
Mechanism-of-Action-Based Development of New Cyclophosphamides
Institute of Biochemistry II, Goethe University Frankfurt Medical School, 60590 Frankfurt, Germany
SynBio 2023, 1(2), 158-171; https://doi.org/10.3390/synbio1020011
Submission received: 11 July 2023
/
Revised: 16 August 2023
/
Accepted: 21 August 2023
/
Published: 24 August 2023
(This article belongs to the Special Issue Feature Paper Collection in Synthetic Biology)
Abstract
:Even more than 60 years after its introduction into the clinic, cyclophosphamide (CP), which belongs to the group of alkylating cytostatics, is indispensable for the treatment of cancer. This is despite the fact that its exact mechanism of action was unknown until a few years ago, and therefore, all attempts to improve the effectiveness of CP failed. The reason for not knowing the mechanism of action was the uncritical transfer of the chemical processes that lead to the formation of the actual alkylating CP metabolite phosphoreamide mustard (PAM) in vitro to in vivo conditions. In vitro—e.g., in cell culture experiments—PAM is formed by β-elimination of acrolein from the pharmacologically active CP metabolite aldophosphamide (ALD). In vivo, on the other hand, it is formed by enzymatic cleavage of ALD by phosphodiesterases (PDE) with the formation of 3-hydroxypropanal (HPA). The discovery of HPA as a cyclophosphamide metabolite, together with the discovery that HPA is a proapoptotic aldehyde and the discovery that the cell death event in therapy with CP is DNA-alkylation-initiated p53-controlled apoptosis, led to the formulation of a mechanism of action of CP and other oxazaphosphorine cytostatics (OX). This mechanism of action is presented here and is confirmed by newly developed CP-like compounds with lower toxicity and an order of magnitude better effectiveness.
1. Introduction
Nitrogen mustard (NM), the oldest cytostatic, was first used as a cytostatic in the 1940s to treat a malignant lymphoma [1]. NM exerts its biological effect by preventing DNA replication by binding DNA and creating crosslinks between DNA strands. The disadvantage of NM is that it not only inhibits the replication of fast-growing tumor cells but also the growth of all fast-growing cells. In order to make NM more tumor-specific and less toxic, Brock [2] developed the concept “active form, transport form”. NM should be chemically masked in such a way that it is transported to the tumor as a non-toxic transport form, where it should then be converted into the active form.
Based on the (wrong) assumption of increased phosphoamidases activity in tumor cells [3]—enzymes that catalyze the cleavage of phosphorus–nitrogen bonds—a number of compounds were synthesized in which NM was chemically masked by a phosphoramide bond. These compounds—according to the idea—were to be cleaved by phosphoamidases in tumor cells into NM and the phosphoric acid residue. Of the substances synthesized according to this specification, CP proved to be the most effective in animal experiments and was introduced into the clinic in Germany under the name “Endoxan” in 1958 [4]. Soon after its introduction into the clinic, it turned out that the idea underlying the development of CP was wrong. It quickly became apparent that Endoxan, as an ineffective prodrug, had to be activated by cytochrome P450 enzymes in the liver.
2. The Metabolism of Cyclophosphamide (Figure 1)
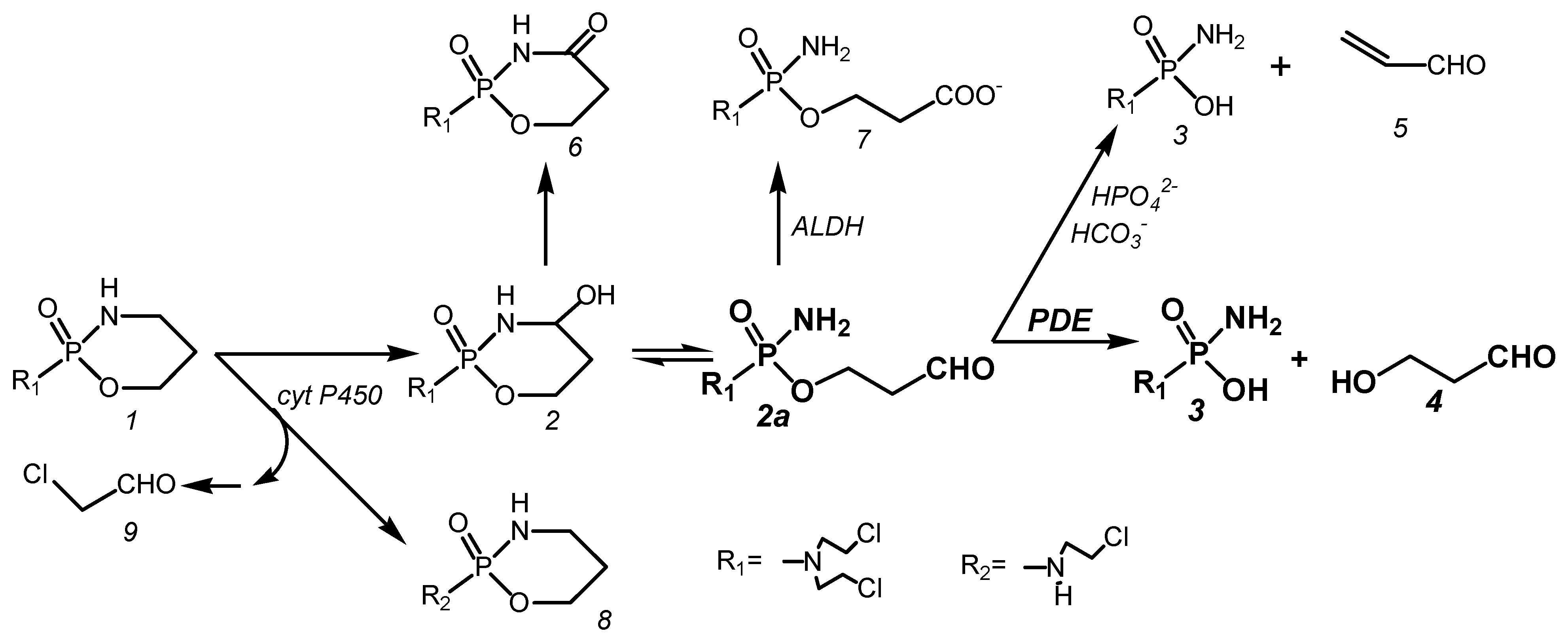
Endoxan, today better known as Cyclophosphamide (CP), is an ineffective prodrug that is hydroxylated in the liver by cytochrome P450 enzymes to 4-hydroxy-cyclophosphamide (CPOH). CPOH forms an equilibrium mixture with its tautomeric aldehyde aldophosphamide (ALD). ALD is the pharmacologically active metabolite from which the DNA-alkylating metabolite phosphoreamide mustard (PAM) is released in vitro by β-elimination of acrolein or in vivo by enzymatic cleavage by phosphodiesterases (PDE) to form 3-hydroxypropanal (HPA). The formation of PAM from CP in vivo is accompanied by side reactions. These are the formation of toxic chloroacetaldehyde by side-chain hydroxylation by cytochrome P450 enzymes, the formation of 4-ketocyclophosphamide from CPOH, and the oxidation of ALD to non-cytotoxic carboxyphosphamide (CARB).
Figure 1.
Metabolism of CP.

At this point, it should be pointed out that for a long time, the formation of PAM in vitro by β-elimination of acrolein was uncritically transferred to in vivo conditions. As a result, the formation of HPA in vivo was simply overlooked in the formation of PAM by enzymatic cleavage. As a consequence, the mechanism of action in which HPA plays the crucial role was not elucidated.
CP (1) is hydroxylated in vivo to CPOH (2) by cyt P450 hydroxylases. CPOH equilibrates with its ring open tautomer ALD (2a). In vitro, ALD becomes PAM (3) by β-elimination of acrolein (5). This reaction is catalyzed by phosphate and bicarbonate ions. In vivo, CPOH is detoxified to 4-ketocyclophosphamide (6), whereas ALD is cleaved by phosphodiesterases into PAM (3) and HPA (4). This reaction competes in vivo with the formation of CARB (7) catalyzed by aldehyde dehydrogenases (ALDH). A side reaction of the formation of CPOH is the formation of dechloro-cyclophosphamide (8) and toxic chloroacetaldehyde (9) by side-chain hydroxylation by cytochrome P450 enzymes.
After intravenous injection of 358 nmol/g CP corresponding to 100 mg/kg in female NMRI mice, 328 nmol/g (92%) is hydroxylated to CPOH. Of the CPOH formed, 266 nmol/g (81%) is detoxified to CARB and ketocyclophosphamide, so that only 19% of the CPOH formed remains for toxic and therapeutic reactions [5].
3. The Mechanism of Action of Cyclophosphamide and Other Oxazaphosphorins
In 1976/1977, Brock [6] and Brock and Hohorst [7] showed that of all CP metabolites, only CPOH has a therapeutic index, like CP. The therapeutic index—the ratio from the amount of a therapeutic agent that causes toxicity to the amount that causes the therapeutic effect—was determined to be greater than 100 for CP and CPOH but only 3.5 for PAM measured in Yoshida ascites sarcoma-bearing rats. Why DNA-alkylating PAM released from CP in vivo has a 30- to 40-fold higher therapeutic index than injected chemically pure PAM was a mystery and remained unconsidered in attempts to improve the efficacy of CP. Instead, efforts to improve the efficacy of CP have focused on concentrating the alkylating phosphoreamide mustard in tumor cells as evidenced by the development of glufosfamide (β-D-glucose-isophosphoreamide mustard) [8], in which I-phosphoreamide mustard (I-PAM), the alkylating function of Ifosfamide (IF), is linked to glucose and which has been synthesized on the basis of the rationale that cancer cells have an increased uptake of glucose.
Three discoveries had to be brought together to elucidate the mechanism of action of CP and other oxazaphosphorines. These were the discovery of HPA as a CP metabolite [9], the discovery that HPA is a proapoptotic aldehyde [10], and the discovery that DNA alkylation by PAM is not the event leading to cell death, but rather the p53-controlled apoptosis initiated by it [11].
HPA—an antibiotic produced by Lactobacillus reuteri and submitted to the culture medium—is also known as reuterin. It has been shown to be active against bacteria viruses and fungi [12]. It is used as a food additive to prevent spoilage by growth of pathogens. Experiments by Iyer [10], who investigated the effects of the supernatant of L. reuteri cultures on tumor necrosis factor (TNF)-activated apoptose signaling pathways in human leukemia cells, showed that reuterin inhibits (i) the formation of the anti-apoptotic proteins Bcl-2 and Bcl-xL and (ii) the TNF-dependent NF-κB activation by inhibition of the translocation of the p65 subunit of NF-κB into the nucleus. The experiments of Iyer et al. further showed that the degradation of IκBα by lack of the ubiquitination of IκBα is suppressed by the supernatant of LR. Thus, by the inhibition of the nuclear translocation of NF-κB, apoptosis is enhanced [13], comparable to the effect often seen in combination chemotherapies including glucocorticoids which inhibit NF-κB.
Schwartz and Waxman [11] investigated the effect of CPOH on the caspase 8 (extrinsic) and caspase 9 (intrinsic) dependent pathways of apoptosis in 9 L tumor cells and 9 L tumor cells transduced with CYP2B6; the latter is a liver P450 enzyme that hydroxylates CP to CPOH. Contrary to other anticancer drugs like doxorubicin and cisplatin, in which activation of caspase 8 is the initial apoptotic event, after application of CPOH [14], activation of caspase 9 was detectable before the activation of caspase 8. In addition, caspase 9 was activated to a greater extent than caspase 8, indicating the p53-mediated apoptotic pathway is triggered by CPOH. This finding is in agreement with the report that caspase-8-specific inhibitors only block cisplatin but not CP-induced apoptosis [15].
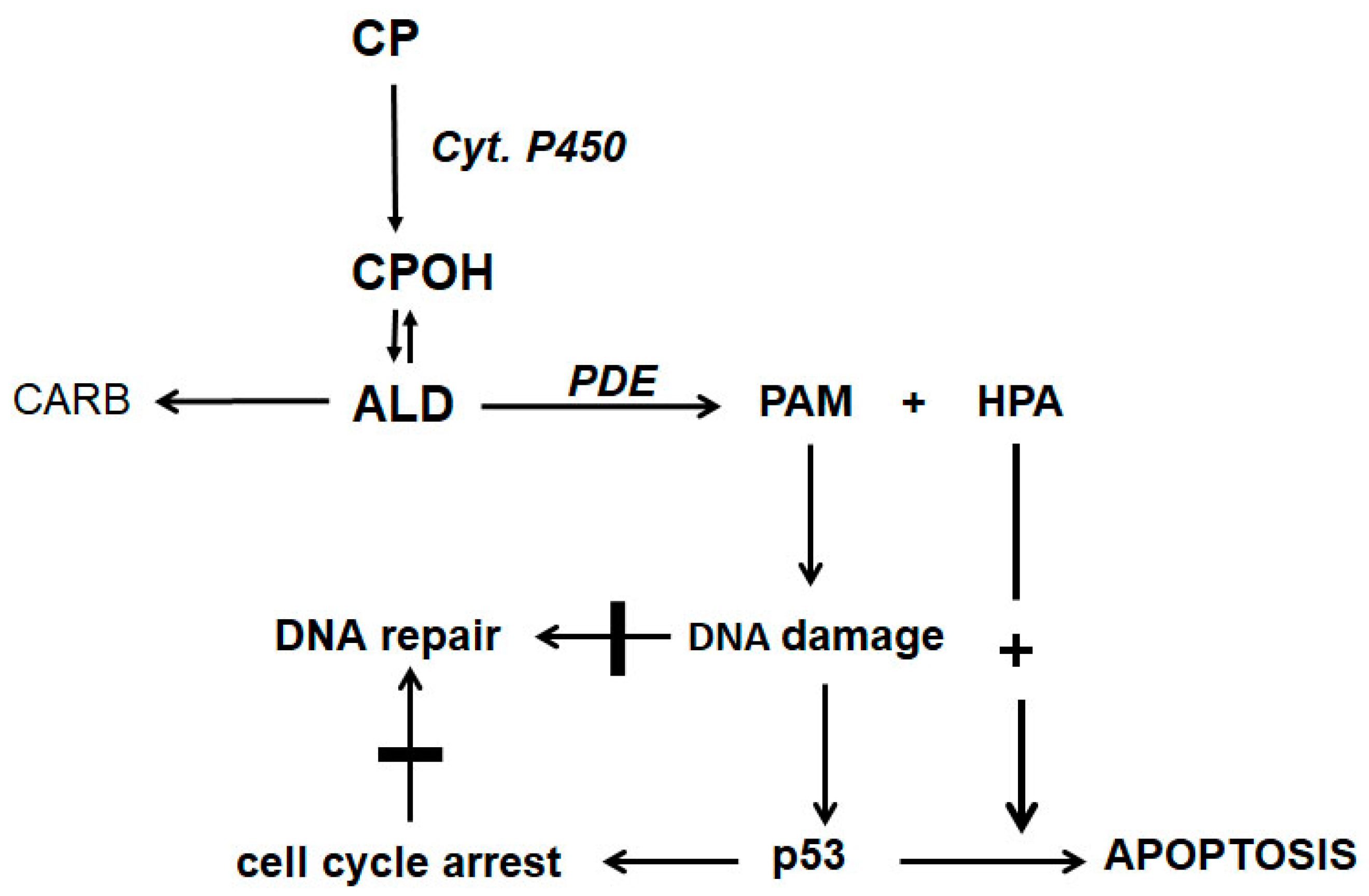
The discovery of HPA as a CP metabolite together with the findings of Iyer et al. [10] and Schwartz and Waxman [11] suggest the scheme for the mechanism of action of CP and other oxazaphosphorins shown in Figure 2.
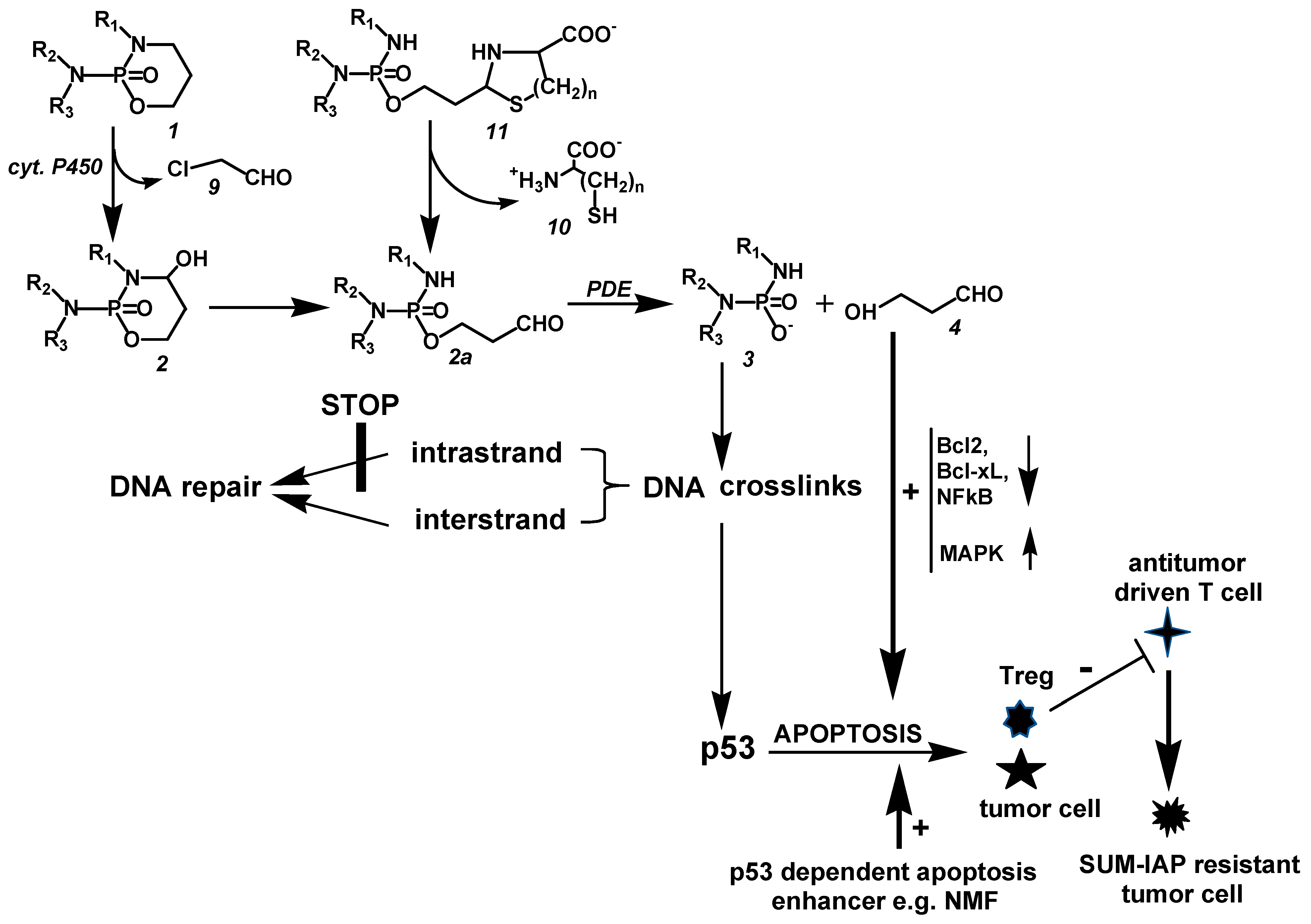
CP is hydroxylated by P450 enzymes in the liver to CPOH, which is in equilibrium with ALD. A part of ALD, the pharmacologic active metabolite, is oxidized by aldehyde dehydrogenases to nontoxic CARB. ALD not oxidized is decomposed by PDE to the alkylating PAM and proapoptotic HPA. PAM damages DNA by alkylation. The alkylated DNA is either repaired immediately or, if this is not possible, the tumor suppressor protein p53 is activated, which induces cell cycle stop to give the cell time to repair the damage. If DNA repair is not possible (indicated by the thick horizontal lines in Figure 2), p53 induces apoptosis, which is, and this is special for oxazaphosphorine cytostatics, enhanced by HPA, which supports apoptosis by inhibiting the antiapoptotic proteins Bcl-2 and Bcl-xL and by inhibiting NF-κB activation. Additionally, HPA promotes apoptosis by enhancing mitogen-activated protein kinase (MAPK) activities by enhancing JNK and p38 phosphorylations.
4. Experimental Confirmation of the Mechanism of Action
If the proposed reaction scheme should apply to the mechanism of action, two conditions must be met. First, ALD is enzymatically cleaved into PAM and HPA. Second, apoptosis assisted by HPA is the event leading to the cell death.
The enzymatic cleavage of ALD is described in the scientific literature [9,16,17], and that it is essential for antitumor efficacy is shown by the experiments by Erven and Skupin on regional perfusion of the tumor-bearing limb in rats [18,19] described in a previous paper [20]. Rats bearing solid growing Yoshida ascites tumor cells transplanted into the m. gastrocnemius were perfused for 30 min via a. epigastrica and v. epigastrica with an oxygenated hemoglobin solution containing CPOH. During perfusion, the blood supply to the leg was disconnected from the body circulation by a tourniquet. No rat was cured by perfusion with the highest tolerable concentration of CPOH. But contrary to therapy by limb perfusion, 50% of rats with the tumor in the same place were cured with a single dose of 125 mg/kg CP administered intraperitoneally. In the CP experiment, the highest concentration of CPOH in blood samples drawn from the a. epigastrica after CP administration was 0.05 μmol/mL, and the area under the curve (auc) was ~3 μmol min compared to a maximum CPOH concentration of 5 μmol/mL and an auc of ~37 μmol min in the perfusion experiment. The result—according to which no therapeutic effect was measured after regional perfusion with oxygenated hemoglobin solution, which did not contain any enzymes that split ALD into PAM and HPA, although the auc of CPOH was 12 times higher than after CP injection—is a strong indication that ALD must be modified enzymatically in vivo.
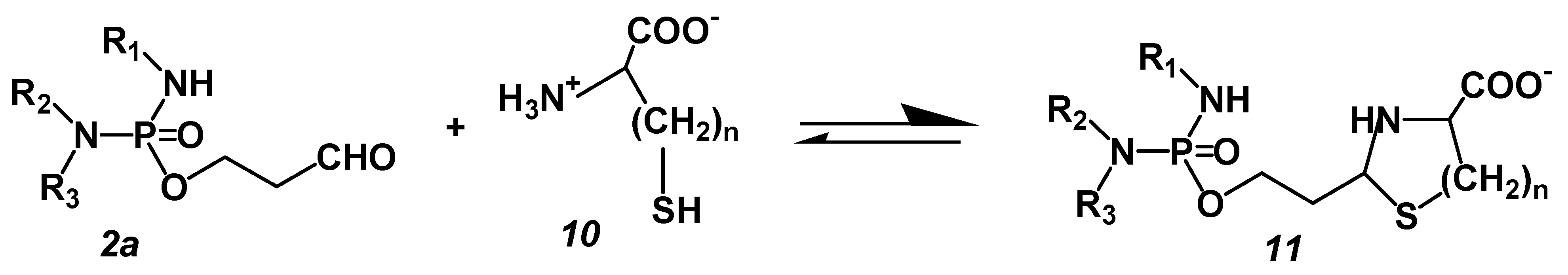
The experiments with the perhydrothiazine derivatives of IALD described below show that HPA-assisted apoptosis is the event leading to cell death. These compounds were chosen because CP as a chance discovery is not optimal for the mechanism of action. The discovery of CP was an accidental finding. Disadvantages are the formation of toxic chloro-acetaldehyde as a byproduct of the hydroxylation reaction in the liver and the formation of the pharmacologically active metabolite ALD from CPOH because CPOH is responsible for that part of CP toxicity that is not due to the alkylating metabolite PAM, e.g., the pronounced urotoxicity of CP but especially of IF. The cause of this CPOH toxicity is the reaction of CPOH with nucleophilic groups, e.g., SH groups of membrane proteins [21]. Because CPOH toxicity would mask apoptosis initiated by DNA alkylation, the following experiments were performed with thiazolidines (TIA) or perhydrothiazines (PT) of I-aldophosphamide (IALD). These compounds are formed when ALD or IALD react with β- or γ-aminothiols like cysteine or homocysteine. They hydrolyze spontaneously, bypassing CPOH or 4-hydroxy-I-fosfamide (IFOH) to ALD or IALD and cysteine or homocysteine (Figure 3). TIA and PT were synthesized as described in detail in [22] and tested for their suitability. They are 7–9 times less toxic than CPOH or IFOH in mice and showed antitumor activity against P388 tumor-bearing CD2F1 mice equivalent to or better than CPOH or IFOH [23].
ALD (2a) reacts with β- or γ-aminothiols such as cysteine (10, n = 1) or homocysteine (10, n = 2) to give TIA or PT, which dissociate in aqueous solution into the starting products ALD/IALD and the thiol.
ALD: R1 = H, R2 = R3 = −CH2CH2Cl, IALD: R1 = R2 = −CH2CH2Cl, R3 = H
According to the mechanism of action scheme (Figure 2), as already said, the event leading to cell death is apoptosis initiated by DNA damage caused by PAM. The apoptosis yield is influenced by the extent of repair of the damaged DNA. Interstrand crosslinks, unlike intrastrand crosslinks, are rapidly repaired by cellular repair systems [24,25] and thus have a lower apoptotic yield than intrastrand crosslinks. The type of DNA strand crosslinking, whether interstrand or intrastrand crosslinking, can be influenced by the alkylating function of an alkylating substance. 2-chloroethyl groups (–CH2CH2Cl) generate easily repairable interstrand crosslinks, whereas 2-mesyl-ethyl groups (–CH2CH2OSO2CH3) generate non- or poorly repairable intrastrand crosslinks (http://www.atdbio.com/content/16/nucleic-acid-drug-interactions (accessed on 10 July 2023)). To test the accuracy of the proposed mechanism of action, I-aldophosphamide-thiazolidine and I-aldophosphamide-perhydrothiazine derivatives with 2-chloroethyl-and 2-mesyl-ethyl groups in the alkylating function were synthesized and tested in therapy experiments in P388 tumor-bearing CD2F1 mice. The Ifosfamide derivatives were chosen because they are easy to synthesize and easy to handle. If the proposed mechanism of action is correct, the mesyl derivatives should show significantly better antitumor efficacy. The results of this experiment are summarized in Table 1. The values in Table 1 are taken from the published paper [23].
The result of the experiment shows that both the thiazolidine and the perhydrothiazine derivatives of IALD with a 2-mesyl-ethyl group in the alkylating function are significantly more effective than the corresponding compounds containing only 2-chloroethyl groups in the alkylating function. Thus, with compound 21, an increase in life span (ILS) of 160% is obtained with no long-term survivors, while with compound 22, which has a 2-mesyl-ethyl group in the alkylating function, an ILS of 237% is obtained with 2/5 long-term survivors. The same is true for thiazolidine derivatives 11 and 12; again, compound 12 with a mesyl group in the alkylating function is clearly more effective. Based on body weight, the doses administered are non-toxic.
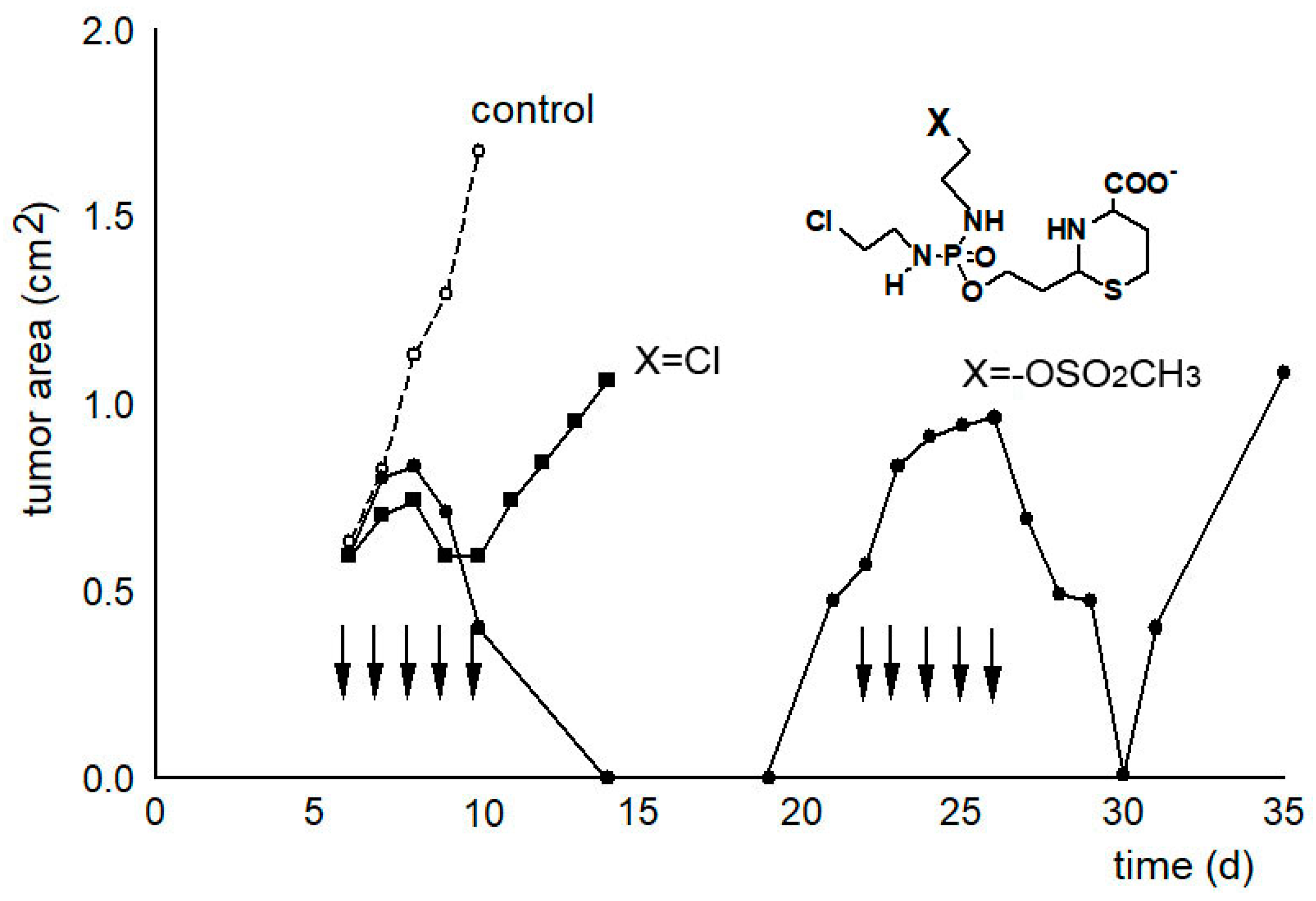
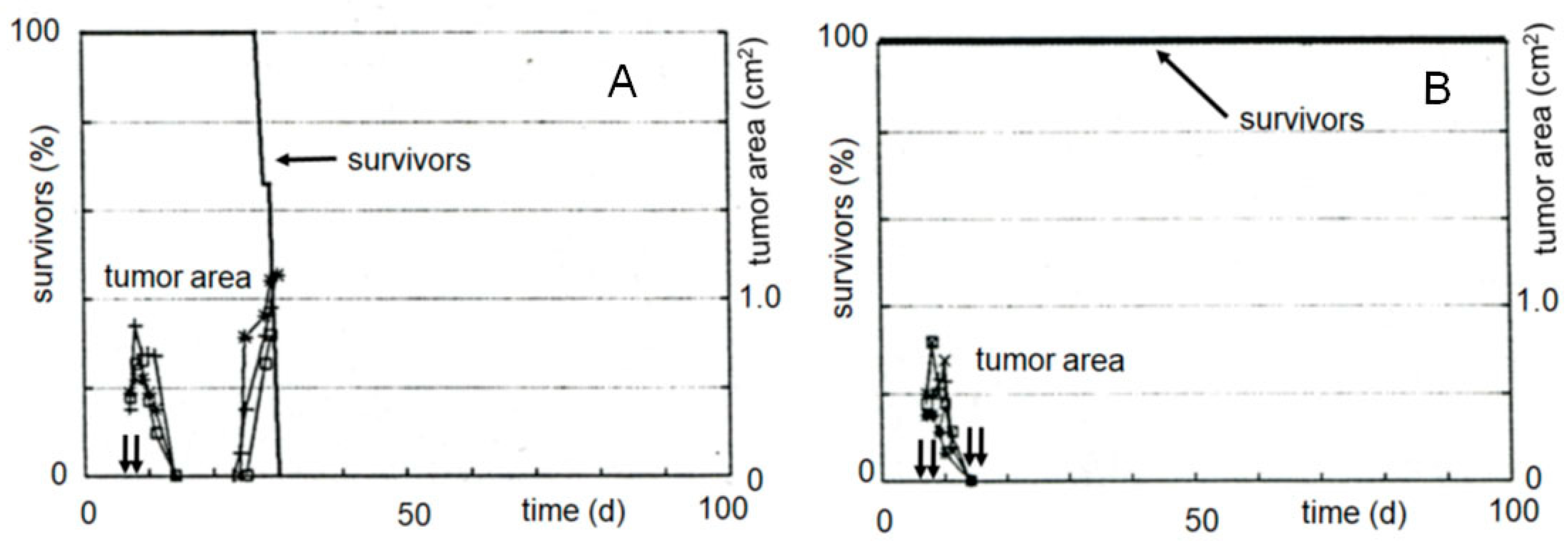
In another experiment, the tumor growth curves were determined as a measure of the therapeutic effectiveness (see Figure 4). While only a slight delay in tumor growth can be measured after therapy with compound 21 called “I-aldophosphamide perhydrothiazine” (IAP, formula see Table 1 or Figure 4), the alkylating function of which only contains 2-chloroethyl groups, the tumor size is suppressed below the detection limit for 7 days after therapy with compound 22 called “sulfonyl methyl-I-aldophosphamide perhydrothiazine” (SUM-IAP). After an interval of 7 days in which the tumor appeared to disappear, the tumor grew again. Re-therapy with SUM-IAP was less successful probably because tumor cells had become resistant to SUM-IAP.
Under the simplifying assumption that the measured tumor area is proportional to the tumor mass (correlation coefficient 0.93) and number of tumor cells and that the tumor cells are in the exponential growth phase, this and additional tumor growth curves were evaluated using the back extrapolation method according to Alexander and Mikulski [26]. From these experiments, it can be concluded that there is a 104–105 times increase in antitumor activity when one chlorine in the IAP molecule is substituted by a mesyl group in SUM-IAP. These results demonstrate that the postulated mechanism of action for OX reflects the real conditions and further shows that the antitumor efficacy of CP or IF is a mixed effect of PAM (IPAM)-initiated apoptosis and direct cytotoxicity of PAM (IPAM). This combination of apoptosis and direct cytotoxicity is shifted towards apoptosis in compounds in which one chlorine atom of the alkylating function is substituted by a mesyl group.
Figure 4.
Tumor growth curves of subcutaneously transplanted P388 tumors in female CD2F1 mice. Therapy with 0.5 mmol/kg (200 mg/kg) IAP (X = Cl) and 0.3 mmol/kg (133 mg/kg) SUM-IAP (X = −OSO2CH3) s.c. on days 7–11 and days 22–26 (arrows), mean of 3 animals, control: tumor growth curves of untreated animals. Figure 4 is an adaptation to the newly postulated mechanism of action of CP of the tumor growth curves from Figure 2 reference [27]. This reference also describes in detail the methodological details that led to the generation of the curve data.
Figure 4.
Tumor growth curves of subcutaneously transplanted P388 tumors in female CD2F1 mice. Therapy with 0.5 mmol/kg (200 mg/kg) IAP (X = Cl) and 0.3 mmol/kg (133 mg/kg) SUM-IAP (X = −OSO2CH3) s.c. on days 7–11 and days 22–26 (arrows), mean of 3 animals, control: tumor growth curves of untreated animals. Figure 4 is an adaptation to the newly postulated mechanism of action of CP of the tumor growth curves from Figure 2 reference [27]. This reference also describes in detail the methodological details that led to the generation of the curve data.

From the experimental results presented so far, the following requirements arise for the development of mechanism-of-action-based oxazaphosphorins: during the formation of the pharmacocologically active metabolite ALD or IALD, the formation of toxic chloroacetaldehyde and CPOH or IFOH must be avoided, and the alkylating function must be such that it produces poorly- or non-repairable DNA damage. A substance that meets these conditions is SUM-IAP, with which the following tests were carried out.
5. Anti-Metastatic Therapy Experiments with SUM-IAP
The therapy experiments with SUM-IAP were carried out on P388 tumor-bearing female CD2F1 mice 7 days after subcutaneous tumor transplantation when the tumor area was about 0.5 cm2.
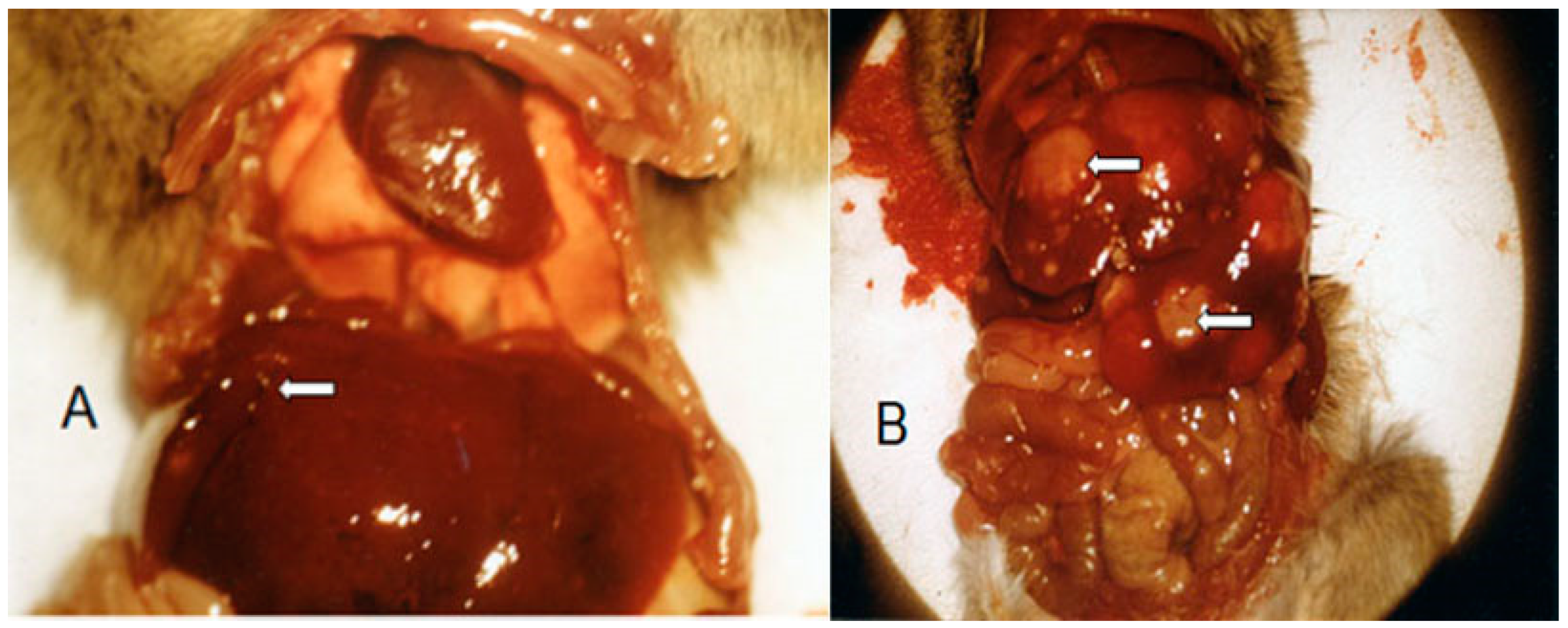
Although the antitumor activity of SUM-IAP compared to IAP was increased 104–105-fold by optimizing the alkylating function with regard to apoptosis initiation, the animals were not cured by treatment with SUM-IAP alone (surviving time > 100 d). The animals died due to growth of metastases between day 40 and day 60 after eradication of the primary tumor. Repeated treatment of the growing metastases with SUM-IAP was—as shown in Figure 4—unsuccessful. Due to the initial treatment, the tumors had become resistant to SUM-IAP. Postmortem examination of sacrificed mice showed that the liver is the starting point for the formation of metastases (see Figure 5A,B).
After subcutaneous transplantation of 106 P388 tumor cells into female CD2F1 mice, the animals were treated on days 7 to 11 with 266 mg/kg SUM-IAP. On days 25 and 50, the animals were sacrificed and examined for metastases. At 25 days after the start of therapy, small metastases can be seen on the liver (white arrow Figure 5A). At 50 days after the start of therapy, the entire abdomen is covered with metastases that kill the animal (Figure 5B).
The situation shown in Figure 5 was the situation in almost all experiments with SUM-IAP alone: eradication of the primary tumor but death of the animals due to metastases approximately between day 30 and day 60. The cause of the formation of metastases in the liver is very likely the detoxification of IALD to ineffective carboxylic acid by aldehyde dehydrogenases in the liver.
6. Therapy with SUM-IAP in Combination with Cis Platinum (CPT)
The assumption that formation of metastasis was due to neutralization of IALD in the liver by aldehyde dehydrogenases was the reason to combine SUM-IAP with CPT which is not detoxified in the liver. A total of 80% of the animals survived the observation period of 100 days in an experiment in which P388 tumor-bearing mice were treated with a SUM-IAP dose eradicating the primary tumor (266 mg/kg d7–11, 21–25) and additionally treated with 1.8 mg/kg CPT on days 13 and 27. But mice of a control group which were only treated with SUM-IAP died between day 52 and 70 due to growth of metastasis. CPT alone was only marginally effective at the administered dose (increase in life span of 67%). For methodological details and evaluation of the experiments, see bibliography [29].
7. Therapy Experiments with SUM-IAP in Combination with the Apoptosis Booster N-Methylformamide (NMF)
NMF was synthesized in 1953 as a solvent for the parenteral administration of water-insoluble substances in experimental chemotherapy. Since NMF was found to be active against S-180-Sarcoma in mice [30], clinical trials have been carried out but were canceled because signs of hepatotoxicity were observed. NMF was chosen for combination therapy with SUM-IAP because anti-metastatic activity of NMF was demonstrated [31].
Mice bearing subcutaneously growing P388 tumors were treated intraperitoneally with 266 mg/kg SUM-IAP on days 7–11. All animals developed visible metastases in the region of lymph nodes of forelegs until day 29. Postmortem examination showed liver metastases in all animals. But a further therapy test with the same dose and schedule of SUM-IAP application but with additional applications of 200 mg/kg NMF (12 injections on day 13 to 24) cured all animals. In the animals, neither visible external metastases nor liver metastases or metastases anywhere else in the abdominal range were found after postmortem examination [29]. The other most remarkable result in the experiment in which SUM-IAP was combined with NMF is lack of any sign of toxicity except a short but reversible decrease in the number of leukocytes after SUM-IAP injection. The body weight of the animal increased steadily during additional therapy with NMF. In control experiments with NMF alone, no or only marginal antitumor activity (ILS 12%) was detected. For methodological details and evaluation of the experiments, see bibliography [29].
Kalyany et al. [32]. investigated the interaction of NMF with commercially available superoxide dismutase 1 (SOD 1) by spectroscopic and molecular modeling studies. Their findings show that NMF causes loss of enzymatic activity due to perturbation of secondary structure of SOD 1. SOD 1 deficiency induces apoptosis associated with O2− accumulation in the cytoplasm and mitochondria, which increases loss of mitochondrial membrane potential and DNA-damage-mediated p53 apoptosis [33]. From this, it can be concluded that NMF is a booster of the p53-dependent apoptosis.
8. Immunological Anti-Metastatic Therapy with SUM-IAP
It is well known that CP has an immune-stimulating effect at low doses. This is due to special sensitivity of T-cell-inhibiting regulatory T cells (Treg) to CPOH. It is shown in the scientific literature that the high sensitivity is due to induction of apoptosis by CPOH and the decreased ability of Treg to repair damaged DNA [34].
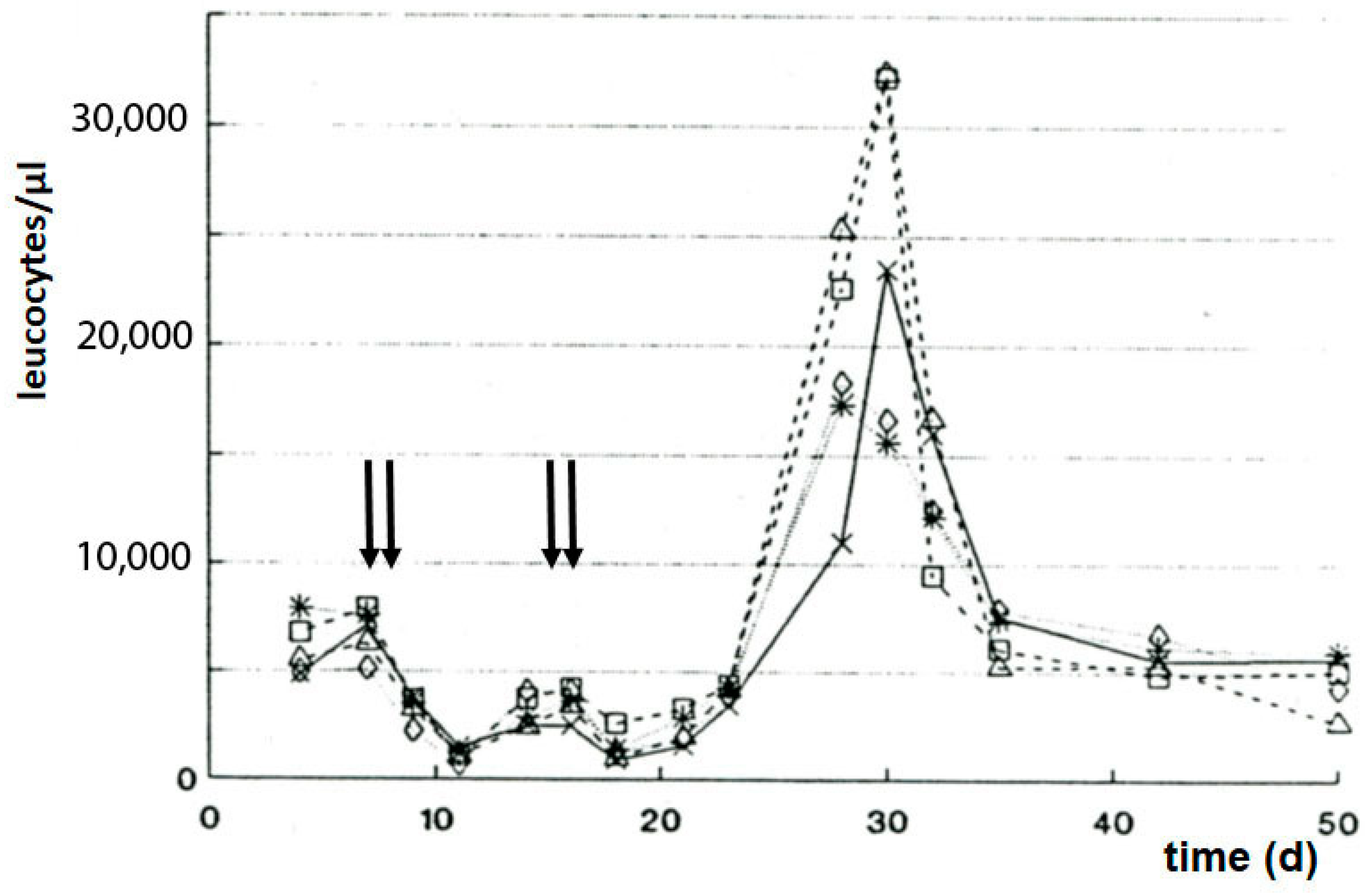
Seven days after subcutaneous P388 tumor transplantation, the mice were treated with a dose of SUM-IAP, which eradicated the transplanted tumor. Twenty-five to thirty days later, all animals had died due to the formation of metastases and regrowth of the primary tumor (Figure 6A). But when the SUM-IAP therapy was repeated on days 13 and 14 after tumor transplantation, no metastases were formed, and no tumor regrowth was observed; all animals survived (Figure 6B). Simultaneously, in the cured animals, a sharp increase in the number of leukocytes was noted (Figure 7). From the result of this experiment, it was concluded that the animals did not survive because the formation of metastases and regrowth of metastasis (resistant to SUM-IAP) was prevented by the cytotoxic effect of the second application of SUM-IAP, but by cytotoxic T cells which could unfold their effects freely because inhibitory Tregs were switched off by SUM-IAP.
Thus, SUM-IAP attacks tumor cells in two different ways: by boosting the DNA-alkylation-mediated apoptosis by HPA and further by apoptotic inhibition of Treg by which it is made possible that cytotoxic T cells can specifically attack tumor cells.
9. Special Feature of Aldophosphamide-Thiazolidine (TIA)
The experimental results shown so far demonstrate the efficacy of the SUM-IAP compound optimized for the OX mechanism of action. When therapeutic experiments were performed on P388 tumor-bearing CD2F1 mice with TIA (Figure 3, compound 11, R1 = H, R2 = R3 = −CH2CH2Cl, n = 1), symptoms were observed indicating intoxication of the central nervous system (CNS) but were avoided by adding 10 mol% cysteine to the injection solution. Subsequent organ distribution experiments showed that TIA was found in all organs including brain [36].
The described observations of symptoms indicating central toxicity in mice after intravenous injection of TIA, the avoidance of this symptoms by adding of L-cys to the solution for injection, and the finding that TIA was detectable in different tissues including the brain caused Jungkamp [37] to investigate the transport of 35S-TIA through cell membranes of Ehrlich ascites cells (EAT). Jungkamp found that TIA enters EAT by a strophanthin-inhibitable Na+ cotransport. The transport of TIA through EAT membranes is inhibited by L-cysteine. Accordingly, TIA is an oxazaphosphorine cytostatic that enters cells via Na+ cotransport. The transport of TIA through the cell membrane is linked to the thiazolidine ring. Evidence of this is that in experiments with aldophosphamide-perhydrothiazine (Figure 3, compound 11, R1 = H, R2 = R3 = −CH2CH2Cl, n = 2), no neurotoxic symptoms were observed. In addition, this aldophosphamide analog only insignificantly inhibits the uptake of L-cysteine in EAT.
10. Summary
Mechanism of Action of CP and Newly Developed CP-like Derivatives (See Figure 8)
CP (1, R1 = H, R2 = R3 = −CH2CH2Cl) is hydroxylated in the liver by Cyt.P450 enzymes to toxic CPOH (2, R1 = H, R2 = R3 = −CH2CH2Cl). A side reaction is side-chain hydroxylation with formation of toxic chloroacetaldehyde (9) and therapeutically ineffective dechloro-cyclophosphamide (1, R1 = R2 = H, R3 = CH2CH2Cl). CPOH becomes ALD (2a, R1 = H, R2 = R3 = −CH2CH2Cl) by ring opening; ALD is enzymatically decomposed by phosphodiesterases (PDE) in alkylating phosphoreamide mustard (PAM 3, R1 = H, R2 = R3 = −CH2CH2Cl) and 3-hydroxypropanal (HPA, 4). PAM causes interstrand DNA crosslinks that are quickly repaired by very effective cellular repair mechanisms. The unrepaired part initiates p53-dependent HPA-enhanced apoptosis.
Figure 8.
Mechanism of action of CP and newly developed CP-like derivatives (for details, see text).
Figure 8.
Mechanism of action of CP and newly developed CP-like derivatives (for details, see text).

Newly developed CP-like derivatives such as SUM-IAP (11, R1 = −CH2CH2OSO2CH3, R2 −CH2CH2Cl, R3 = H, n = 2) dissociate bypassing the formation of toxic CPOH and toxic chloroacetaldehyde (9) to SUM-IALD (2a, R1 = −CH2CH2OSO2CH3, R2 −CH2CH2Cl, R3 = H). SUM-IALD is enzymatically cleaved by PDE into SUM-IPAM (3, R1 = −CH2CH2OSO2CH3, R2 −CH2CH2Cl, R3 = H) and HPA. SUM-IPAM generates DNA intrastrand crosslinks that cannot be repaired by the cell’s own repair mechanisms. Therefore, the HPA enhanced p53-dependent apoptosis triggered by intrastrand crosslinks is many times greater than the apoptosis initiated by IF or CP and can be additionally enhanced by p53-dependent apoptosis enhancers such as NMF. In addition to tumor cells, regulatory T cells (Treg) that block antitumor-driven T cells are switched off by apoptosis so that antitumor-driven T cells can attack SUM-IAP-resistant tumor cells.
11. Discussion
There is hardly any scientific discipline in which the increase in knowledge over the last 50 years has been as great as in biochemistry. As a result, biochemical phenomena that could not be explained 10 or 20 years ago can be explained today. A good example of this is CP and other OX. When CP was introduced into the clinic in the late 1950s, little was known about what distinguished cancer cells from normal cells except that they grow faster and are therefore more sensitive than normal cells to DNA-damaging substances. Thus, it is not surprising that the mechanism of action of CP could not be explained for a long time. CP was classified in the group of substances damaging DNA by alkylation. But all attempts to make CP a better “alkylating substance” were in vain. The reason for this was that the event leading to cell death is not only DNA damage by alkylation, as was assumed for a long time due to limited biochemical knowledge, but apoptosis initiated by DNA alkylation. Only with the recognition of apoptosis as a general biological principle of regulation of cell homeostasis and the concurrence of three discoveries in metabolism and mechanism of action of CP was it possible to identify p53-regulated apoptosis initiated by DNA alkylation as the cause of cell death caused by CP. The discoveries are the discovery of HPA as a CP metabolite [9], the discovery of HPA as a proapoptotic agent [10], and the finding that the event leading to cell death is DNA damage-induced apoptosis [11]. The scheme for the mechanism of action formulated on the basis of these three findings allows—as the model substance SUM-IAP shows—the development of new cyclophosphamide derivatives with lower toxicity, an order of magnitude better antitumor effectiveness, and new application possibilities.
The difference in antitumor efficacy against P388 tumor-bearing mice between the accidentally found I-aldophosphamide (I-ALD) released from IAP and the modified SUM-I-aldophosphamide (SUM-I-ALD) specifically developed according to the postulated mechanism of action, released from SUM-IAP, shown in Figure 4, impressively confirms the correctness of the postulated mechanism of action, because due to irreparable DNA damage, SUM-IPAM results in a higher apoptotic yield and therefore is orders of magnitude more potent than I-ALD, which causes easily repairable DNA damage. At this point, it must be explicitly stated that SUM-IAP is many times less cytotoxic than IAP against P388 mouse leukemia cells in vitro. By incubation with 5μM IAP, about 25 times more P388 mouse leukemia cells die due to direct cytotoxicity than by incubation with the same concentration SUM-IAP [27].
Another important result of the experiments with SUM-IAP is that SUM-IAP in high antitumor doses prevents the formation of metastasis of the transplanted P388 tumor in mice by immunological mechanisms whereby the mice are cured by monotherapy with SUM-IAP. It is true that CP in low doses also stimulates the immune defense against tumors by inactivating regulatory T cells (Treg), which block antitumor-directed cytotoxic T cells. However, low doses of CP and the resulting weak apoptosis only inactivate Tregs, but not tumor cells, which are eradicated after CP injection by combined PAM-initiated apoptosis and direct, for the whole body toxic, PAM cytotoxicity. In SUM-IAP, this mixed effect is shifted in favor of less toxic apoptosis. By SUM-IAP, all rapidly proliferating cells—tumor cells and subpopulations of T cells—are attacked. The cells recover at different rates. The fastest recovering cells are surviving tumor cells and T effector cells, but less good are the Tregs, whose inhibitory influence on tumor-directed effector cells ceases, so that antitumor T cells can proliferate and specifically attack the remaining tumor cells.
It has long been evident that there is a correlation between the immune system and tumor growth because it had long been known from clinical observations that some tumors regress in the presence of bacterial infections. Based on the observations about the influence “welche heftige Erysipele auf die Rückbildung von Geschwulsten haben” (which violent erysipelas have on the regression of tumors), W. Busch undertook a specific healing attempt in 1865 to cure a patient “mit einem gewaltigen Sarcome der Halsdrüsen” (with an enormous sarcoma of the neck glands). The patient’s skin over the tumor was injured with a cauterizing iron. In the course of the prompt infection by streptococci, which was accompanied by fever, the tumor regressed within 2 weeks [38]. Further attempts to regress tumors by artificial infections were described by Coley in 1893, who injected tumor patients with dead bacteria [39]. Subsequently, the experiments were forgotten. One reason for this may have been that tumor therapy by stimulation of the immune system could not be reproduced in animal experiments because the detection of tumor-specific antigens, which are recognized as foreign by the immune system of the host organism, was not possible with the experimental animals available at that time. It was not possible to distinguish between immune reactions of one mouse against normal tissue and tumor tissue of another mouse. Only after Gross proved the existence of tumor-specific antigens by transplantation experiments in syngeneic mice in 1943 could attempts to treat tumors by the body’s own immunological mechanisms be resumed [40].
Today, we are on the cusp of an era in which immunological tumor therapy is becoming increasingly likely. A tool to achieve this goal can be substances such as SUM-IAP. They can both eradicate the tumor cell through apoptosis and suppress the inhibitory influence of Tregs on T effector cells directed against the tumor.
Other new applications for thiazolidin and perhydrothiazin derivatives of ALD and IALD are emerging for the therapy with apoptosis booster like NMF and for therapy of tumors of the CNS because the thiazolidin derivatives cross the blood–brain barrier.
Funding
This study was funded by the Bundesministerium für Forschung und Technologie.
Conflicts of Interest
The author declare no conflict of interest.
References
- Goodman, L.S.; Wintrobe, M.M. Nitrogen mustard therapy; use of methyl-bis (beta-chloroethyl) amine hydrochloride and tris (beta-chloroethyl) amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. J. Am. Med. Assoc. 1946, 132, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Brock, N. Transport-und Wirkform als chemotherapeutisches Prinzip in der Tumortherapie. Z. Für Krebsforsch. 1957, 62, 9–24. [Google Scholar] [CrossRef]
- Meyer, J.; Weinmann, J.P. Phosphamidase Content of Normal and Pathologic Tissues of the Oral Cavity. J. Histochem. Cytochem. 1953, 1, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Arnold, H.; Bourseaux, F.; Brock, N. Chemotherapeutic action of a cyclic nitrogen mustard phosphamide ester (B 518-ASTA) in experimental tumours of the rat. Nature 1958, 181, 931. [Google Scholar] [CrossRef] [PubMed]
- Voelcker, G.; Haeglsperger, R. Pharmacokinetics of cyclophosphamide and cyclophosphamide metabolites in the mouse and their influence on the therapeutic effect of “activated” cyclophosphamide (4-hydroxycyclophosphamide) (author’s transl). Arzneimittelforschung 1982, 32, 639–647. [Google Scholar]
- Brock, N. Comparative pharmacologic study in vitro and in vivo with cyclophosphamide (NSC-26271), cyclophosphamide metabolites, and plain nitrogen mustard compounds. Cancer Treat. Rep. 1976, 60, 301–308. [Google Scholar]
- Brock, N.; Hohorst, H.J. The problem of specificity and selectivity of alkylating cytostatics: Studies on N-2-chlorethylamido-oxazaphosphorines. Z. Für Krebsforsch. 1977, 88, 185–215. [Google Scholar] [CrossRef]
- Seker, H.; Bertram, B.; Bürkle, A.; Kaina, B.; Pohl, J.; Koepsell, H.; Wiesser, M. Mechanistic aspects of the cytotoxic activity of glufosfamide, a new tumour therapeutic agent. Br. J. Cancer 2000, 82, 629. [Google Scholar] [CrossRef]
- Voelcker, G. Enzyme Catalyzed Decomposition of 4-Hydroxycyclophosphamide. Open Conf. Proc. J. 2017, 8, 44–51. [Google Scholar] [CrossRef]
- Iyer, C.; Kosters, A.; Sethi, G.; Kunnumakkara, A.B.; Aggarwal, B.B.; Versalovic, J. Probiotic Lactobacillus reuteri promotes TNF-induced apoptosis in human myeloid leukemia-derived cells by modulation of NF-kappaB and MAPK signalling. Cell. Microbiol. 2008, 10, 1442–1452. [Google Scholar] [CrossRef]
- Schwartz, P.S.; Waxman, D.J. Cyclophosphamide induces caspase 9-dependent apoptosis in 9 L tumor cells. Mol. Pharmacol. 2001, 60, 1268–1279. [Google Scholar] [CrossRef]
- Cleusix, V.; Lacroix, C.; Vollenweider, S.; Duboux, M.; Le Blay, G. Inhibitory activity spectrum of reuterin produced by Lactobacillus reuteri against intestinal bacteria. BMC Microbiol. 2007, 12, 101. [Google Scholar] [CrossRef]
- Wu, M.; Lee, H.; Bellas, R.E.; Schauer, S.L.; Arsura, M.; Katz, D.; FitzGerald, M.J.; Rothstein, T.L.; Sherr, D.H.; Sonenshein, G.E. Inhibition of NF-kappaB/Rel induces apoptosis of murine B cells. EMBO J. 1996, 2, 4682–4690. [Google Scholar] [CrossRef]
- Seki, K.; Yoshikawa, H.; Shiiki, K.; Hamada, Y.; Akamatsu, N.; Tasaka, K. Cisplatin (CDDP) specifically induces apoptosis via sequential activation of caspase-8, -3 and -6 in osteosarcoma. Cancer Chemother. Harmacol. 2000, 45, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Susin, S.A.; Kroemer, G.; Debatin, K.M. Molecular ordering of apoptosis induced by anticancer drugs in neuroblastoma cells. Cancer Res. 1998, 58, 4453–4460. [Google Scholar] [PubMed]
- Bielicki, L.; Voelcker, G.; Hohorst, H.J. Enzymatic toxicogenation of “activated” cyclophosphamide by 3′-5′exonucleases. J. Cancer Res. Clin. Oncol. 1983, 105, 27–29. [Google Scholar] [CrossRef]
- Bielicki, L.; Voelcker, G.; Hohorst, H.J. Activated cyclophosphamide: An enzyme-mechanism-based suicige inactivator of DNA polymerase/3′-5′exonuclease. J. Cancer Res. Clin. Oncol. 1984, 107, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Erven, G. Vergleichende Untersuchungen Über Die Antitumorwirkung von Stabilisierten, Aktivierten Cyclophosphamid und cis Platin am Modell des Tumortragenden Rattenbeins Durch Regionale Perfusion. Ph.D. Dissertation, Justus-Liebig-Universität, Gießen, Germeny, 1986. [Google Scholar]
- Skupin, W. Die Regionale Perfusion des Tumortragenden Rattenbeins mit Stabilisierten, Sktivierten Cyclophosphamiden in Kombination mit Protektorthiolen. Ph.D. Dissertation, JustusLiebig-Universität, Gießen, Germeny, 1985. [Google Scholar]
- Voelcker, G. Mechanism of Action of Oxazaphosphorine Cytostatics Demonstrated by Regional Perfusion of the Tumor Bearing Limb in Rats with 4-Hydroxycyclophosphamide. EC Pharmacol. Toxicol. 10.3 2022. [Google Scholar]
- Voelcker, G. Causes and possibilities to circumvent cyclophosphamide. Toxic. Anti-Cancer Drugs 2020, 31, 617–622. [Google Scholar] [CrossRef]
- Zimmermann, J.; Bauer, H.H.; Hohorst, H.J.; Voelcker, G. Synthesis of I-aldofosfamide-perhydrothiazines. Arzneimittelforschung 2000, 50, 843–847. [Google Scholar] [CrossRef]
- Voelcker, G.; Hohorst, H.-J. Structure/activity studies with thiazolidinyl- and perhydrothizinyl-phosphamide ester. J. Cancer Res. Clin. Oncol. 1998, 124, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Povirk, L.F.; Shuker, D.E. DNA damage and mutagenesis induced by nitrogen mustards. Mutat. Res. 1994, 318, 205–226. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T.; Hiraku, Y.; Oikawa, S.; Mizutani, H.; Kojima, M.; Kawanishi, S. DNA intrastrand cross- at 5′-GA-3′ sequence formed by busulfan and its role in the cytotoxic effect. Cancer Sci. 2004, 95, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Alexander, P.; Mikulski, Z. Differences in the Response of Leukaemia Cells in Tissue Culture to Nitrogen Mustard and to Dimethyl Myleran. Biochem. Pharmacol. 1961, 5, 275–282. [Google Scholar] [CrossRef]
- Voelcker, G.; Pfeiffer, B.; Schnee, A.; Hohorst, H.J. Increased antitumour activity of mesyl-I-aldophosphamide-perhydrothiazine, in vivo but not in vitro, compared to I-aldophosphamide-perhydrothiazine. Cancer Res. Clin. Oncol. 2000, 126, 74–78. [Google Scholar]
- Voelcker, G. Mechanism of action of oxazaphosphorine cytostatics and antimetatatic experimental therapy with SUM-IAP a new Ifosfamide derivative adapted to the mechanism of action. Am. J. Med. Clin. Res. Rev. 2023, 2, 1–12. [Google Scholar]
- Voelcker, G. Enhancement of antitumor activity of the oxazaphosphorine cytostatic SUM-IAP by N-methylformamide. J. Cancer Res. Clin. Oncol. 2016, 142, 1183–1189. [Google Scholar] [CrossRef]
- Clarke, C.A.; Philips, S.F.; Sternber, S.S. Effects of N-methylformamideand related compounds in sarcoma 180. Proc. Soc. Exp. Biol. Med. 1953, 84, 203–207. [Google Scholar] [CrossRef]
- Iwakawa, M.; Tofilon, P.J.; Hunter, N.; Stephens, L.C.; Milas, L. Antitumor and antimetastatic activity of the differentiating agent N-methylformamide in murine tumor systems. Clin. Exp. Metastasis 1987, 5, 289–300. [Google Scholar] [CrossRef]
- Kalyani, D.; Jyothi, K.; Sivaprakasam, C.; Nachiappan, V. Spectroscopic N-methylformamide with superoxide dismutase. Mol. Biomol. Spectrosc. 2014, 124, 148–152. [Google Scholar] [CrossRef]
- Watanabe, K.; Shibuya, S.; Koyama, H.; Ozawa, Y.; Toda, T.; Yokote, K.; Shimizu, T. Sod1 loss induces intrinsic superoxide accumulation leading to p53-mediated growth arrest and apoptosis. Int. J. Mol. Sci. 2013, 14, 10998–11010. [Google Scholar] [CrossRef] [PubMed]
- Heylmann, D.; Bauer, M.; Becker, H.; van Gool, S.; Bacher, N.; Steinbrink, K.; Kaina, B. Human CD4+CD25+ regulatory T cells are sensitive to low dose cyclophosphamide: Implications for the immune response. PLoS ONE 2013, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Voelcker, G. Immunostimulating and cancer-reductive experimental therapy with the oxazaphosphorine cytostatic SUM-IAP. Anticancer Drugs 2018, 29, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Voelcker, G. Aldophosphamide-thiazolidine (NSC-613060) an oxazaphosphorine cytostatic that crosses the blood brain barrier. Anticancer Drugs 2021, 32, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Jungkamp, W. Aktive Anreicherung von Aldophosphamidthiazolidincarbonsäure in Tierischen Zellen Untersuchungen an Ehrlich-ascitestumorzellen. Ph.D. Dissertation, Johann Wolfgang Goethe Universität, Frankfurt am Main, Germany, 1996. [Google Scholar]
- Busch, W. Verhandlungen ärztlicher Gesellschaften. Berl. Klin. Wschr. 1868, 5, 125–127. [Google Scholar] [CrossRef]
- Coley, W.B. The treatment of malignant tumors by repeated inoculations of crysipelas, with a report of 10 original cases. Am. J. Med. Sci. 1893, 105, 488–511. [Google Scholar] [CrossRef]
- Gross, L. Intradermal Immunization of C3H mice against a sarcoma that originated in an animal of the same line. Cancer Res. 1943, 3, 326–333. [Google Scholar]
Figure 2.
Mechanism of action of CP and other OX.

Figure 3.
Formation and hydrolysis of TIA (11, n = 1) and PT (11, n = 2).

Figure 5.
Metastases (white arrows) in the abdominal cavity of mice 25 (A) and 50 (B) days after therapy with SUM-IAP. The illustration has already been published in the literature citation [28].
Figure 5.
Metastases (white arrows) in the abdominal cavity of mice 25 (A) and 50 (B) days after therapy with SUM-IAP. The illustration has already been published in the literature citation [28].

Figure 6.
Survival and tumor growth curves of female CD2F1 mice after subcutaneous transplantation of 106 P388 murine leukemia cells and subcutaneous therapy with SUM-IAP. (A): 2 times 666 mg/kg SUM-IAP on days 7 and 8 (arrows); (B): 4 times 666 mg/kg on days 7, 8, 14, and 15 (arrows). Individual values of 5 mice. The mean value curves and methodological details can be found in the literature citation [35]. The different lines in the figure indicate the probability of survival in 20% steps and the tumor size in 0.5 cm2 steps.
Figure 6.
Survival and tumor growth curves of female CD2F1 mice after subcutaneous transplantation of 106 P388 murine leukemia cells and subcutaneous therapy with SUM-IAP. (A): 2 times 666 mg/kg SUM-IAP on days 7 and 8 (arrows); (B): 4 times 666 mg/kg on days 7, 8, 14, and 15 (arrows). Individual values of 5 mice. The mean value curves and methodological details can be found in the literature citation [35]. The different lines in the figure indicate the probability of survival in 20% steps and the tumor size in 0.5 cm2 steps.

Figure 7.
Number of leucocytes in blood of female CD2F1 mice bearing subcutaneously transplanted solid growing P388 tumors following therapy with 666 mg/kg SUM-IAP on days 7, 8, 14, and 15 (arrows). Individual values of 5 mice. The mean value curves and methodological details can be found in the literature citation [35]. The different lines in the figure indicate five thousand steps of the leukocyte count.
Figure 7.
Number of leucocytes in blood of female CD2F1 mice bearing subcutaneously transplanted solid growing P388 tumors following therapy with 666 mg/kg SUM-IAP on days 7, 8, 14, and 15 (arrows). Individual values of 5 mice. The mean value curves and methodological details can be found in the literature citation [35]. The different lines in the figure indicate five thousand steps of the leukocyte count.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Antitumor activity and toxicity of thiazolidines (n = 1) and perhydrothiazines (n = 2) of I-aldophosphamide. I-aldophosphamid thiazolidine compounds (n = 1): subcutaneous administration of 0.26 mmol/kg (corresponding to 100 mg/kg compound 11), days 1–5. I-aldophosphamide-perhydrothiazine (n = 2): subcutaneous administration of 0.51 mmol/kg (corresponding to 200 mg/kg compound 21) days 1–5, female CD2F1 mice, intraperitoneal transplantation of 106 P388 mouse leukemia cells on day 0.
Table 1.
Antitumor activity and toxicity of thiazolidines (n = 1) and perhydrothiazines (n = 2) of I-aldophosphamide. I-aldophosphamid thiazolidine compounds (n = 1): subcutaneous administration of 0.26 mmol/kg (corresponding to 100 mg/kg compound 11), days 1–5. I-aldophosphamide-perhydrothiazine (n = 2): subcutaneous administration of 0.51 mmol/kg (corresponding to 200 mg/kg compound 21) days 1–5, female CD2F1 mice, intraperitoneal transplantation of 106 P388 mouse leukemia cells on day 0.
 | ||||
| X | ILS 1 (%) | LTS 2 (%) | ΔG 3 (g) | |
| n = 1 | ||||
| 11 | Cl | 94 | 0/5 | 0 |
| 12 | −OSO2CH3 | 180 | 1/5 | 0 |
| n = 2 | ||||
| 21 (IAP) | Cl | 160 | 0/5 | −1 |
| 22 (SUM-IAP) | −OSO2CH3 | 237 | 2/5 | 0 |
1: ILS increase in life span, 2: LTS long time survivors (surviving time > 100 d), 3: ΔG difference in body weight at day 0 and the lowest body weight after drug administration.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Voelcker, G. Mechanism-of-Action-Based Development of New Cyclophosphamides. SynBio 2023, 1, 158-171. https://doi.org/10.3390/synbio1020011
AMA Style
Voelcker G. Mechanism-of-Action-Based Development of New Cyclophosphamides. SynBio. 2023; 1(2):158-171. https://doi.org/10.3390/synbio1020011
Chicago/Turabian StyleVoelcker, Georg. 2023. "Mechanism-of-Action-Based Development of New Cyclophosphamides" SynBio 1, no. 2: 158-171. https://doi.org/10.3390/synbio1020011