
Multi-Omics Analysis of NFE2L2-Altered TCGA-Cervical Squamous Cell Carcinoma Patients †


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
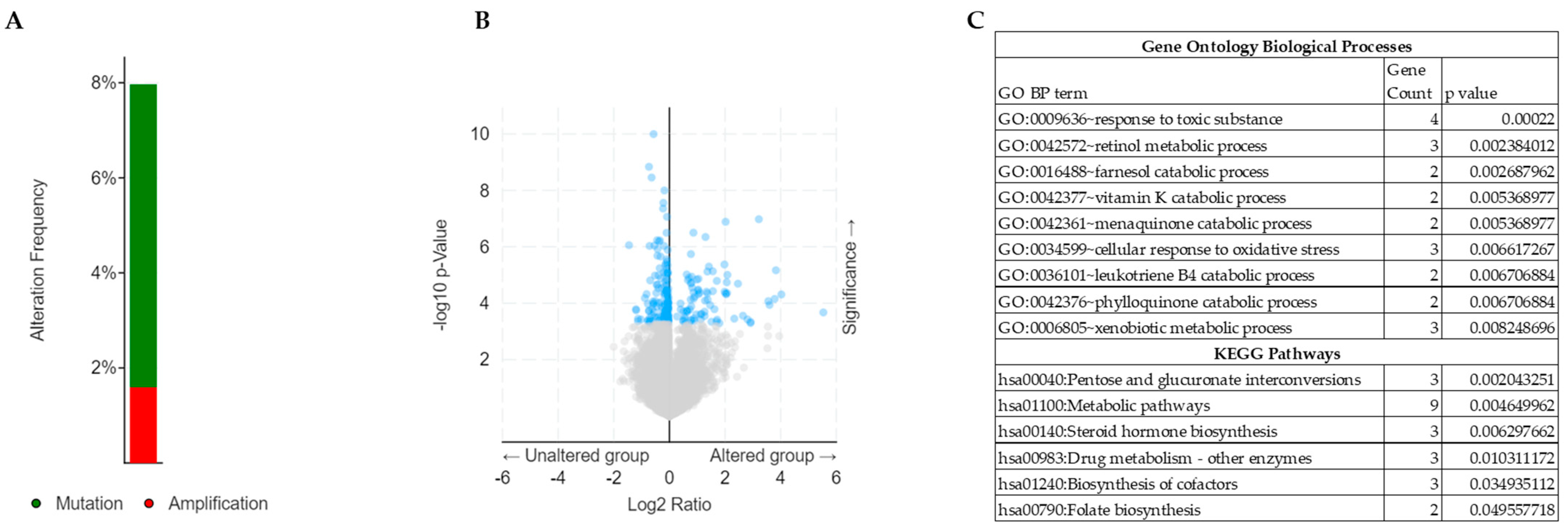
2.1. Identification of Genetic Alterations of NRF2 in TCGA-CSCC
2.2. Analysis of Differentially Expressed Genes (DEGs) in NRF2-Altered TCGA-CSCC
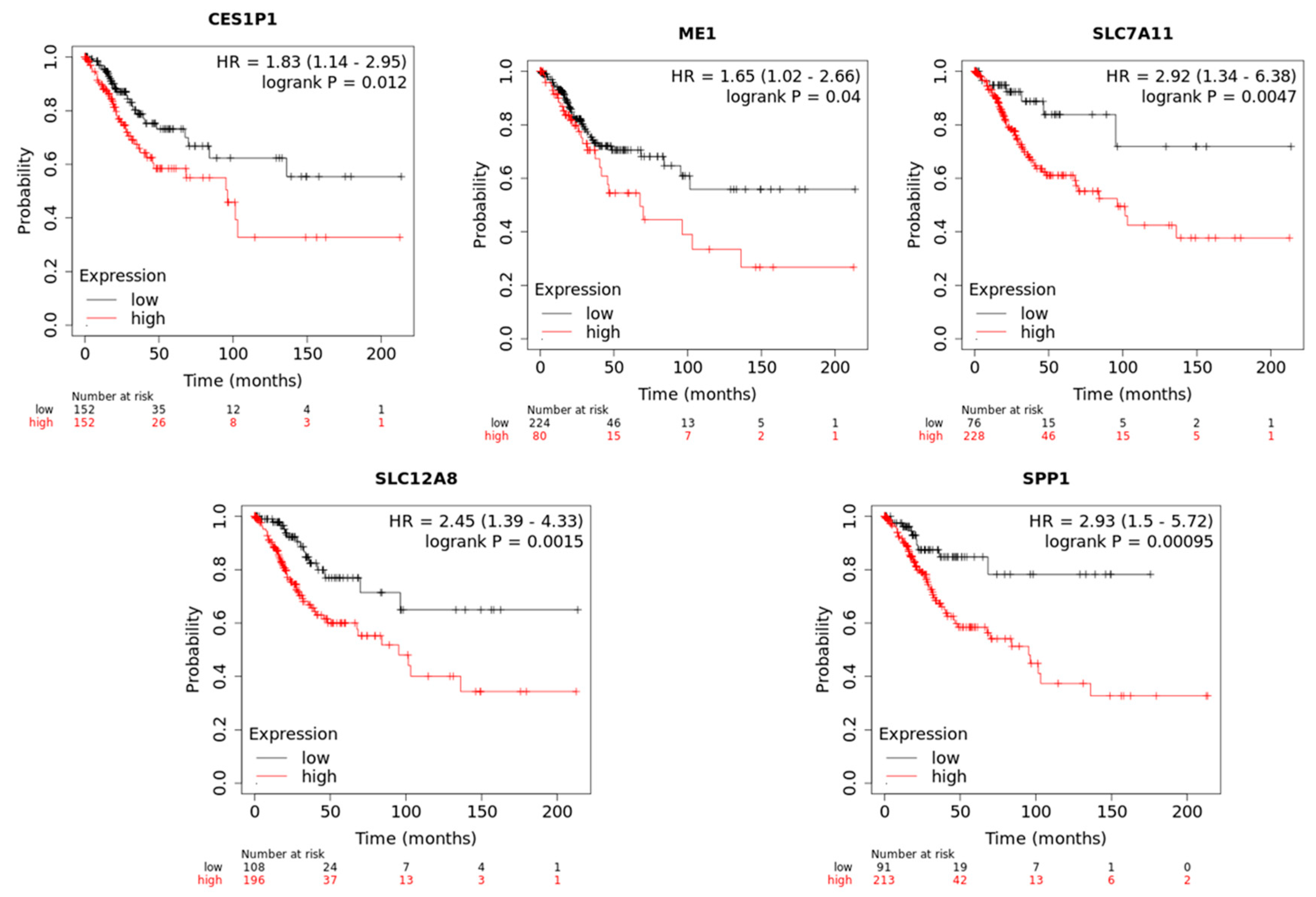
2.3. Functional Annotation and Survival Analysis
2.4. Identification of NRF2 Binding Sites by In Silico Analysis
3. Results and Discussions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjose, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e296. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Namani, A.; Zheng, Z.; Wang, X.J.; Tang, X. Systematic Identification of Multi Omics-based Biomarkers in KEAP1 Mutated TCGA Lung Adenocarcinoma. J. Cancer 2019, 10, 6813–6821. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Namani, A.; Cui, Q.Q.; Wu, Y.; Wang, H.; Wang, X.J.; Tang, X. NRF2-regulated metabolic gene signature as a prognostic biomarker in non-small cell lung cancer. Oncotarget 2017, 8, 69847–69862. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Munkacsy, G.; Gyorffy, B. Pancancer survival analysis of cancer hallmark genes. Sci. Rep. 2021, 11, 6047. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Huang, C.H. LASAGNA-Search 2.0: Integrated transcription factor binding site search and visualization in a browser. Bioinformatics 2014, 30, 1923–1925. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramisetti, S.V.; Namani, A. Multi-Omics Analysis of NFE2L2-Altered TCGA-Cervical Squamous Cell Carcinoma Patients. Med. Sci. Forum 2023, 20, 1. https://doi.org/10.3390/IECC2023-14223
Ramisetti SV, Namani A. Multi-Omics Analysis of NFE2L2-Altered TCGA-Cervical Squamous Cell Carcinoma Patients. Medical Sciences Forum. 2023; 20(1):1. https://doi.org/10.3390/IECC2023-14223
Chicago/Turabian StyleRamisetti, Sri Vidya, and Akhileshwar Namani. 2023. "Multi-Omics Analysis of NFE2L2-Altered TCGA-Cervical Squamous Cell Carcinoma Patients" Medical Sciences Forum 20, no. 1: 1. https://doi.org/10.3390/IECC2023-14223