
Elucidation of Functional Genes Associated with Long Chain-Polyunsaturated Fatty Acids (LC-PUFAs) Metabolism in Oleaginous Diatom Phaeodactylum tricornutum


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Retrieval and Metabolic Pathway Analysis
2.2. Transcriptome Analysis
2.3. Validation of RNA-Seq Data by Quantitative Real-Time PCR (qPCR)
2.4. Construction of Phylogenetic Trees
2.5. Prediction of Subcellular Localization, Motifs and Domain Architecture
2.6. Subcellular Networking and Pathway Enrichment Analysis
2.7. Statistical Analysis
3. Results
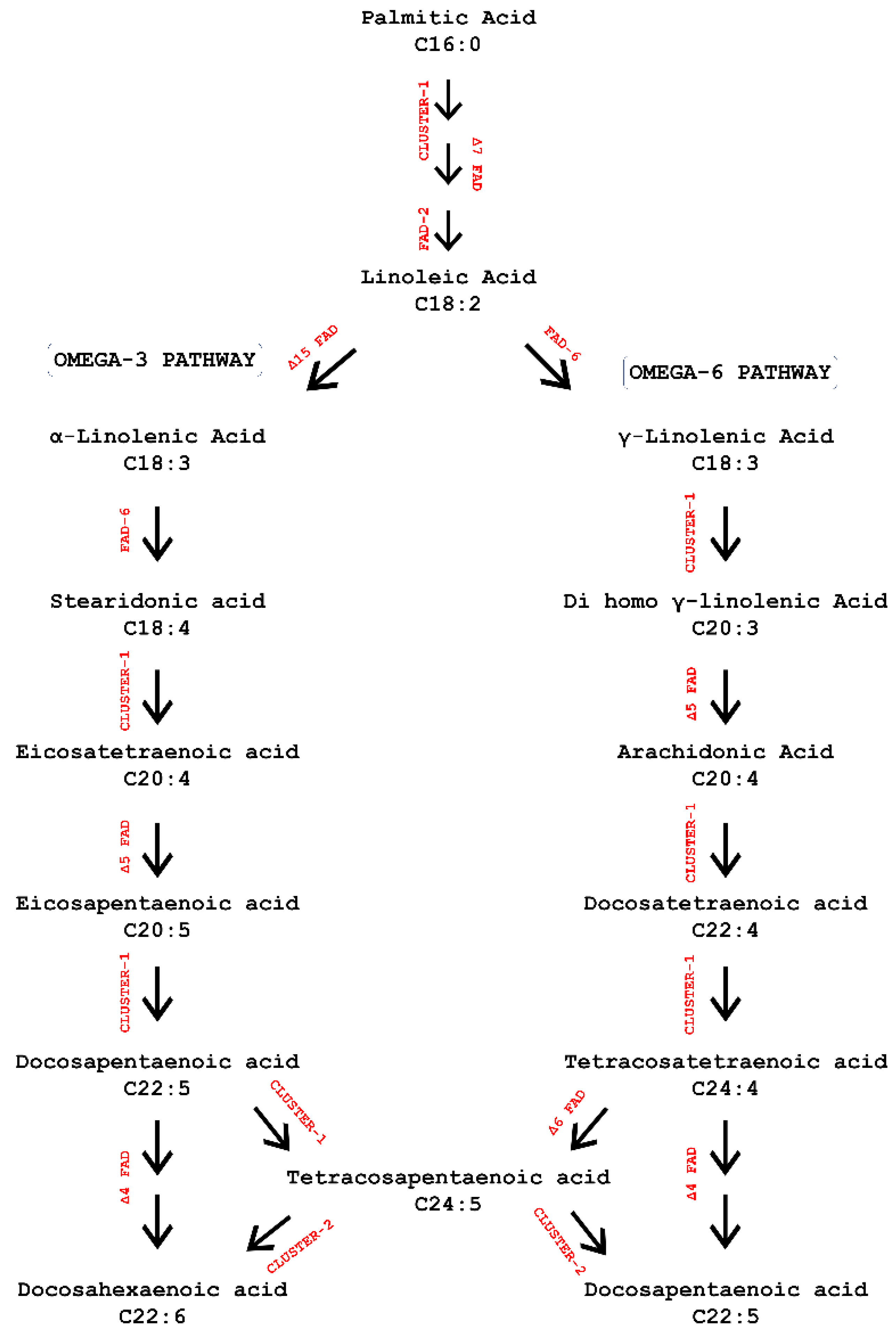
3.1. Gene Mining and Analysis in Microalgal Lineage
3.2. Functional Characterization of LC-PUFAs
3.2.1. Temporal Expression Profile
3.2.2. Molecular Analysis of LC-PUFAs Genes Expression in P. tricornutum
3.3. Organization Studies
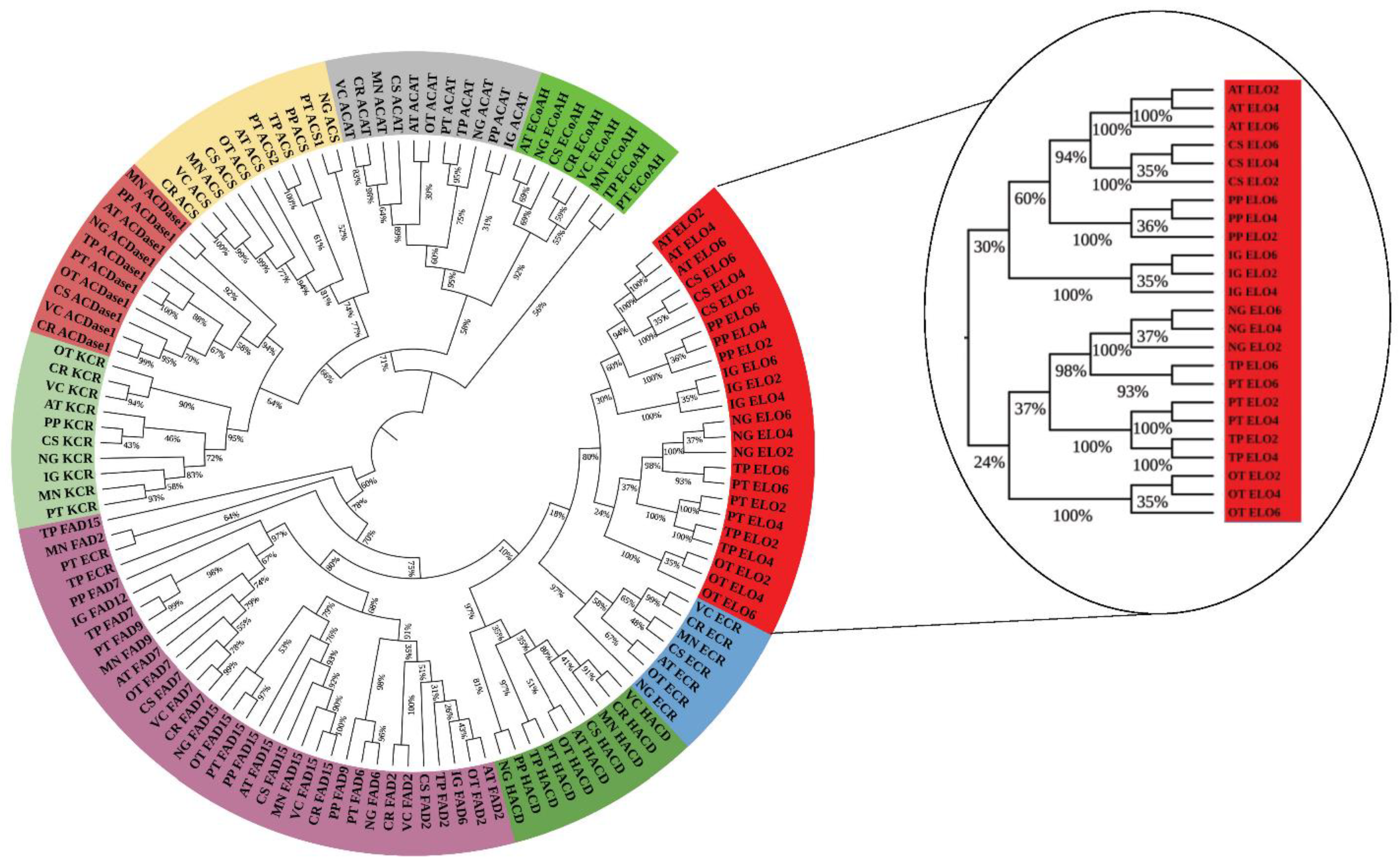
3.3.1. Phylogenomic Analysis
3.3.2. Subcellular Localization
3.3.3. Identification of Motifs and Domains
3.3.4. Subcellular Networking and Pathway Enrichment Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Chen, Y.; Xu, C.; Vaidyanathan, S. Microalgae: A robust green bio-bridge between energy and environment. Crit. Rev. Biotechnol. 2018, 38, 351–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, M.L.; Haslam, R.P.; Napier, J.A.; Sayanova, O. Metabolic engineering of Phaeodactylum for the enhanced accumulation of omega-3 long chain polyunsaturated fatty acids. Metab. Eng. 2014, 22, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Vaezi, R.; Napier, J.A.; Sayanova, O. Identification and functional characterization of genes encoding omega-3 polyunsaturated fatty acid biosynthetic activities from unicellular microalgae. Mar. Drugs 2013, 11, 5116–5129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hultberg, M.; Jönsson, H.L.; Bergstrand, K.-J.; Carlsson, A.S. Impact of light quality on biomass production and fatty acid content in the microalga Chlorella vulgaris. Bioresour. Technol. 2014, 159, 465–467. [Google Scholar] [CrossRef]
- Meesapyodsuk, D.; Qiu, X. Biosynthetic mechanism of very long chain polyunsaturated fatty acids in Thraustochytrium sp. 26185. J. Lipid Res. 2016, 57, 1854–1864. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Sommerfeld, M.; Jarvis, E.; Ghirardi, M.; Posewitz, M.; Seibert, M.; Darzins, A. Microalgal triacylglycerols as feedstocks for biofuel production: Perspectives and advances. Plant Mol. Biol. 2008, 54, 621–639. [Google Scholar] [CrossRef]
- Goncalves, E.C.; Wilkie, A.C.; Kirst, M.; Rathinasabapathi, B. Metabolic regulation of triacylglycerol accumulation in the green algae: Identification of potential targets for engineering to improve oil yield. Plant Biotechnol. J. 2016, 14, 1649–1660. [Google Scholar] [CrossRef]
- Hulatt, C.J.; Smolina, I.; Dowle, A.; Kopp, M.; Vasanth, G.K.; Hoarau, G.G.; Wijffels, R.H.; Kiron, V. Proteomic and transcriptomic patterns during lipid remodeling in Nannochloropsis gaditana. Int. J. Mol. Sci. 2020, 21, 6946. [Google Scholar] [CrossRef]
- Remmers, I.M.; Martens, D.E.; Wijffels, R.H.; Lamers, P.P. Dynamics of triacylglycerol and EPA production in Phaeodactylum tricornutum under nitrogen starvation at different light intensities. PLoS ONE 2017, 12, e0175630. [Google Scholar] [CrossRef] [Green Version]
- Blifernez-Klassen, O.; Chaudhari, S.; Klassen, V.; Wördenweber, R.; Steffens, T.; Cholewa, D. Metabolic survey of Botryococcus braunii: Impact of the physiological state on product formation. PLoS ONE 2018, 13, e0198976. [Google Scholar] [CrossRef]
- Vadivelan, G.; Venkateswaran, G. Production and enhancement of omega-3 fatty acid from Mortierella alpina CFR-GV15: Its food and therapeutic application. Biomed. Res. Int. 2014, 2014, 657414. [Google Scholar] [CrossRef] [Green Version]
- Cerón García, M.C.; Sánchez Mirón, A.; Fernández Sevilla, J.M.; Molina Grima, E.; García Camacho, F. Mixotrophic growth of the microalga Phaeodactylum tricornutum: Influence of different nitrogen and organic carbon sources on productivity and biomass composition. Process Biochem. 2005, 40, 297–305. [Google Scholar] [CrossRef]
- Cui, Y.; Thomas-Hall, S.R.; Schenk, P.M. Phaeodactylum tricornutum microalgae as a rich source of omega-3 oil: Progress in lipid induction techniques towards industry adoption. Food Chem. 2019, 297, 124937. [Google Scholar] [CrossRef]
- Hamilton, M.L.; Powers, S.; Napier, J.A.; Sayanova, O. Heterotrophic Production of Omega-3 Long-Chain Polyunsaturated Fatty Acids by Trophically Converted Marine Diatom Phaeodactylum tricornutum. Mar. Drugs 2016, 14, 53. [Google Scholar] [CrossRef] [Green Version]
- Haslam, R.P.; Hamilton, M.L.; Economou, C.K.; Smith, R.; Hassall, K.L.; Napier, J.A.; Sayanova, O. Overexpression of an endogenous type 2 diacylglycerol acyltransferase in the marine diatom Phaeodactylum tricornutum enhances lipid production and omega-3 long-chain polyunsaturated fatty acid content. Biotechnol. Biofuels 2020, 13, 87. [Google Scholar] [CrossRef]
- Kadalag, N.L.; Pawar, P.R.; Prakash, G. Co-cultivation of Phaeodactylum tricornutum and Aurantiochytrium limacinum for polyunsaturated omega-3 fatty acids production. Bioresour. Technol. 2022, 346, 126544. [Google Scholar] [CrossRef]
- Zhu, B.-H.; Tu, C.-C.; Shi, H.-P.; Yang, G.-P.; Pan, K.-H. Overexpression of endogenous delta-6 fatty acid desaturase gene enhances eicosapentaenoic acid accumulation in Phaeodactylum tricornutum. Process Biochem. 2017, 57, 43–49. [Google Scholar] [CrossRef]
- Mühlroth, A.; Li, K.; Røkke, G.; Winge, P.; Olsen, Y.; Hohmann-Marriott, M.F.; Vadstein, O.; Bones, A.M. Pathways of lipid metabolism in marine algae, co-expression network, bottlenecks and candidate genes for enhanced production of EPA and DHA in species of Chromista. Mar. Drugs 2013, 11, 4662–4697. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Yuan, W.; Ma, Y.; Balamurugan, S.; Li, H.-Y.; Fu, S.; Wu, L. Harnessing the lipogenic potential of δ6-desaturase for simultaneous hyperaccumulation of lipids and polyunsaturated fatty acids in Nannochloropsis oceanica. Front. Mar. Sci. 2019, 6, 682. [Google Scholar] [CrossRef]
- Diao, J.; Song, X.; Guo, T.; Wang, F.; Chen, L.; Zhang, W. Cellular engineering strategies toward sustainable omega-3 long chain polyunsaturated fatty acids production: State of the art and perspectives. Biotechnol. Adv. 2020, 40, 107497. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Li-Beisson, Y.; Shorrosh, B.; Beisson, F.; Andersson, M.X.; Arondel, V.; Bates, P.D.; Baud, S.; Bird, D.; Debono, A.; Durrett, T.P.; et al. Acyl-lipid metabolism. Arab. Book 2010, 8, e0133. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general-purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 2015, 43, 566–570. [Google Scholar] [CrossRef]
- Rio, D.C.; Ares, M., Jr.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 2010, 2010, 5439. [Google Scholar] [CrossRef]
- Verbruggen, H.; Theriot, E.C. Building trees of algae: Some advances in phylogenetic and evolutionary analysis. Eur. J. Phycol. 2008, 43, 229–252. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. CABIOS 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Mount, D.W. Maximum parsimony method for phylogenetic prediction. CSH Protoc. 2008, 2008, 32. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Emanuelsson, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef]
- Horton, P.; Park, K.-J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, 585–587. [Google Scholar] [CrossRef] [Green Version]
- Chou, K.-C.; Shen, H.-B. A new method for predicting the subcellular localization of eukaryotic proteins with both single and multiple sites: Euk-mploc 2.0. PLoS ONE 2010, 5, e9931. [Google Scholar] [CrossRef]
- Yu, C.S.; Chen, Y.C.; Lu, C.H.; Hwang, J.K. Prediction of protein subcellular localization. Proteins 2006, 64, 643–651. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, 202–208. [Google Scholar] [CrossRef]
- Sigrist, C.J.; de Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and continuing developments at PROSITE. Nucleic Acids Res. 2013, 41, 344–347. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Chen, D. Phylogeny, functional annotation, and protein interaction network analyses of the Xenopus tropicalis basic helix-loop-helix transcription factors. Biomed. Res. Int. 2013, 2013, 145037. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, 447–452. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Robertson, R.; Guihéneuf, F.; Schmid, M.; Stengel, D.; Fitzgerald, G.; Ross, P.; Stanton, C. Algae-Derived Polyunsaturated Fatty Acids: Implications for Human Health. In Polyunsaturated Fatty Acids: Sources, Antioxidant Properties and Health Benefits; Catalá, A., Ed.; Nova Sciences: Hauppauge, NY, USA, 2013; pp. 1–54. [Google Scholar]
- Shanab, S.M.M.; Hafez, R.M.; Fouad, A.S. A review on algae and plants as potential source of arachidonic acid. J. Adv. Res. 2018, 11, 3–13. [Google Scholar] [CrossRef]
- Harwood, J.L. Algae: Critical sources of very long-chain polyunsaturated fatty acids. Biomolecules 2019, 9, 708. [Google Scholar] [CrossRef] [Green Version]
- Molino, A.; Martino, M.; Larocca, V.; Di Sanzo, G.; Spagnoletta, A.; Marino, T.; Karatza, D.; Iovine, A.; Mehariya, S.; Musmarra, D. Eicosapentaenoic acid extraction from Nannochloropsis gaditana using carbon dioxide at super critical conditions. Mar. Drugs 2019, 17, 132. [Google Scholar] [CrossRef] [Green Version]
- Pudney, A.; Gandini, C.; Economou, C.K.; Smith, R.; Goddard, P.; Napier, J.A.; Spicer, A.; Sayanova, O. Multifunctionalizing the marine diatom Phaeodactylum tricornutum for sustainable co-production of omega-3 long chain polyunsaturated fatty acids and recombinant phytase. Sci. Rep. 2019, 9, 11444. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.K.; Niu, Y.F.; Ma, Y.H.; Xue, J.; Zhang, M.H.; Yang, W.D.; Liu, J.S.; Lu, S.H.; Guan, Y.; Li, H.Y. Molecular and cellular mechanisms of neutral lipid accumulation in diatom following nitrogen deprivation. Biotechnol. Biofuels 2013, 6, 67. [Google Scholar] [CrossRef]
- Levitan, O.; Dinamarca, J.; Zelzion, E.; Lun, D.S.; Guerra, L.T.; Kim, M.K.; Kim, J.; Van Mooy, B.A.S.; Bhattacharya, D.; Falkowski, P.G. Remodeling of intermediate metabolism in the diatom Phaeodactylum tricornutum; under nitrogen stress. Proc. Natl. Acad. Sci. 2015, 112, 412–417. [Google Scholar] [CrossRef] [Green Version]
- McFarlane, H.E.; Shin, J.J.H.; Bird, D.A.; Samuels, A.L. Arabidopsis abcg transporters, which are required for export of diverse cuticular lipids, dimerize in different combinations. Plant Cell 2010, 22, 3066–3075. [Google Scholar] [CrossRef] [Green Version]
- Mashek, D.G.; Li, L.O.; Coleman, R.A. Long-chain acyl-CoA synthetases and fatty acid channeling. Future Lipidol. 2007, 2, 465–476. [Google Scholar] [CrossRef] [Green Version]
- Peng, K.-T.; Zheng, C.-N.; Xue, J.; Chen, X.-Y.; Yang, W.-D.; Liu, J.-S.; Bai, W.; Li, H.-Y. Delta 5 fatty acid desaturase upregulates the synthesis of polyunsaturated fatty acids in the marine diatom Phaeodactylum tricornutum. J. Agric. Food Chem. 2014, 62, 8773–8776. [Google Scholar] [CrossRef]
- Degraeve-Guilbault, C.; Gomez, R.E.; Lemoigne, C.; Pankansem, N.; Morin, S.; Tuphile, K.; Joubès, J.; Jouhet, J.; Gronnier, J.; Suzuki, I.; et al. Plastidic Δ6 fatty-acid desaturases with distinctive substrate specificity regulate the pool of c18-PUFAs in the ancestral picoalga Ostreococcus tauri. Plant Physiol 2020, 184, 82–96. [Google Scholar] [CrossRef]
- Bowler, C.; Allen, A.E.; Badger, J.H.; Grimwood, J.; Jabbari, K.; Kuo, A.; Maheswari, U.; Martens, C.; Maumus, F.; Otillar, R.P.; et al. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 2008, 456, 239–244. [Google Scholar] [CrossRef] [Green Version]
- Dönnes, P.; Höglund, A. Predicting protein subcellular localization: Past, present, and future. Genom. Proteom. Bioinform. 2004, 2, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.Q.; Burger, G. Unite and conquer: Enhanced prediction of protein subcellular localization by integrating multiple specialized tools. BMC Bioinform. 2007, 8, 420. [Google Scholar] [CrossRef] [Green Version]
- Misra, N.; Panda, P.K.; Parida, B.K. Genome-wide identification and evolutionary analysis of algal LPAT genes involved in TAG biosynthesis using bioinformatics approaches. Mol. Biol. Rep. 2014, 41, 8319–8332. [Google Scholar] [CrossRef]
- He, M.; Qin, C.-X.; Wang, X.; Ding, N.-Z. Plant unsaturated fatty acids: Biosynthesis and regulation. Front. Plant Sci. 2020, 11, 390. [Google Scholar] [CrossRef] [Green Version]
- Jonasdottir, S.H. Fatty acid profiles and production in marine phytoplankton. Mar. Drugs 2019, 17, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manan, S.; Ahmad, M.Z.; Zhang, G.; Chen, B.; Haq, B.U.; Yang, J.; Zhao, J. Soybean LEC2 regulates subsets of genes involved in controlling the biosynthesis and catabolism of seed storage substances and seed development. Front. Plant Sci. 2017, 8, 1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li-Beisson, Y.; Beisson, F.; Riekhof, W. Metabolism of acyl-lipids in Chlamydomonas reinhardtii. Plant J. 2015, 82, 504–522. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Benning, C. Lipid metabolism in microalgae distinguishes itself. Curr. Opin. Biotechnol. 2013, 24, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Merchant, S.S.; Kropat, J.; Liu, B.; Shaw, J.; Warakanont, J. TAG, you’re it! Chlamydomonas as a reference organism for understanding algal triacylglycerol accumulation. Curr. Opin. Biotechnol. 2012, 23, 352–363. [Google Scholar]
- Lavell, A.A.; Benning, C. Cellular Organization and Regulation of Plant Glycerolipid Metabolism. Plant Cell Physiol. 2019, 60, 1176–1183. [Google Scholar] [CrossRef]
- Marchive, C.; Nikovics, K.; To, A.; Lepiniec, L.; Baud, S. Transcriptional regulation of fatty acid production in higher plants: Molecular bases and biotechnological outcomes. Eur. J. Lipid Sci. Technol. 2014, 116, 1332–1343. [Google Scholar] [CrossRef]
- Dehghan Nayeri, F.; Yarizade, K. Bioinformatics study of delta-12 fatty acid desaturase 2 (FAD2) gene in oilseeds. Mol. Biol. Rep. 2014, 41, 5077–5087. [Google Scholar] [CrossRef]
- Mitchell, A.G.; Martin, C.E. A novel cytochrome b5-like domain is linked to the carboxyl terminus of the Saccharomyces cerevisiae delta-9 fatty acid desaturase. J. Biol. Chem. 1995, 270, 29766–29772. [Google Scholar] [CrossRef] [Green Version]
- Thiyagarajan, S.; Arumugam, M.; Kathiresan, S. Identification and Functional Characterization of Two Novel Fatty Acid Genes from Marine Microalgae for Eicosapentaenoic Acid Production. Appl. Biochem. Biotechnol. 2020, 190, 1371–1384. [Google Scholar] [CrossRef]
- Li, N.; Xu, C.; Li-Beisson, Y.; Philippar, K. Fatty acid and lipid transport in plant cells. Trends Plant Sci. 2016, 21, 145–158. [Google Scholar] [CrossRef]
- Yu, S.Y.; Li, H.; Tong, M.; Ouyang, L.L.; Zhou, Z.G. Identification of a Δ6 fatty acid elongase gene for arachidonic acid biosynthesis localized to the endoplasmic reticulum in the green microalga Myrmecia incisa Reisigl. Gene 2012, 493, 219–227. [Google Scholar] [CrossRef]
- Tatzer, V.; Zellnig, G.; Kohlwein, S.D.; Schneiter, R. Lipid-dependent subcellular relocalization of the acyl chain desaturase in yeast. Mol. Biol. Cell 2002, 13, 4429–4442. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Fu, Y.; Shi, Y.; Li, Y.; Kou, Y.; Mao, X.; Liu, J. Functional characterization of long-chain acyl-coa synthetase gene family from the oleaginous alga Chromochloris zofingiensis. J. Agric. Food Chem. 2020, 68, 4473–4484. [Google Scholar] [CrossRef]
- Li-Beisson, Y.; Thelen, J.J.; Fedosejevs, E.; Harwood, J.L. The lipid biochemistry of eukaryotic algae. Prog. Lipid Res. 2019, 74, 31–68. [Google Scholar] [CrossRef]
- Groot, P.H.; Scholte, H.R.; Hülsmann, W.C. Fatty acid activation: Specificity, localization and function. Adv. lipid Res. 1976, 14, 75–126. [Google Scholar]
- Domergue, F.; Abbadi, A.; Ott, C.; Zank, T.K.; Zähringer, U.; Heinz, E. Acyl carriers used as substrates by the desaturases and elongases involved in very long-chain polyunsaturated fatty acids biosynthesis reconstituted in yeast. J. Biol. Chem. 2003, 278, 35115–35126. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Ning, K.; Zeng, X.; Luo, Y.; Wang, D.; Hu, J.; Li, J.; Xu, H.; Huang, J.; Wan, M.; et al. Genomic foundation of starch-to-lipid switch in oleaginous Chlorella spp. Plant Physiol. 2015, 169, 2444–2461. [Google Scholar] [CrossRef] [Green Version]
- Kannangara, R.; Branigan, C.; Liu, Y.; Penfield, T.; Rao, V.; Mouille, G.; Höfte, H.; Pauly, M.; Riechmann, J.L.; Broun, P. The transcription factor win1/shn1 regulates cutin biosynthesis in Arabidopsis thaliana. Plant Cell 2007, 19, 1278–1294. [Google Scholar] [CrossRef] [Green Version]
- Raffaele, S.; Vailleau, F.; Léger, A.; Joubès, J.; Miersch, O.; Huard, C.; Blée, E.; Mongrand, S.; Domergue, F.; Roby, D. A MYB transcription factor regulates very-long-chain fatty acid biosynthesis for activation of the hypersensitive cell death response in Arabidopsis. Plant Cell 2008, 20, 752–767. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.B.; Suh, M.C. Cuticular wax biosynthesis is up-regulated by the MYB94 transcription factor in Arabidopsis. Plant Cell Physiol. 2015, 56, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Zhang, Y.; Meng, H.; Li, S.; Wang, S.; Xiao, Z.; Chang, P.; Zhang, X.; Li, Q.; Guo, L.; et al. Characterization of fatty acid exporters involved in fatty acid transport for oil accumulation in the green alga Chlamydomonas reinhardtii. Biotechnol. Biofuels 2019, 12, 14. [Google Scholar] [CrossRef]
- Rehmanji, M.; Nesamma, A.A.; Khan, N.J.; Fatma, T.; Jutur, P.P. Media engineering in marine diatom Phaeodactylum tricornutum employing cost-effective substrates for sustainable production of high-value renewables. Biotechnol. J. 2022, 17, e2100684. [Google Scholar] [CrossRef]
- Kareya, M.S.; Mariam, I.; Shaikh, K.M.; Nesamma, A.A.; Jutur, P.P. Photosynthetic carbon partitioning and metabolic regulation in response to very-low and high CO2 in Microchloropsis gaditana NIES 2587. Front. Plant Sci. 2020, 11, 981. [Google Scholar] [CrossRef]
- Guerra, L.T.; Levitan, O.; Frada, M.; Sun, J.; Falkowski, P.; Dismukes, G. Regulatory branch points affecting protein and lipid biosynthesis in the diatom Phaeodactylum tricornutum. Biomass Bioenerg. 2013, 59, 306–315. [Google Scholar] [CrossRef]
- Shaikh, K.M.; Nesamma, A.A.; Abdin, M.Z.; Jutur, P.P. Molecular profiling of an oleaginous trebouxiophycean alga Parachlorella kessleri subjected to nutrient deprivation for enhanced biofuel production. Biotechnol. Biofuels 2019, 12, 182. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Paliwal, C.; Nesamma, A.A.; Narula, A.; Jutur, P.P. Nutrient deprivation mobilizes the production of unique tocopherols as a stress-promoting response in a new indigenous isolate Monoraphidium sp. Front. Mar. Sci. 2020, 7, 575817. [Google Scholar] [CrossRef]
- Brown, M.R. The amino-acid and sugar composition of 16 species of microalgae used in mariculture. J. Exp. Mar. Biol. Ecol. 1991, 145, 79–99. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehmanji, M.; Kumar, A.; Nesamma, A.A.; Khan, N.J.; Fatma, T.; Jutur, P.P. Elucidation of Functional Genes Associated with Long Chain-Polyunsaturated Fatty Acids (LC-PUFAs) Metabolism in Oleaginous Diatom Phaeodactylum tricornutum. Hydrobiology 2022, 1, 451-468. https://doi.org/10.3390/hydrobiology1040027
Rehmanji M, Kumar A, Nesamma AA, Khan NJ, Fatma T, Jutur PP. Elucidation of Functional Genes Associated with Long Chain-Polyunsaturated Fatty Acids (LC-PUFAs) Metabolism in Oleaginous Diatom Phaeodactylum tricornutum. Hydrobiology. 2022; 1(4):451-468. https://doi.org/10.3390/hydrobiology1040027
Chicago/Turabian StyleRehmanji, Mohammed, Ashish Kumar, Asha Arumugam Nesamma, Nida Jamil Khan, Tasneem Fatma, and Pannaga Pavan Jutur. 2022. "Elucidation of Functional Genes Associated with Long Chain-Polyunsaturated Fatty Acids (LC-PUFAs) Metabolism in Oleaginous Diatom Phaeodactylum tricornutum" Hydrobiology 1, no. 4: 451-468. https://doi.org/10.3390/hydrobiology1040027