Regeneration: Focus on Antioxidant Enzymes. J. Muscle. Res. Cell Motil. 2015, 36, 377–393. [Google Scholar] [CrossRef]
- Senf, S.M.; Howard, T.M.; Ahn, B.; Ferreira, L.F.; Judge, A.R. Loss of the Inducible Hsp70 Delays the Inflammatory Response to Skeletal Muscle Injury and Severely Impairs Muscle Regeneration. PLoS ONE 2013, 8, e62687. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Lou, J.; Li, Y.; Qian, P.; He, W.; Hao, Y.; Xue, T.; Li, Y.; Song, Y.-H. Satellite Cell-Specific Deletion of Cipc Alleviates Myopathy in Mdx Mice. Cell Rep. 2022, 39, 110939. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.-C.; Ryu, S.; Hao, R.; Wang, B.; Kapur, M.; Fan, C.-M.; Yao, T.-P. HDAC4 Promotes Pax7-Dependent Satellite Cell Activation and Muscle Regeneration. EMBO Rep. 2014, 15, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Da, W.; Zhao, Y.; Wang, T.; Xu, H.; Shu, B.; Gao, X.; Shi, Q.; Ma, Y.; Zhang, Y.; et al. Role of Skeletal Muscle Satellite Cells in the Repair of Osteoporotic Fractures Mediated by β-Catenin. J. Cachexia Sarcopenia Muscle 2022, 13, 1403–1417. [Google Scholar] [CrossRef]
- Boyer, J.G.; Huo, J.; Han, S.; Havens, J.R.; Prasad, V.; Lin, B.L.; Kass, D.A.; Song, T.; Sadayappan, S.; Khairallah, R.J.; et al. Depletion of Skeletal Muscle Satellite Cells Attenuates Pathology in Muscular Dystrophy. Nat. Commun. 2022, 13, 2940. [Google Scholar] [CrossRef] [PubMed]
- Mobley, C.B.; Vechetti, I.J.; Valentino, T.R.; McCarthy, J.J. CORP: Using Transgenic Mice to Study Skeletal Muscle Physiology. J. Appl. Physiol. 2020, 128, 1227–1239. [Google Scholar] [CrossRef]
- Bloemberg, D.; Quadrilatero, J. Caspase Activity and Apoptotic Signaling in Proliferating C2C12 Cells Following Cisplatin or A23187 Exposure. Data Brief 2016, 7, 1024–1030. [Google Scholar] [CrossRef]
- Bloemberg, D.; McDonald, E.; Dulay, D.; Quadrilatero, J. Autophagy Is Altered in Skeletal and Cardiac Muscle of Spontaneously Hypertensive Rats. Acta Physiol. 2014, 210, 381–391. [Google Scholar] [CrossRef]
- McMillan, E.M.; Paré, M.-F.; Baechler, B.L.; Graham, D.A.; Rush, J.W.E.; Quadrilatero, J. Autophagic Signaling and Proteolytic Enzyme Activity in Cardiac and Skeletal Muscle of Spontaneously Hypertensive Rats Following Chronic Aerobic Exercise. PLoS ONE 2015, 10, e0119382. [Google Scholar] [CrossRef]
- McMillan, E.M.; Quadrilatero, J. Differential Apoptosis-Related Protein Expression, Mitochondrial Properties, Proteolytic Enzyme Activity, and DNA Fragmentation between Skeletal Muscles. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2011, 300, R531–R543. [Google Scholar] [CrossRef]
- Bloemberg, D.; Quadrilatero, J. Rapid Determination of Myosin Heavy Chain Expression in Rat, Mouse, and Human Skeletal Muscle Using Multicolor Immunofluorescence Analysis. PLoS ONE 2012, 7, e35273. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atg7fl/fl | Atg7fl/fl:HSAMCM | |
|---|---|---|
| Body Weight (g) | 26.9 ± 0.52 | 25.1 ± 0.47 |
| Absolute Muscle Weight (mg): | ||
| Gastrocnemius | 123.6 ± 1.23 | 106.48 ± 0.89 * |
| Tibialis Anterior | 43.9 ± 0.75 | 39.8 ± 0.61 * |
| Soleus | 7.6 ± 0.25 | 7.4 ± 0.26 |
| Plantaris | 16.7 ± 0.42 | 14.2 ± 0.32 * |
| Relative Muscle Weight (mg/g): | ||
| Gastrocnemius | 4.5 ± 0.15 | 4.2 ± 0.16 * |
| Tibialis Anterior | 1.6 ± 0.11 | 1.5 ± 0.10 |
| Soleus | 0.28 ± 0.04 | 0.29 ± 0.06 |
| Plantaris | 0.62 ± 0.05 | 0.50 ± 0.06 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, F.A.; Campbell, T.; Bloemberg, D.; Chapman, S.; Quadrilatero, J. Degradative Signaling in ATG7-Deficient Skeletal Muscle Following Cardiotoxin Injury. Muscles 2023, 2, 299-316. https://doi.org/10.3390/muscles2030023
Rahman FA, Campbell T, Bloemberg D, Chapman S, Quadrilatero J. Degradative Signaling in ATG7-Deficient Skeletal Muscle Following Cardiotoxin Injury. Muscles. 2023; 2(3):299-316. https://doi.org/10.3390/muscles2030023
Chicago/Turabian StyleRahman, Fasih Ahmad, Troy Campbell, Darin Bloemberg, Sarah Chapman, and Joe Quadrilatero. 2023. "Degradative Signaling in ATG7-Deficient Skeletal Muscle Following Cardiotoxin Injury" Muscles 2, no. 3: 299-316. https://doi.org/10.3390/muscles2030023