
Renal and Urinary Tract Involvement in Fibrosclerosing or Fibroinflammatory Diseases: A Narrative Review
Abstract
:1. Introduction
2. IgG4-Related Kidney Disease
3. Retroperitoneal Fibrosis
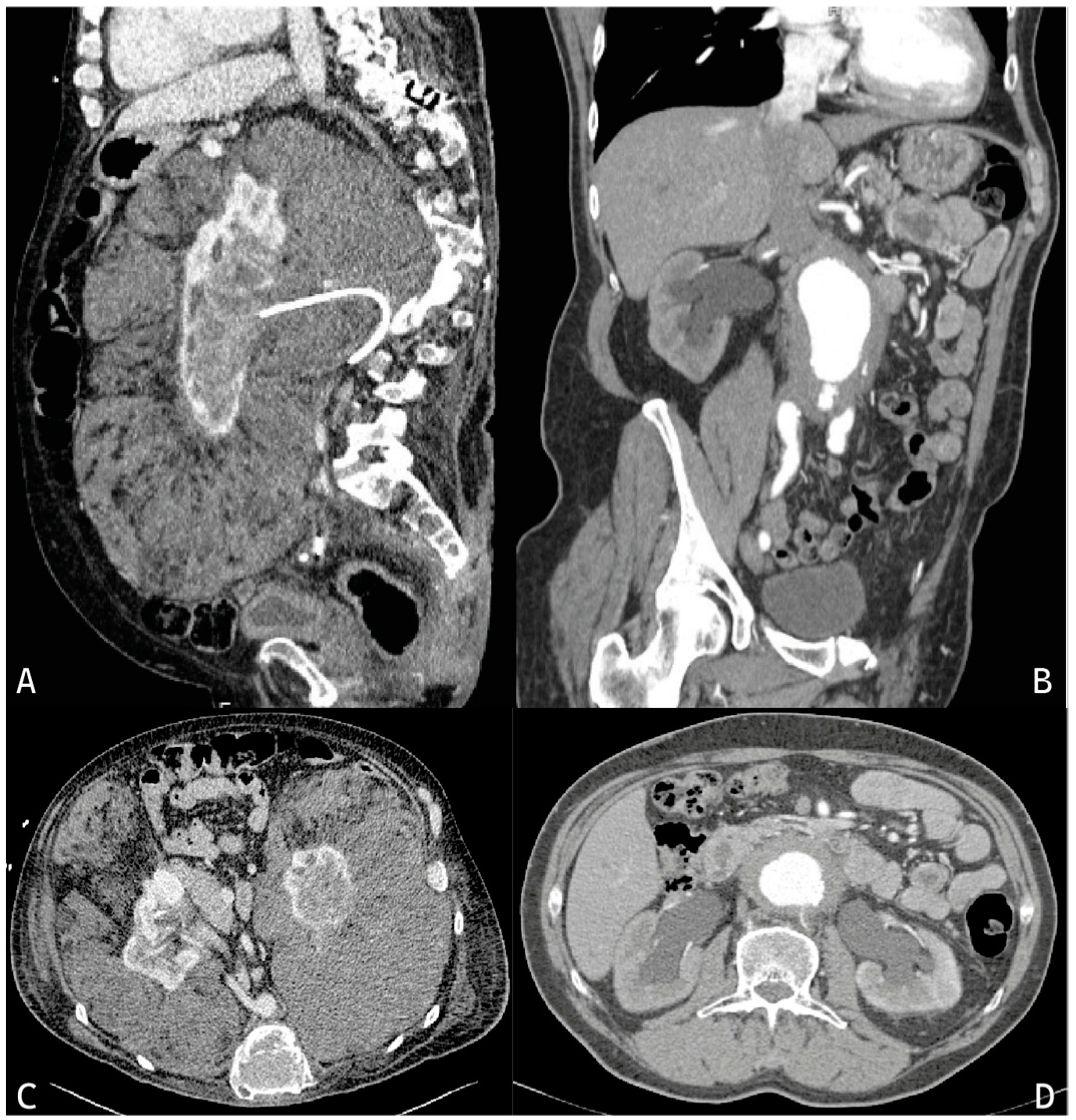
4. Erdheim–Chester Disease
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| FD | fibrosclerosing diseases |
| IgG4-RD | IgG4-related disease |
| RPF | retroperitoneal fibrosis |
| ECD | Erdheim–Chester disease |
| AKI | acute kidney injury |
| CKD | chronic kidney disease |
| ESKD | end-stage kidney disease |
| IgG4-RKD | IgG4-related kidney disease |
| IgG4-TIN | IgG4-tubular interstitial nephritis |
| MRI | magnetic resonance imaging |
| CT | computed tomography |
| FDG-PET | 18F-fluorodeoxyglucose positron emission tomography |
| TBM | tubular basement membrane |
| MN | membranous nephropathy |
| PLA2R | anti-phospholipase A2 receptor |
| IRF | idiopathic retroperitoneal fibrosis |
| CNS | central nervous system |
References
- Kawano, M.; Saeki, T. IgG4-related kidney disease—An update. Curr. Opin. Nephrol. Hypertens 2015, 24, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, A.; Vaglio, A. Chronic periaortitis: A fibro-inflammatory disorder. Best. Pract. Res. Clin. Rheumatol. 2009, 23, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Vaglio, A.; Maritati, F. Idiopathic Retroperitoneal Fibrosis. J. Am. Soc. Nephrol. 2016, 27, 1880–1889. [Google Scholar] [CrossRef] [PubMed]
- Haroche, J.; Cohen-Aubart, F.; Amoura, Z. Erdheim-Chester disease. Blood 2020, 135, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Maritati, F.; Peyronel, F.; Vaglio, A. IgG4-related disease: A clinical perspective. Rheumatology 2020, 59, iii123–iii131. [Google Scholar] [CrossRef] [PubMed]
- Raglianti, V.; Rossi, G.M.; Vaglio, A. Idiopathic retroperitoneal fibrosis: An update for nephrologists. Nephrol. Dial. Transplant. 2021, 36, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.M.; Emmi, G.; Corradi, D.; Urban, M.L.; Maritati, F.; Landini, F.; Galli, P.; Palmisano, A.; Vaglio, A. Idiopathic Mediastinal Fibrosis: A Systemic Immune-Mediated Disorder. A Case Series and a Review of the Literature. Clin. Rev. Allergy Immunol. 2017, 52, 446–459. [Google Scholar] [CrossRef]
- Rossi, G.M.; Rocco, R.; Accorsi Buttini, E.; Marvisi, C.; Vaglio, A. Idiopathic retroperitoneal fibrosis and its overlap with IgG4-related disease. Intern. Emerg. Med. 2017, 12, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Gianfreda, D.; Nicastro, M.; Galetti, M.; Alberici, F.; Corradi, D.; Becchi, G.; Baldari, G.; De Filippo, M.; Ferretti, S.; Moroni, G.; et al. Sirolimus plus prednisone for Erdheim-Chester disease: An open-label trial. Blood 2015, 126, 1163–1171. [Google Scholar] [CrossRef]
- Corradi, D.; Maestri, R.; Palmisano, A.; Bosio, S.; Greco, P.; Manenti, L.; Ferretti, S.; Cobelli, R.; Moroni, G.; Dei Tos, A.P.; et al. Idiopathic retroperitoneal fibrosis: Clinicopathologic features and differential diagnosis. Kidney Int. 2007, 72, 742–753. [Google Scholar] [CrossRef]
- Corradi, D.; Nicastro, M.; Vaglio, A. Immunoglobulin G4-related disease: Some missing pieces in a still unsolved complex puzzle. Cardiovasc. Pathol. 2016, 25, 90–92. [Google Scholar] [CrossRef]
- Lang, D.; Zwerina, J.; Pieringer, H. IgG4-related disease: Current challenges and future prospects. Ther. Clin. Risk Manag. 2016, 12, 189–199. [Google Scholar] [CrossRef]
- Martorana, D.; Marquez, A.; Carmona, F.D.; Bonatti, F.; Adorni, A.; Urban, M.L.; Maritati, F.; Accorsi Buttini, E.; Marvisi, C.; Palmisano, A.; et al. A large-scale genetic analysis reveals an autoimmune origin of idiopathic retroperitoneal fibrosis. J. Allergy Clin. Immunol. 2018, 142, 1662–1665. [Google Scholar] [CrossRef]
- Vaglio, A.; Salvarani, C.; Buzio, C. Retroperitoneal fibrosis. Lancet 2006, 367, 241–251. [Google Scholar] [CrossRef]
- Peyronel, F.; Vaglio, A. IgG4-Related Kidney Disease. Clin. J. Am. Soc. Nephrol. 2023, 18, 994–996. [Google Scholar] [CrossRef]
- Umehara, H.; Okazaki, K.; Masaki, Y.; Kawano, M.; Yamamoto, M.; Saeki, T.; Matsui, S.; Sumida, T.; Mimori, T.; Tanaka, Y.; et al. A novel clinical entity, IgG4-related disease (IgG4RD): General concept and details. Mod. Rheumatol. 2012, 22, 1–14. [Google Scholar] [CrossRef]
- Kamisawa, T.; Zen, Y.; Pillai, S.; Stone, J.H. IgG4-related disease. Lancet 2015, 385, 1460–1471. [Google Scholar] [CrossRef]
- Inoue, D.; Yoshida, K.; Yoneda, N.; Ozaki, K.; Matsubara, T.; Nagai, K.; Okumura, K.; Toshima, F.; Toyama, J.; Minami, T.; et al. IgG4-related disease: Dataset of 235 consecutive patients. Medicine 2015, 94, e680. [Google Scholar] [CrossRef]
- Katz, G.; Stone, J.H. Clinical Perspectives on IgG4-Related Disease and Its Classification. Annu. Rev. Med. 2022, 73, 545–562. [Google Scholar] [CrossRef]
- Wallace, Z.S.; Naden, R.P.; Chari, S.; Choi, H.; Della-Torre, E.; Dicaire, J.F.; Hart, P.A.; Inoue, D.; Kawano, M.; Khosroshahi, A.; et al. The 2019 American College of Rheumatology/European League Against Rheumatism Classification Criteria for IgG4-Related Disease. Arthritis Rheumatol 2020, 72, 7–19. [Google Scholar] [CrossRef]
- Kawano, M.; Yamada, K. IgG4-Related Kidney Disease and IgG4-Related Retroperitoneal Fibrosis. Semin. Liver Dis. 2016, 36, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Yamamoto, M.; Saeki, T.; Mizushima, I.; Matsui, S.; Fujisawa, Y.; Hara, S.; Takahashi, H.; Nomura, H.; Kawa, S.; et al. New clues to the nature of immunoglobulin G4-related disease: A retrospective Japanese multicenter study of baseline clinical features of 334 cases. Arthritis Res. Ther. 2017, 19, 262. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.D.R.; Cargill, T.; Goodchild, G.; Oliveira, B.; Rodriguez-Justo, M.; Pepper, R.; Connolly, J.; Salama, A.; Webster, G.; Barnes, E.; et al. Clinical Manifestations and Long-term Outcomes of IgG4-Related Kidney and Retroperitoneal Involvement in a United Kingdom IgG4-Related Disease Cohort. Kidney Int. Rep. 2019, 4, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Wallace, Z.S.; Deshpande, V.; Mattoo, H.; Mahajan, V.S.; Kulikova, M.; Pillai, S.; Stone, J.H. IgG4-Related Disease: Clinical and Laboratory Features in One Hundred Twenty-Five Patients. Arthritis Rheumatol. 2015, 67, 2466–2475. [Google Scholar] [CrossRef] [PubMed]
- Saeki, T.; Kawano, M.; Nagasawa, T.; Ubara, Y.; Taniguchi, Y.; Yanagita, M.; Nishi, S.; Nagata, M.; Hisano, S.; Yamaguchi, Y.; et al. Validation of the diagnostic criteria for IgG4-related kidney disease (IgG4-RKD) 2011, and proposal of a new 2020 version. Clin. Exp. Nephrol. 2021, 25, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.; Saeki, T.; Nakashima, H.; Nishi, S.; Yamaguchi, Y.; Hisano, S.; Yamanaka, N.; Inoue, D.; Yamamoto, M.; Takahashi, H.; et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin. Exp. Nephrol. 2011, 15, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Teng, F.; Lu, H.; Zheng, K.; Chen, G.; Wen, Y.; Liu, Z.; Peng, L.; Huo, L.; Zeng, X.; Zhang, W.; et al. Urinary System Manifestation of IgG4-Related Disease: Clinical, Laboratory, Radiological, and Pathological Spectra of a Chinese Single-Centre Study. J. Immunol. Res. 2020, 2020, 5851842. [Google Scholar] [CrossRef] [PubMed]
- Raissian, Y.; Nasr, S.H.; Larsen, C.P.; Colvin, R.B.; Smyrk, T.C.; Takahashi, N.; Bhalodia, A.; Sohani, A.R.; Zhang, L.; Chari, S.; et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J. Am. Soc. Nephrol. 2011, 22, 1343–1352. [Google Scholar] [CrossRef]
- Oh, J.W.; Rha, S.E.; Choi, M.H.; Oh, S.N.; Youn, S.Y.; Choi, J.I. Immunoglobulin G4-related Disease of the Genitourinary System: Spectrum of Imaging Findings and Clinical-Pathologic Features. Radiographics 2020, 40, 1265–1283. [Google Scholar] [CrossRef]
- Kawano, M.; Saeki, T.; Ubara, Y.; Matsui, S. Recent advances in IgG4-related kidney disease. Mod. Rheumatol. 2023, 33, 242–251. [Google Scholar] [CrossRef]
- Saeki, T.; Nishi, S.; Imai, N.; Ito, T.; Yamazaki, H.; Kawano, M.; Yamamoto, M.; Takahashi, H.; Matsui, S.; Nakada, S.; et al. Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int. 2010, 78, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, Y.; Mizushima, I.; Yamada, K.; Yamamoto, M.; Saeki, T.; Matsui, S.; Tsuge, S.; Hara, S.; Ito, K.; Fujii, H.; et al. Hypocomplementemia is related to elevated serum levels of IgG subclasses other than IgG4 in IgG4-related kidney disease. Mod. Rheumatol. 2021, 31, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Saeki, T.; Kawano, M.; Mizushima, I.; Yamamoto, M.; Wada, Y.; Nakashima, H.; Homma, N.; Tsubata, Y.; Takahashi, H.; Ito, T.; et al. The clinical course of patients with IgG4-related kidney disease. Kidney Int. 2013, 84, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.; Watanabe, H.; Asano, T.; Sato, S.; Takagi, T.; Kobayashi, H.; Ohira, H. Possible participation of IgG4 in the activation of complement in IgG4-related disease with hypocomplementemia. Mod. Rheumatol. 2016, 26, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; He, D.; Zhao, L.; Liang, S.; Liang, D.; Xu, F.; Zhang, M.; Zhu, X.; Chen, H.; Xie, H.; et al. Role of complement system in patients with biopsy-proven immunoglobulin G4-related kidney disease. Hum. Pathol. 2018, 81, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Lu, H.; Zhou, J.; Zhang, P.; Li, J.; Liu, Z.; Wu, D.; Zhang, S.; Yang, Y.; Bai, W.; et al. Clinical characteristics and outcome of IgG4-related disease with hypocomplementemia: A prospective cohort study. Arthritis Res. Ther. 2021, 23, 102. [Google Scholar] [CrossRef] [PubMed]
- Saeki, T.; Nagasawa, T.; Ubara, Y.; Taniguchi, Y.; Yanagita, M.; Nishi, S.; Nagata, M.; Yamaguchi, Y.; Saito, T.; Nakashima, H.; et al. Comparison of clinicopathological features between patients with and without hypocomplementemia in IgG4-related kidney disease. Nephrol. Dial. Transplant. 2023, 38, 1053–1056. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Gao, J.; Zhang, X.; Liu, A.; Wang, Z.; Wang, Z.; Chi, X.; Shi, Q.; Wang, Y.; Yang, F.; et al. Disparities between IgG4-related kidney disease and extrarenal IgG4-related disease in a case-control study based on 450 patients. Sci. Rep. 2021, 11, 10397. [Google Scholar] [CrossRef]
- Ishikawa, T.; Kawashima, H.; Ohno, E.; Iida, T.; Suzuki, H.; Uetsuki, K.; Yashika, J.; Yamada, K.; Yoshikawa, M.; Gibo, N.; et al. Clinical characteristics and long-term prognosis of autoimmune pancreatitis with renal lesions. Sci. Rep. 2021, 11, 406. [Google Scholar] [CrossRef]
- Khosroshahi, A.; Wallace, Z.S.; Crowe, J.L.; Akamizu, T.; Azumi, A.; Carruthers, M.N.; Chari, S.T.; Della-Torre, E.; Frulloni, L.; Goto, H.; et al. International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease. Arthritis Rheumatol. 2015, 67, 1688–1699. [Google Scholar] [CrossRef]
- Saeki, T.; Kawano, M.; Mizushima, I.; Yamamoto, M.; Wada, Y.; Ubara, Y.; Nakashima, H.; Ito, T.; Yamazaki, H.; Narita, I.; et al. Recovery of renal function after glucocorticoid therapy for IgG4-related kidney disease with renal dysfunction. Clin. Exp. Nephrol. 2016, 20, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Ogata, S.; Ozeki, T.; Takahashi, K.; Tsuboi, N.; Maruyama, S.; Inaguma, D.; Hasegawa, M.; Yuzawa, Y.; Hayashi, H. Long-term changes in renal function after treatment initiation and the importance of early diagnosis in maintaining renal function among IgG4-related tubulointerstitial nephritis patients in Japan. Arthritis Res. Ther. 2020, 22, 261. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, I.; Yamamoto, M.; Inoue, D.; Nishi, S.; Taniguchi, Y.; Ubara, Y.; Matsui, S.; Yasuno, T.; Nakashima, H.; Takahashi, H.; et al. Factors related to renal cortical atrophy development after glucocorticoid therapy in IgG4-related kidney disease: A retrospective multicenter study. Arthritis Res. Ther. 2016, 18, 273. [Google Scholar] [CrossRef] [PubMed]
- McMahon, B.A.; Novick, T.; Scheel, P.J.; Bagnasco, S.; Atta, M.G. Rituximab for the Treatment of IgG4-Related Tubulointerstitial Nephritis: Case Report and Review of the Literature. Medicine 2015, 94, e1366. [Google Scholar] [CrossRef] [PubMed]
- Goldoni, M.; Bonini, S.; Urban, M.L.; Palmisano, A.; De Palma, G.; Galletti, E.; Coggiola, M.; Buzio, C.; Mutti, A.; Vaglio, A. Asbestos and smoking as risk factors for idiopathic retroperitoneal fibrosis: A case-control study. Ann. Intern. Med. 2014, 161, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, A.; Maritati, F.; Vaglio, A. Chronic Periaortitis: An Update. Curr. Rheumatol. Rep. 2018, 20, 80. [Google Scholar] [CrossRef] [PubMed]
- Greco, P.; Vaglio, A.; Corradi, D.; Cobelli, R.; Zompatori, M.; Buzio, C. Tuberculosis as a trigger of retroperitoneal fibrosis. Clin. Infect. Dis. 2005, 41, e72–e75. [Google Scholar] [CrossRef] [PubMed]
- Fenaroli, P.; Maritati, F.; Vaglio, A. Into Clinical Practice: Diagnosis and Therapy of Retroperitoneal Fibrosis. Curr. Rheumatol. Rep. 2021, 23, 18. [Google Scholar] [CrossRef]
- Ramshaw, A.L.; Roskell, D.E.; Parums, D.V. Cytokine gene expression in aortic adventitial inflammation associated with advanced atherosclerosis (chronic periaortitis). J. Clin. Pathol. 1994, 47, 721–727. [Google Scholar] [CrossRef]
- Kermani, T.A.; Crowson, C.S.; Achenbach, S.J.; Luthra, H.S. Idiopathic retroperitoneal fibrosis: A retrospective review of clinical presentation, treatment, and outcomes. Mayo Clin. Proc. 2011, 86, 297–303. [Google Scholar] [CrossRef]
- Scheel, P.J., Jr.; Feeley, N. Retroperitoneal fibrosis: The clinical, laboratory, and radiographic presentation. Medicine (Baltimore) 2009, 88, 202–207. [Google Scholar] [CrossRef] [PubMed]
- van Bommel, E.F.H.; Jansen, I.; Hendriksz, T.R.; Aarnoudse, A. Idiopathic retroperitoneal fibrosis: Prospective evaluation of incidence and clinicoradiologic presentation. Medicine 2009, 88, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Moriconi, D.; Giannese, D.; Capecchi, R.; Cupisti, A.; Barsotti, S.; Morganti, R.; Orsitto, E.; Gaetano Tavoni, A.; Francesca Egidi, M. Risk factors for relapse and long-term outcome of idiopathic retroperitoneal fibrosis. Clin. Exp. Nephrol. 2019, 23, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Gianfreda, D.; Superchi, E.; Peyronel, F.; Mazzariol, M.; Vaglio, A. Chronic periaortitis: A clinical approach. Rev. Med. Interne 2023, 44, 79–84. [Google Scholar] [CrossRef]
- Vaglio, A.; Diamond, E.L. Erdheim-Chester disease: The “targeted” revolution. Blood 2017, 130, 1282–1284. [Google Scholar] [CrossRef] [PubMed]
- Emile, J.F.; Abla, O.; Fraitag, S.; Horne, A.; Haroche, J.; Donadieu, J.; Requena-Caballero, L.; Jordan, M.B.; Abdel-Wahab, O.; Allen, C.E.; et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016, 127, 2672–2681. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, L.; Hervier, B.; Neel, A.; Hamidou, M.A.; Kahn, J.E.; Wechsler, B.; Perez-Pastor, G.; Blomberg, B.; Fuzibet, J.G.; Dubourguet, F.; et al. CNS involvement and treatment with interferon-alpha are independent prognostic factors in Erdheim-Chester disease: A multicenter survival analysis of 53 patients. Blood 2011, 117, 2778–2782. [Google Scholar] [CrossRef]
- Kumar, P.; Singh, A.; Gamanagatti, S.; Kumar, S.; Chandrashekhara, S.H. Imaging findings in Erdheim-Chester disease: What every radiologist needs to know. Pol. J. Radiol. 2018, 83, e54–e62. [Google Scholar] [CrossRef]
- Wu, Z.; Jiang, G.L.; Tang, Y.; Jiang, C.; Sun, L.L.; Li, N.; Jin, Z.Y.; Sun, H. Urinary involvement in Erdheim-Chester disease: Computed tomography imaging findings. Abdom. Radiol. 2021, 46, 4324–4331. [Google Scholar] [CrossRef]
- Gianfreda, D.; Palumbo, A.A.; Rossi, E.; Buttarelli, L.; Manari, G.; Martini, C.; De Filippo, M.; Vaglio, A. Cardiac involvement in Erdheim-Chester disease: An MRI study. Blood 2016, 128, 2468–2471. [Google Scholar] [CrossRef]
- Papo, M.; Emile, J.F.; Maciel, T.T.; Bay, P.; Baber, A.; Hermine, O.; Amoura, Z.; Haroche, J. Erdheim-Chester Disease: A Concise Review. Curr. Rheumatol. Rep. 2019, 21, 66. [Google Scholar] [CrossRef] [PubMed]
- Vaglio, A.; Palmisano, A.; Alberici, F.; Maggiore, U.; Ferretti, S.; Cobelli, R.; Ferrozzi, F.; Corradi, D.; Salvarani, C.; Buzio, C. Prednisone versus tamoxifen in patients with idiopathic retroperitoneal fibrosis: An open-label randomised control trial. Lancet 2011, 378, 338–346. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| IgG4-TIN | IRF | ECD | |
|---|---|---|---|
| Renal and urinary clinical manifestations |
|
|
|
| Constitutional symptoms | Rarely | Frequently | Frequently |
| Renal involvement |
|
|
|
| Ureteral involvement | If associated IgG4-RPF |
| Mono or bilateral ab extrinseco obstruction of proximal portion |
| Other organs involvement | Pancreas, bile ducts, retroperitoneum, mediastinum, orbit, salivary glands, lymph nodes, etc. | Abdominal and/or thoracic aorta (lateral and anterior side), periaortic arteries, inferior cava, iliac vessels | Circumferential abdominal and/or thoracic aorta (“coated aorta”), periaortic arteries, bone, heart, lungs, orbits, CNS, glands, skin |
| Laboratory findings | Hypocomplementemia Elevated serum IgG and IgG4 levels Elevated total IgE CRP levels in range | Elevated ESR and CPR levels | Elevated ESR and CPR levels |
| TC scan findings |
|
|
|
| MRI findings |
|
|
|
| PET scan findings | Increased uptake | Increased uptake in active stages | Increased uptake in active stages |
| Main histological findings |
|
| Non-Langerhans histiocytes (CD68+/CD1a− “foamy” macrophages); lymphocytes. |
| Therapy |
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, G.M.; Pala, C.; Gianfreda, D. Renal and Urinary Tract Involvement in Fibrosclerosing or Fibroinflammatory Diseases: A Narrative Review. Rheumato 2024, 4, 1-12. https://doi.org/10.3390/rheumato4010001
Rossi GM, Pala C, Gianfreda D. Renal and Urinary Tract Involvement in Fibrosclerosing or Fibroinflammatory Diseases: A Narrative Review. Rheumato. 2024; 4(1):1-12. https://doi.org/10.3390/rheumato4010001
Chicago/Turabian StyleRossi, Giovanni Maria, Chiara Pala, and Davide Gianfreda. 2024. "Renal and Urinary Tract Involvement in Fibrosclerosing or Fibroinflammatory Diseases: A Narrative Review" Rheumato 4, no. 1: 1-12. https://doi.org/10.3390/rheumato4010001