1. Introduction
Hereditary angioedema (HAE) is a clinical condition characterized by recurrent episodes of swelling that may affect the subcutaneous tissues of the extremities, trunk, face, or upper airways or the gastrointestinal mucosae or genitalia. It is a rare condition that affects 1:50,000 to 1:100,000 people in the general population and is determined by mutations of a specific gene called
SERPING1, which codifies for a protein called C1 inhibitor, a regulatory component of the classical complement pathway that also shows anti-enzymatic activity that regulates the activation of several proteases included in the coagulation, fibrinolytic, and contact system pathways [
1]. It is now clear that the specific pathogenesis of angioedema attacks resides in a lack of the regulatory activity of C1 inhibitor on the contact system enzymes pre-kallikrein and Factor XII, which normally undergo a mutual activation after modifications that occur on the endothelial surface [
2,
3]. Kallikrein acts on high molecular weight kininogen and generates a nonapeptide called bradykinin. This latter vasoactive peptide binds specifically to B2 receptors, G-protein coupled receptors that are constitutively expressed on endothelial cells. B2 receptors induce intracellular transduction mechanisms that, through steric rearrangement and destruction of VE-cadherin, produce an increased permeability of the endothelial layer to plasma fluids and the ultimate translocation of liquid from the intravascular space to the subcutaneous site of the derma. HAE patients do not show an increased risk of bleeding or thrombosis [
4,
5].
HAE is a genetic disease with an autosomal dominant pattern of transmission, and almost all patients show a heterozygous state, with one functioning allele. Homozygous HAE patients have been described but are very rare [
6]. Typically, patients will have reduced circulating antigenic (type I) or functional (type II) levels of C1 inhibitor [
7]. Due to the pattern of genetic transmission, probands have generally vertical or transversal familial transmission of the disease. However, frequent de novo mutations of the
SERPING1 gene (up to 25% of all cases) must be kept in mind for those cases that do not show a familial pattern. Mutations of the
SERPING1 gene, especially those that produce a type I phenotype (85% of cases), are diverse and include missense, non-sense, frameshift insertion or deletion or slicing defects which are spread all over the gene domains [
8]; while mutations that produce a type II phenotype (15% of cases), are generally missense mutations localized in exon 8 [
9]; overall, almost 750 different mutations have been reported [
10].
In recent years, a different form of hereditary angioedema has been identified (called type III or hereditary angioedema with normal C1 inhibitor), where the genetic defect does not involve
SERPING1; different genes have been implicated like Factor XII, angiopoietin-1, and plasminogen [
10].
Laboratory diagnosis includes assays for C1 inhibitor antigenic and functional levels, C4, and potentially a genetic assessment of the SERPING1 gene. Circulating and functional levels of C1 inhibitor are generally well below 50% of normal (different from the estimate one could make in consideration of the presence of a functioning allele) probably due to trans-inhibition of the normal allele by the mutated allele, and also by a putative increased clearance of the C1 inhibitor–protease complex; as a matter of fact, C1 inhibitor is a suicide anti-protease, which means that it is destroyed after binding to the target protease.
From a clinical point of view, the disease onset generally occurs in childhood, but a proper diagnosis can be delayed by as long as 10 years [
11]. Angioedema episodes are asymmetric pale, non-pitting, and non-pruritic localized edema without urticaria, which are recurrent with patient-by-patient frequency variability and a duration, if untreated, that may last up to 48–72 h for each attack. For many patients, about 50% of their attacks are cutaneous and about 50% are abdominal; localization to the intestinal mucosae produces severe pain that may mimic an acute abdomen and require unnecessary surgery. Localization of an edema attack to the upper airways is rare, and less than 1% of all attacks involve the larynx, but it may represent a potentially life-threatening phenomenon, because it may cause asphyxiation. Triggering factors of attacks can be identified in several cases [
12], but many attacks have a spontaneous appearance. A frequent reported factor is psychologic stress, and trauma, infections, medical procedures (surgery, endoscopy or dental procedures), and fatigue, are triggering factors [
12]. Some drugs that may provoke angioedema attacks in normal subjects [
13] may also favor the recurrence of angioedema attacks in HAE patients; these are especially represented by exogenous estrogens, ACE inhibitors, or NSAIDs [
13]. Gestation may have a different impact on the frequency and severity of HAE attacks. In some cases there is an increase and in some other cases there is a reduction of frequency/severity. Delivery can be also carried out by the vaginal route and generally is not complicated [
14].
Overall, the disease is highly disabling if not properly treated.
2. State of the Art Treatment of Hereditary Angioedema
The objective of treatment of HAE is to reduce the number and severity of attacks and to restitute a normal quality of life to the patients. According to the last WAO/EAACI recommendations these objectives can be reached with approved therapies [
15]. However, there are still some unmet needs for which several drugs are undergoing active testing and will probably enrich the therapeutic armamentarium towards the end of this decade.
The mainstay of the actual HAE therapies includes drugs for the treatment of single attacks with an on-demand basis and prophylactic treatments for both short-term pre-procedural prophylaxis (i.e., before surgery or dental procedures) or long-term prophylaxis aimed at preventing HAE attacks and potentially completely abrogating the occurrence of attacks. For on-demand treatment there are currently three drugs approved by the FDA and EMA and one more drug approved only by the FDA [
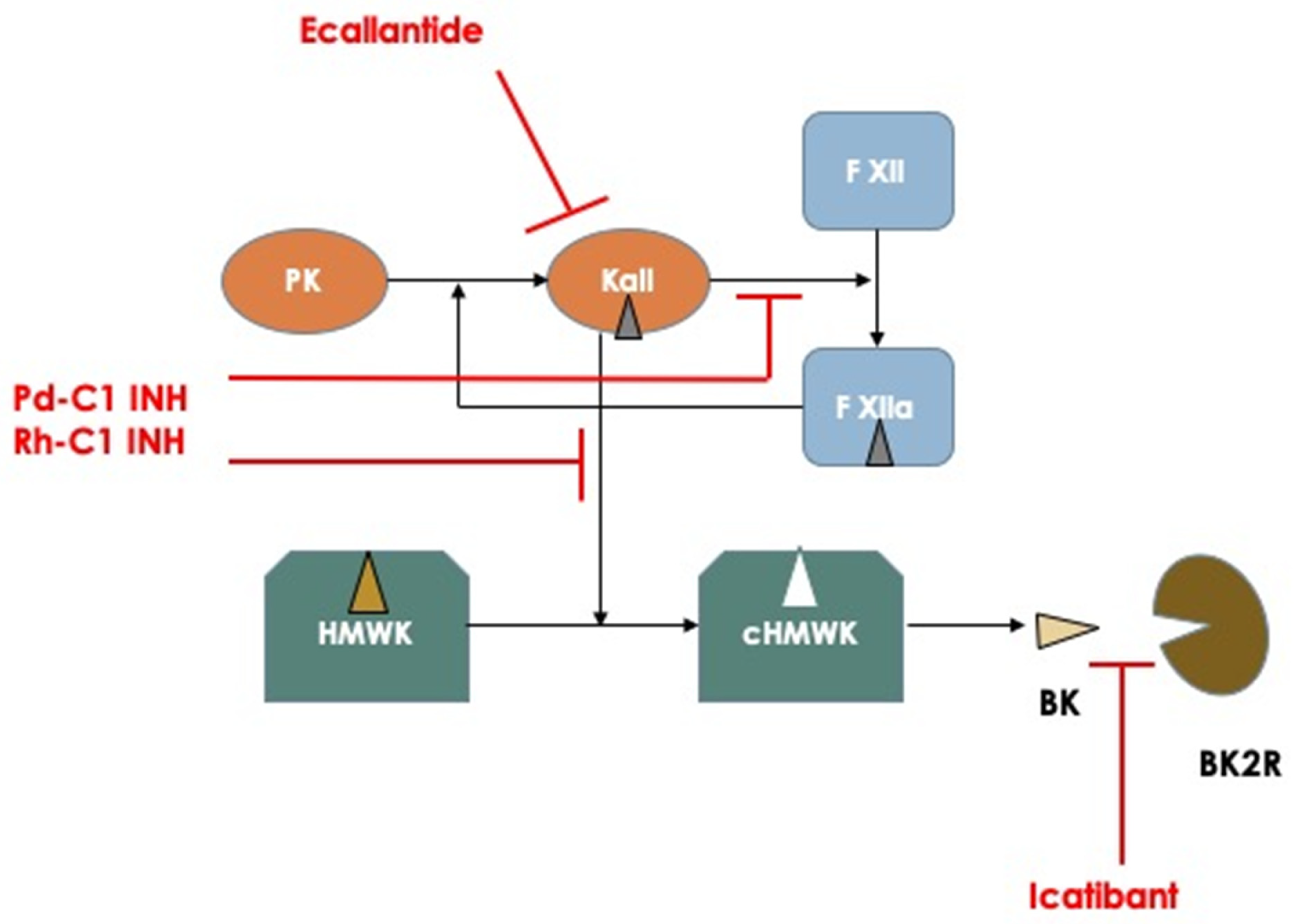
16]. The first three are plasma-derived C1 inhibitor for IV injection (Berinert, CSL Behring, Marburg, Germany), recombinant human C1 inhibitor (Ruconest, Pharming, Leiden, The Netherlands), and icatibant (Firazyr, Takeda Pharmaceutical International AG, Dublin, Ireland), a specific antagonist of bradykinin binding to its receptor B2R, that can be administered by subcutaneous injection. Ecallantide (Kalbitor, Takeda) is the fourth drug approved only by the FDA; this is a polypeptide inhibitor of plasma kallikrein, administered subcutaneously. All these drugs have a different pharmacodynamic profile (
Figure 1) and have been proved effective in halting the evolution of HAE attacks, including laryngeal attacks, in RCT studies, thus accelerating the resolution of an attack and providing relief shortly after administration and complete resolution of the attack in a few hours after injection. Moreover, post-registration registries have shown a consistent effect on the repetitive use and safety of these drugs over a long period of time [
17].
For short-term prophylaxis, the only approved drug is plasma-derived C1 inhibitor (Bering, CSL Behring; Cinryze, Takeda), which is generally administered intravenously shortly (1 h) before the procedure. Due to the long half-life of plasma-derived C1 inhibitor—up to 32 h—a single injection of the proper dosage is sufficient to provide an adequate coverage even for long surgical procedures. Alternatively, an old approach for short term prophylaxis consisted of the administration of attenuated androgens, like danazol, at high dosage (i.e., 600 mg/day in three doses) for a period of 5 days before the procedure. The effect of danazol in preventing HAE attacks is not completely understood, but probably consists of a combination of a stimulation of C1 inhibitor synthesis and an increase in circulating metalloprotease that normally operate a catabolism of bradykinin. Attenuated androgens are no longer recommended as first line therapies, due to the high rate of adverse effects associated with the use of these drugs.
In the last 10–15 years there has been an increased use of long-term prophylaxis for HAE attacks, especially in patients that present with frequent attacks (>3–4/month) and severe attacks (laryngeal or abdominal). Owing to the availability of novel therapeutic options that include plasma-derived C1 inhibitor and a monoclonal antibody that targets activated plasma kallikrein, the last updated version of the WAO/EAACI guidelines on the management of HAE recommend that the goals of treatment are to achieve total control of the disease and to normalize patient’s lives [
15]. The available therapeutic options then include plasma-derived C1 inhibitor for IV infusion (Cinryze, Takeda) or for subcutaneous administration at a higher dosage (Haegarda or Berinert sc, CSL Behring); both types of preparations need a twice-weekly infusion of each drug and can obtain a reduction in the attack rate of between 50% to a maximum of 88% for the subcutaneous C1 inhibitor preparation. Another more recent option for long-term prophylaxis is represented by lanadelumab, a humanized monoclonal antibody against activated plasma kallikrein, which is administered subcutaneously once every two weeks in the induction and once every 28 days after stabilization of the therapeutic response or after 6 months if the patient is attack-free. Lanadelumab can abate the attack rate by up to 73–87% [
18]. Finally, an oral inhibitor of activated plasma kallikrein has recently been developed; berotralstat (Orladeyo, BioCryst Pharmaceutical, Dublin, Ireland) can be administered once a day for long-term prophylaxis. It can reduce attack frequency by 45–50% with a tendency toward increased efficacy for longer treatment periods. In
Table 1 there is a synthesis of the main characteristics of actually available drugs for the treatment of HAE patients.
According to the efficacy of available treatments that allow the efficient management of attacks or reduce the frequency of attacks in long-term prophylaxis, but cannot offer complete disease control or normalize patients’ lives, there are some unmet needs that can be met with new drugs. For this reason, several pharmaceutical companies are actively developing new therapeutic options for HAE patients. In the next part of this paper, we will review all the novel drugs that are undergoing active testing for efficacy and safety in HAE patients.
3. New Drugs for the Treatment of Hereditary Angioedema
Moving from the actual scenario of the treatment of hereditary angioedema, one can envision some possible unmet needs in the therapy for this specific condition: (1) the possibility to have an effective and rapid drug for treating on demand acute attacks that should allow the avoidance of any type of injective drug; (2) an effective oral drug for long-term prophylaxis with an efficiency of attack abatement comparable to the available injective molecules; (3) an injective drug for long-term prophylaxis with extended efficacy (2–3 months or more); (4) a definitive cure for the disease, possibly by genetic therapy.
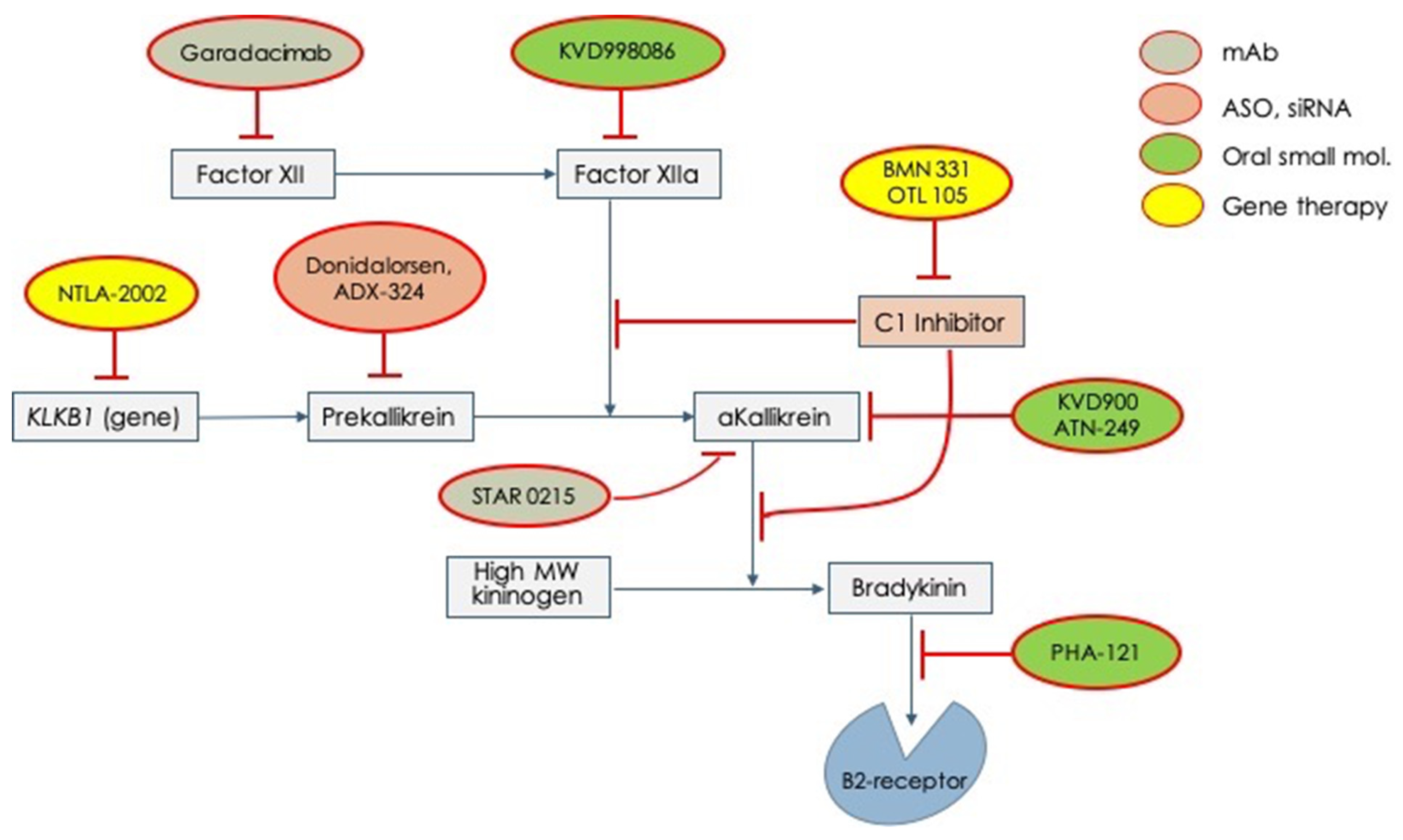
Given these unmet needs, the pharmaceutical industry has undertaken a rush to try to fill all these needs. Therefore, several molecules have begun clinical experimentation and are actually under active testing in different phases (
Table 2). In
Figure 2, all the new tested drugs are shown according to their pharmacodynamic target. Several new molecules are undergoing experimentation and they have been developed with different pharmacologic technologies that include: (1) orally active, small inhibiting molecules, (2) monoclonal antibodies, (3) antisense oligonucleotide for RNA translation blockade, and (4) genetic technology for genome editing. According to the targets for which the different drugs have been engineered, we can indicate: (1) activated factor XII, (2) prekallikrein gene, (3) activated plasma kallikrein, (4) Bradykinin B2-receptor, (5)
SERPING1 gene (genetic therapy), and (6) prekallikrein gene (genetic therapy)
Figure 2.
4. New Medications for the On-Demand Treatment of Hereditary Angioedema Attacks
According to the WAO/EAACI guidelines on the management of HAE, angioedema attacks should be treated as soon as possible, regardless of the involved manifestation site; in fact, the sooner therapy is administered, the faster the attack resolves and the lower is the severity of symptoms [
15].
Therapeutic options have to be tailored from patient to patient according to efficacy and tolerability, owing to the inter-individual response variability. Moreover, training patients for self-administration is of pivotal importance in reducing delay in treating attacks and in consideration of the fact that the currently available medications are administered by intravenous or subcutaneous routes [
16].
Studies on new drugs are ongoing, focusing on oral administration in order to alleviate patient discomfort.
Sebetralstat (KVD900) (KalVista Pharmaceutical, Ltd., Porton Down, Wiltshire, UK), an oral plasma kallikrein inhibitor, has been tested in a phase 2, two-part trial. In part one, the pharmacokinetics, pharmacodynamics, and safety of a single 600 mg dose of sebetralstat were assessed. In part 2, eligible patients were randomized to one cross-over sequence drug placebo or vice versa, to treat two consecutive attacks with sebetralstat 600 mg or a placebo. Primary end-point was the rate of treatment with conventional drugs in the 12 h post-attack onset. The pharmacodynamic study demonstrated that plasma samples obtained from 15 min up to 4 h after sebetralstat administration showed almost complete inhibition of kallikrein, with an ex vivo test. Among the 68 treated patients in part 2, in patients treated with sebetralstat, time to use of conventional drugs was significantly longer than time to use after placebo (>12 h, compared to 8.0 h). No serious adverse effects or study interruption for adverse effects were recorded [
19].
A phase 3 study to assess efficacy and safety of sebetralstat is ongoing.
Deucrictibant (Pharvaris, Leiden, The Netherlands) is an orally bioavailable B2 receptor antagonist, more potent and longer lived than icatibant in vivo. RAPIDe-1 (dose ranging study) is complete and RAPIDe-2 (extension study) is in the recruitment phase. Preliminary data have shown that deucrictibant at variable dosages of 10, 20, and 30 mg can induce a significant reduction of attack symptom scores, evaluated by a visual analog scale for skin pain, skin swelling, and abdominal pain. Even with the lowest dose of 10 mg, on a total of 147 attacks enrolled and treated by double-blind versus placebo, deucrictibant proved significantly more efficacious than the placebo in reducing VAS-3 after 4 h of onset; also, patients treated had less need for rescue medications compared to the placebo (18.9, 10.7, and 6.5% for 10, 20, and 30 mg, respectively; 60.8% for the placebo). The drug was well-tolerated with an occurrence of adverse events <5% in treated patients.
5. New Medications for Long-Term Prophylaxis (LTP)
Long-term prophylaxis (LTP) consists of prophylactic treatment, and involves ongoing or maintenance treatment with the objective of reducing the occurrence, intensity, and duration of HAE attacks.
The authors of the international WAO/EAACI guideline recommend that long-term prophylaxis (LTP) should be considered when patients experience significant life events that are linked to heightened disease activity. The 2021 revision and update highlight the importance of complete control of the disease, with the aim of normalizing patients’ lives [
15].
Beyond the almost consolidated experience with lanadelumab and plasma-derived C1-INH, in a short time we will have new drugs, some already approved for the treatment of HAE attacks or short-term or long-term prophylaxis, and others still in experimentation.
5.1. Berotralstat
Berotralstat (BCX7353) (BioCryst Pharmaceuticals) was the first oral prophylactic treatment for HAE that received orphan status designation in both the European Union (EU) and the United States (USA). It is an oral, second-generation, synthetic, small-molecule plasma kallikrein inhibitor taken once daily for the prevention of HAE attacks. It inhibits kallikrein hyperactivity, thus decreasing pathological bradykinin production.
The phase 1 clinical trial (NCT02448264) demonstrated the safety of the drug [
20]. The findings from the APeX trials (APeX-1 NCT02870972, APeX-2 NCT03485911, APeX-S NCT03472040, APeX-J NCT03873116) highlighted the effectiveness of berotralstat as a long-term prophylactic treatment for individuals with HAE. The results demonstrated a decrease in the frequency of attacks and the use of on-demand therapies.
The phase 3 APeX-2 study included 120 patients with type I or type II HAE, randomized into one of three parallel treatment arms for the 24-week treatment period: 110 mg berotralstat, 150 mg berotralstat, or a placebo by oral administration once daily [
21]. Both doses of berotralstat resulted in a significant reduction in the attack rate. Specifically, the attack rates were 1.65, 1.31, and 2.35 attacks per month for the 110 mg dose, 150 mg dose, and placebo, respectively.
The most common adverse events observed with berotralstat included abdominal pain, vomiting, diarrhea, and back pain. Importantly, there were no serious adverse events related to the drug. In the open label extension study, patients—even those taking the placebo—were randomized to therapy with 110 mg or 150 mg berotralstat. The 150 mg berotralstat group reported a mean monthly attack rate reduction from 2.9 (1.50) at the baseline to 1.5 (1.43) at month 1, 1.3 (1.57) at month 6, and 1.0 (1.34) at month 12 [
22]. The optimal risk–benefit balance was identified at a daily dosage of 150 mg. Moreover, ongoing research is investigating the potential of oral berotralstat in managing acute angioedema attacks (NCT03240133).
5.2. Garadacimab
Garadacimab (CSL Behring) is a novel treatment for LTP in patients affected by HAE that targets factor XII (FXIIa); HAE patients experiment with a dysregulation of the contact system of coagulation and an exacerbation of the kallikrein–kinin activity.
Garadacimab (CSL312) is a fully human monoclonal IgG4 antibody against FXIIa, and has been tested in the prevention of HAE attacks in patients with a severe course of the disease. In February 2023, the results of the pivotal phase 3 study VANGUARD were published [
23]; they followed the encouraging results of the phase 2 trial [
24]. The scientific staff involved tested the efficacy of treatment with garadacimab in patients (>12 yo) affected by type I or type II HAE. Among 80 patients screened, 65 were eligible and were randomized 3:2 to receive either treatment or a placebo for a total of 6 months. The subcutaneous antibody was administered monthly (first month in clinic, loading dose of 400 mg, subsequently by subcutaneous self-administration of 200 mg), patients were matched for age and severity of the disease. During the treatment period, the mean numbers of attacks/months were significantly lower in patients treated with garadacimab (0.27, 95% CI 0.05 to 0.49) than in the placebo group (2.01, 1.44 to 2.57;
p < 0.0001). The drug was well-tolerated, and Factor XIIa inhibition was not associated with increased risks of thrombotic or hemorrhagic events.
The results of phase 2 and 3 trial support the use of garadacimab as an effective drug to prevent HAE attacks in patients with a severe course of the disease (>1 episodes/months). For this reason, in the near future, this antibody might increase the availability of specific treatment for LTP. In the future, as experimented already for other subcutaneously administered medications, the prolonged efficacy of treatment (administration intervals up to 45–60 days) might increase the sustainability of these high-cost medical therapies.
5.3. ATN-249
ATN-249 (Attune Pharmaceuticals, New York, NY, USA) represents an innovative and potent small-molecule plasma kallikrein inhibitor, under study for oral prophylactic treatment of HAE.
In preclinical and ex vivo studies, ATN-249 showed exceptional selectivity, surpassing a 2000-fold specificity in normal human plasma to inhibit plasma kallikrein when compared to other closely related serine proteases such as thrombin, factor Xa, factor VIIa, and tissue plasminogen activator [
25]. In comparison to pd-C1 INH, a non-specific kallikrein inhibitor, ATN-249 demonstrated a remarkable, greater than 10-fold higher inhibition of plasma kallikrein and contact system activation.
In a randomized, double-blind, placebo-controlled, single-ascending dose phase 1 study of ATN-249 (ACTRN12618000430235), which involved 48 healthy male participants, the outcomes indicated a dose-dependent inhibition of plasma kallikrein activity. Additionally, the study demonstrated a promising safety profile for ATN-249 [
26].
5.4. Donidalorsen
Donidalorsen (IONIS-PKK-LRx) (IONIS Pharmaceuticals, Inc. Carlsbad, CA, USA) is a 2′-O-methoxyethyl-modified antisense oligonucleotide (MOE-ASO) conjugated to a triantennary N-acetylgalactosamine (GalNAc3). This molecule, designed for subcutaneous use, inhibits the production of plasma prekallikrein through a mechanism involving ribonuclease (RNase) H1–mediated degradation of plasma prekallikrein messenger RNA (mRNA). The GalNAc3 conjugation to the antisense molecule plays a crucial role in promoting uptake into hepatic parenchymal cells, which are the primary site of plasma prekallikrein production. This conjugation is a strategic enhancement that facilitates the efficient delivery and internalization of the antisense molecule into the liver cells. By successfully reaching and being absorbed by hepatic parenchymal cells, the antisense molecule can exert more effectively its inhibitory effects on plasma prekallikrein production, ultimately contributing to the intended therapeutic outcome [
27].
The phase 1 trial involved 32 healthy volunteers and the phase 2 trial involved 20 patients with hereditary angioedema with C1 inhibitor deficiency [
28]. In this trial, patients were randomized to receive for 17 weeks donidalorsen (80 mg) or a placebo, with one dose administered every 4 weeks. Fourteen patients received donidalorsen and six received the placebo. The study results showed a reduction of about 90% in the number of the mean monthly rate of angioedema attacks in the group of patients treated with donidalorsen (0.23) compared to the placebo (2.21). Additionally, the incidence of mild-to-moderate adverse events was 71% among patients receiving donidalorsen and 83% among those receiving the placebo.
The phase 3 trial (NCT05392114) is currently underway, with an expected number of participants of 144. The study includes two group of patients: OLE participants (Open-Label Extension), participants from another study with donidalorsen, who continue their participation in an open-label extension of this study, and new participants with previous HAE prophylactic therapy (lanadelumab, berotralstat, or a C1-esterase inhibitor (C1-INH)). The study duration differs slightly for the two groups: for OLE participants, approximately 70 weeks of active study participation; for other participants: approximately 76 weeks of active study participation.
All participants will enter an extended treatment period, during which they will receive donidalorsen for an additional duration of up to 104 weeks. This extensive treatment period aims to evaluate the efficacy, safety, and long-term outcomes of donidalorsen in the management of HAE, providing valuable insights into its potential as a therapeutic option for participants with HAE-1 and HAE-2. It is estimated that the study will be completed in 2027.
5.5. Deucrictibant
Deucrictibant (PHA-121, Pharvaris) is a synthetic, small-molecule, competitive bradykinin B2 receptor antagonist designed for oral formulation. There are two formulations of the drug: one for long-term prophylaxis of HAE (PHVS719) and one for on-demand therapy for acute attacks (PHVS416).
In vitro and ex vivo preclinical studies showed that the new molecule has a potency 20–29 times greater than icatibant [
29].
PHVS719, the extended-release formulation of PHA-121, has the pharmacological characteristics to be used as LTP. Pharvaris has completed patient enrollment for its proof-of-concept CHAPTER-1 clinical trial that will evaluate the safety and effectiveness of deucrictibant as a prophylactic, or preventive, treatment for adults with hereditary angioedema.
From the first data, PHA-121 demonstrated promising pharmacological properties, including rapid action, predictable pharmacokinetics, and enhanced potency as a bradykinin B2 receptor antagonist, all while maintaining a good safety profile with mild adverse events that were no more frequent than those in the placebo group. These findings suggest that PHA-121 may hold potential as a therapeutic option for conditions involving bradykinin-mediated responses [
29].
In the phase 2 CHAPTER-1 study (NCT05047185) the involved centers are recruiting about 30 patients who will receive a low (10 mg) or high (20 mg) dose of PHVS719, or a placebo, twice daily. The main objective of this trial is to evaluate the incidence of investigator-confirmed HAE attacks over a period of 12 weeks, or approximately 3 months (Dose-ranging Study of Oral
PHA-022121 for Prophylaxis Against Angioedema Attacks in Patients With Hereditary Angioedema Type I or Type II,
https://clinicaltrials.gov/study/NCT05047185 (accessed on 10 October 2023)). The study is expected to be completed in 2026.
5.6. STAR-0215
STAR-0215 (Astria Therapeutics Inc., Boston, MA, USA) is a novel, long-acting monoclonal antibody inhibitor of plasma kallikrein for the potential treatment of HAE. It is an innovative IgG1 monoclonal antibody meticulously engineered to exert potent and durable (≥3 month) inhibition on the enzymatic activity of plasma kallikrein. This inhibition effectively blocks the release of bradykinin from. HMWK (High Molecular Weight Kininogen). One of the distinguishing features of STAR-0215 is its specific binding to a unique allosteric site. This design element enhances its selectivity, ensuring a strong affinity for the active form of the enzyme while exhibiting minimal to no binding to the pro-enzyme prekallikrein. This quality is of paramount importance as it helps to avoid undesired interactions and potential target-mediated clearance, especially in the presence of higher levels of circulating prekallikrein [
30].
Overall, the carefully engineered design and attributes of STAR-0215 position it as a promising candidate for the effective modulation of bradykinin-related responses. This characterization brings to light its potential as a significant breakthrough in therapeutic intervention for conditions associated with bradykinin dysregulation. STAR-0215 was designed for subcutaneous administration every 3 or 6 months, according to minimal physiologically-based pharmacokinetic model simulations [
30].
In the phase 1a trial (NCT05477160), a randomized, double-blind, and placebo-controlled design was employed to assess the safety, pharmacokinetics (PK), and pharmacodynamics (PD) of STAR-0215. It involved 25 healthy adult subjects, each of whom received a single subcutaneous administration of one of three dose levels: 100 mg, 300 mg, or 600 mg of STAR-0215, or a placebo. The subjects were closely monitored for safety, pharmacokinetics (PK), and pharmacodynamics (PD) for a total duration of 224 days. The blinded safety analysis revealed that STAR-0215 was well-tolerated across all administered dose levels. The most prevalent treatment-related adverse event was mild (Grade 1), consisting of self-resolving injection site reactions, primarily presenting as localized site redness. There were no significant alterations in liver enzymes or coagulation parameters, and no occurrences of serious adverse events or necessitated discontinuations. Within the 300 and 600 mg dose cohorts, both pharmacokinetic (PK) and pharmacodynamic (PD) outcomes consistently indicated clinical benefits for up to 3 months. Moreover, the estimated half-life of STAR-0215 was notably extended, reaching up to 110 days. The rapid and sustained maintenance of drug levels associated with clinical benefits supports the potential for administering STAR-0215 at intervals of 3 months or even less frequently [
31].
The phase 1b/2 ALPHA-STAR trial (NCT05695248) is evaluating STAR-0215 in HAE patients. It will consider the safety, tolerability, clinical activity, pharmacokinetics, pharmacodynamics, and immunogenicity of the subcutaneous administration of STAR-0215 in participants with type I or type II HAE in three dose cohorts: the first group will receive one dose of STAR-0215; the second and third will receive two sequential doses. The trial is ongoing, with proof-of-concept results expected in mid-2024 (A Study of STAR-0215 in Participants With Hereditary Angioedema
https://clinicaltrials.gov/study/NCT05695248 (accessed on 10 October 2023)).
6. Genetic Therapies for the Curation of Hereditary Angioedema
The ultimate objective of any novel therapy for treating hereditary angioedema is curation of the disease. This can be obtained by manipulating the genome by inserting a new SERPING1 gene that will compensate for the mutated allele and, therefore, will restitute a condition similar to normality with two alleles producing a normal amount of the C1 inhibitor protein and, at the bottom, abrogating the predisposing condition for recurrence of angioedema attacks. The other modality of genetic therapy that is being explored for curation involves the manipulation of a different target gene, without the insertion of a new SERPING1 gene. A gene target is KLKB1, which codifies for plasma prekallikrein, the precursor of activated plasma kallikrein, a central enzyme in the generation of the ultimate mediator of angioedema attacks: bradykinin.
BioMarin pharmaceutical (San Raphael, CA, USA) is developing an adenovirus-based vector to carry the wild-type SERPING1 gene within the genome of HAE patients, which could determine a curation of the disease with one-time gene therapy. The specific product BMN 331 is under experimentation [HAErmony-1] with a phase 1–2 single arm, dose escalation, open label trial that is testing AAV5 hSERPING1 in patients with HAE type I or II [NCT05121376]; the gene sequence is under the control of a liver-specific promoter, so that the specific protein can be produced and released only by hepatocytes. Patients will receive a single IV injection of the BMN 331 at a dose that is selected from three doses and will be followed-up for 5 years thereafter, monitoring efficacy (levels of plasma C1 inhibitor) and safety. Completion of the study is estimated for November 2028, and at the moment no information has been released by the manufacturer.
The alternative approach is to use CRISP-Cas9 technology to perform gene editing of the
KLKB1 gene, that codifies for prekallikrein. NTLA-2002 (Intellia Therapeutics, Inc., Cambridge, MA, USA) contains genetic material that, once included in the genome, will deactivate the
KLKB1 gene, producing a significant reduction of circulating plasma levels of prekallikrein. Some interim data from the phase 1 and 2 trial [NCT05120830] were presented at the American College of Allergy, Asthma and Immunology in November 2022. They included data from 10 enrolled patients that had received 25, 50, or 75 mg IV injections of NTLA-2002. After 32 weeks, patients presented a reduction of plasma prekallikrein by 64–92% according to different dosages. Patients showed an attack-free period lasting up to almost 1 year, with a global reduction in the attack rate by 95%. Adverse effects were only mild, and the treatment showed a good safety profile [
32].
7. Conclusions
HAE may significantly affect the quality of life of patients. Nowadays, available drugs may allow for good management of the disease process in terms of efficacy and safety; however, there are some issues that may represent unmet needs for this specific pathologic condition. At the same time, according to the latest guidelines, the objective of HAE treatment is to restitute a normal life to patients. The more extensive application of long-term prophylaxis goes in this direction. Nevertheless, different new drugs are under active testing that will hopefully give an adequate answer to these unmet needs. Medications under trial include different molecules that target differently critical molecular aspects of HAE pathogenesis and could possibly be more effective and easier to administer compared to the drugs available. Finally, in the future, some pharmacological approaches that include gene editing and genetic therapy could represent a definitive cure for the disease.
Author Contributions
Conceptualization, V.M.; methodology, validation, V.M., B.C., M.G., A.M. and L.R.; investigation, resources, data curation, V.M., B.C., M.G., A.M. and L.R.; writing—original draft preparation, V.M., B.C., M.G., A.M. and L.R.; writing—review and editing, V.M.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
This study did not require ethical approval.
Informed Consent Statement
Not Applicable.
Data Availability Statement
No research data for this work are available.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Longhurst, H.; Cicardi, M. Hereditary angio-oedema. Lancet 2012, 379, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Reshef, A.; Kidon, M.; Leibovich, I. The Story of Angioedema: From Quincke to Bradykinin. Clin. Rev. Allergy Immunol. 2016, 51, 121–139. [Google Scholar] [CrossRef] [PubMed]
- Zuraw, B.L.; Christiansen, S.C. HAE Pathophysiology and Underlying Mechanisms. Clin. Rev. Allergy Immunol. 2016, 51, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Joseph, K.; Tholanikunnel, T.E.; Kaplan, A.P. Treatment of episodes of hereditary angioedema with C1 inhibitor: Serial assessment of observed abnormalities of the plasma bradykinin-forming pathway and fibrinolysis. Ann. Allergy Asthma Immunol. 2010, 104, 50–54. [Google Scholar] [CrossRef]
- Reshef, A.; Zanichelli, A.; Longhurst, H.; Relan, A.; Hack, C.E. Elevated D-dimers in attacks of hereditary angioedema are not associated with increased thrombotic risk. Allergy 2015, 70, 506–513. [Google Scholar] [CrossRef]
- Bafunno, V.; Divella, C.; Sessa, F.; Tiscia, G.L.; Castellano, G.; Gesualdo, L.; Margaglione, M.; Montinaro, V. De novo homozygous mutation of the C1 inhibitor gene in a patient with hereditary angioedema. J. Allergy Clin. Immunol. 2013, 132, 748–750.e3. [Google Scholar] [CrossRef] [PubMed]
- Germenis, A.E.; Speletas, M. Genetics of Hereditary Angioedema Revisited. Clin. Rev. Allergy Immunol. 2016, 51, 170–182. [Google Scholar] [CrossRef]
- Pappalardo, E.; Caccia, S.; Suffritti, C.; Tordai, A.; Zingale, L.C.; Cicardi, M. Mutation screening of C1 inhibitor gene in 108 unrelated families with hereditary angioedema: Functional and structural correlates. Mol. Immunol. 2008, 45, 3536–3544. [Google Scholar] [CrossRef]
- Zahedi, R.; Aulak, K.S.; Eldering, E.; Davis, A.E., 3rd. Characterization of C1 inhibitor-Ta. A dysfunctional C1INH with deletion of lysine 251. J. Biol. Chem. 1996, 271, 24307–24312. [Google Scholar] [CrossRef]
- Santacroce, R.; D’Andrea, G.; Maffione, A.B.; Margaglione, M.; d’Apolito, M. The Genetics of Hereditary Angioedema: A Review. J. Clin. Med. 2021, 10, 2023. [Google Scholar] [CrossRef]
- Zanichelli, A.; Magerl, M.; Longhurst, H.; Fabien, V.; Maurer, M. Hereditary angioedema with C1 inhibitor deficiency: Delay in diagnosis in Europe. Allergy Asthma Clin. Immunol. 2013, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Caballero, T.; Maurer, M.; Longhurst, H.J.; Aberer, W.; Bouillet, L.; Fabien, V.; IOS Study Group. Triggers and Prodromal Symptoms of Angioedema Attacks in Patients With Hereditary Angioedema. J. Investig. Allergol. Clin. Immunol. 2016, 26, 383–386. [Google Scholar] [CrossRef]
- Montinaro, V.; Cicardi, M. ACE inhibitor-mediated angioedema. Int. Immunopharmacol. 2020, 78, 106081. [Google Scholar] [CrossRef]
- Montinaro, V.; Castellano, G. Management of pregnancy and vaginal delivery by C1 inhibitor concentrate in two hereditary angioedema twins. Clin. Immunol. 2010, 136, 456–457. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Magerl, M.; Betschel, S.; Aberer, W.; Ansotegui, I.J.; Aygören-Pürsün, E.; Banerji, A.; Bara, N.A.; Boccon-Gibod, I.; Bork, K.; et al. The international WAO/EAACI guideline for the management of hereditary angioedema-The 2021 revision and update. Allergy 2022, 77, 1961–1990. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, S.C.; Zuraw, B.L. Hereditary angioedema: On-demand treatment of angioedema attacks. Allergy Asthma Proc. 2020, 41, S26–S29. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Aberer, W.; Caballero, T.; Bouillet, L.; Grumach, A.S.; Botha, J.; Andresen, I.; Longhurst, H.J.; IOS Study Group. The Icatibant Outcome Survey: 10 years of experience with icatibant for patients with hereditary angioedema. Clin. Exp. Allergy 2022, 52, 1048–1058. [Google Scholar] [CrossRef]
- Zanichelli, A.; Montinaro, V.; Triggiani, M.; Arcoleo, F.; Visigalli, D.; Cancian, M. Emerging drugs for the treatment of hereditary angioedema due to C1-inhibitor deficiency. Expert. Opin. Emerg. Drugs 2022, 27, 103–110. [Google Scholar] [CrossRef]
- Aygören-Pürsün, E.; Zanichelli, A.; Cohn, D.M.; Cancian, M.; Hakl, R.; Kinaciyan, T.; Magerl, M.; Martinez-Saguer, I.; Stobiecki, M.; Farkas, H.; et al. An investigational oral plasma kallikrein inhibitor for on-demand treatment of hereditary angioedema: A two-part, randomised, double-blind, placebo-controlled, crossover phase 2 trial. Lancet 2023, 401, 458–469. [Google Scholar] [CrossRef]
- Aygören-Pürsün, E.; Bygum, A.; Grivcheva-Panovska, V.; Magerl, M.; Graff, J.; Steiner, U.C.; Fain, O.; Huissoon, A.; Kinaciyan, T.; Farkas, H.; et al. Oral Plasma Kallikrein Inhibitor for Prophylaxis in Hereditary Angioedema. N. Engl. J. Med. 2018, 379, 352–362. [Google Scholar] [CrossRef]
- Zuraw, B.; Lumry, W.R.; Johnston, D.T.; Aygören-Pürsün, E.; Banerji, A.; Bernstein, J.A.; Christiansen, S.C.; Jacobs, J.S.; Sitz, K.V.; Gower, R.G.; et al. Oral once-daily berotralstat for the prevention of hereditary angioedema attacks: A randomized, double-blind, placebo-controlled phase 3 trial. J. Allergy Clin. Immunol. 2021, 148, 164–172.e9. [Google Scholar] [CrossRef]
- Wedner, H.J.; Aygören-Pürsün, E.; Bernstein, J.; Craig, T.; Gower, R.; Jacobs, J.S.; Johnston, D.T.; Lumry, W.R.; Zuraw, B.L.; Best, J.M.; et al. Randomized Trial of the Efficacy and Safety of Berotralstat (BCX7353) as an Oral Prophylactic Therapy for Hereditary Angioedema: Results of APeX-2 Through 48 Weeks (Part 2). J. Allergy Clin. Immunol. Pract. 2021, 9, 2305–2314.e4. [Google Scholar] [CrossRef]
- Craig, T.J.; Reshef, A.; Li, H.H.; Jacobs, J.S.; Bernstein, J.A.; Farkas, H.; Yang, W.H.; Stroes, E.S.G.; Ohsawa, I.; Tachdjian, R.; et al. Efficacy and safety of garadacimab, a factor XIIa inhibitor for hereditary angioedema prevention (VANGUARD): A global, multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2023, 401, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Craig, T.; Magerl, M.; Levy, D.S.; Reshef, A.; Lumry, W.R.; Martinez-Saguer, I.; Jacobs, J.S.; Yang, W.H.; Ritchie, B.; Aygören-Pürsün, E.; et al. Prophylactic use of an anti-activated factor XII monoclonal antibody, garadacimab, for patients with C1-esterase inhibitor-deficient hereditary angioedema: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2022, 399, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Kalfus, I.; McDonald, A.; Qian, S. Potency, selectivity, and exposure evaluation of ATN-249, a new oral kallikrein inhibitor for hereditary angioedema. J. Allergy Clin. Immunol. 2017, 139, AB378. [Google Scholar] [CrossRef]
- Kalfus, I.; Offman, E.; McDonald, A. Pharmacokinetics and safety of ATN-249, a novel oral plasma kallikrein inhibitor for hereditary angioedema. In Proceedings of the Western Society of Allergy, Asthma, and Immunology 2019 Annual Scientific Session, Maui, HI, USA, 20–24 January 2019. [Google Scholar]
- Crooke, S.T.; Baker, B.F.; Xia, S.; Yu, R.Z.; Viney, N.J.; Wang, Y.; Tsimikas, S.; Geary, R.S. Integrated Assessment of the Clinical Performance of GalNAc3-Conjugated 2′-O-Methoxyethyl Chimeric Antisense Oligonucleotides: I. Human Volunteer Experience. Nucleic Acid Ther. 2019, 29, 16–32. [Google Scholar] [CrossRef]
- Fijen, L.M.; Riedl, M.A.; Bordone, L.; Bernstein, J.A.; Raasch, J.; Tachdjian, R.; Craig, T.; Lumry, W.R.; Manning, M.E.; Alexander, V.J.; et al. Inhibition of Prekallikrein for Hereditary Angioedema. N. Engl. J. Med. 2022, 386, 1026–1033. [Google Scholar] [CrossRef]
- Lesage, A.; Gibson, C.; Knolle, J.; Groen, K.; Crabbé, R.; Lu, P. Development of PHVS719: An oral extended-release bradykinin B2 receptor antagonist to prevent hereditary Angioedema attacks. In Proceedings of the ACAAI Annual Scientific Meeting 2022, Louisville, KY, USA, 10–14 November 2022. [Google Scholar]
- Bedian, V.; Biris, N.; Omer, C.; Chung, J.K.; Fuller, J.; Dagher, R.; Chandran, S.; Harwin, P.; Kiselak, T.; Violin, J.; et al. STAR-0215 is a Novel, Long-Acting Monoclonal Antibody Inhibitor of Plasma Kallikrein for the Potential Treatment of Hereditary Angioedema. J. Pharmacol. Exp. Ther. 2023, 387, 214–225. [Google Scholar] [CrossRef]
- Morabito, C.; Stevens, C.; Chung, J.-K.; Dagher, R.; Bista, P.; Bernard, K.; Gustafson, P.; Gunsior, M.; Nichols, A. Initial Results from a Phase 1 Single Ascending Dose Clinical Trial of STAR-0215, an Investigational Long-Acting Monoclonal Antibody Plasma Kallikrein Inhibitor for Hereditary Angioedema (HAE), in Healthy Subjects Followed for at Least 3 Months. In Proceedings of the 2023 AAAAI Annual Meeting, San Antonio, TX, USA, 24–27 February 2023. [Google Scholar]
- Longhurst, H.; Fijen, L.M.; Lindsay, K.; Butler, J.; Golden, A.; Maag, D.; Xu, Y.; Cohn, D.M. In vivo CRISP/Cas 9 editing of KLKB1 in patients with hereditary angioedema: A first in-human study. In Proceedings of the ACAAI Annual Scientific Meeting 2022, Louisville, KY, USA, 10–14 November 2022. [Google Scholar]
| Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).






{kind=link}
{kind=link}