
Pharmacogenetic Perspective for Optimal Gout Management


Abstract
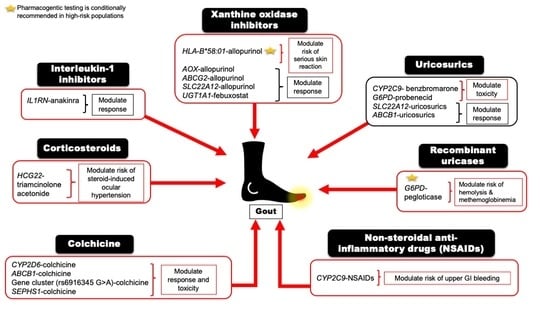
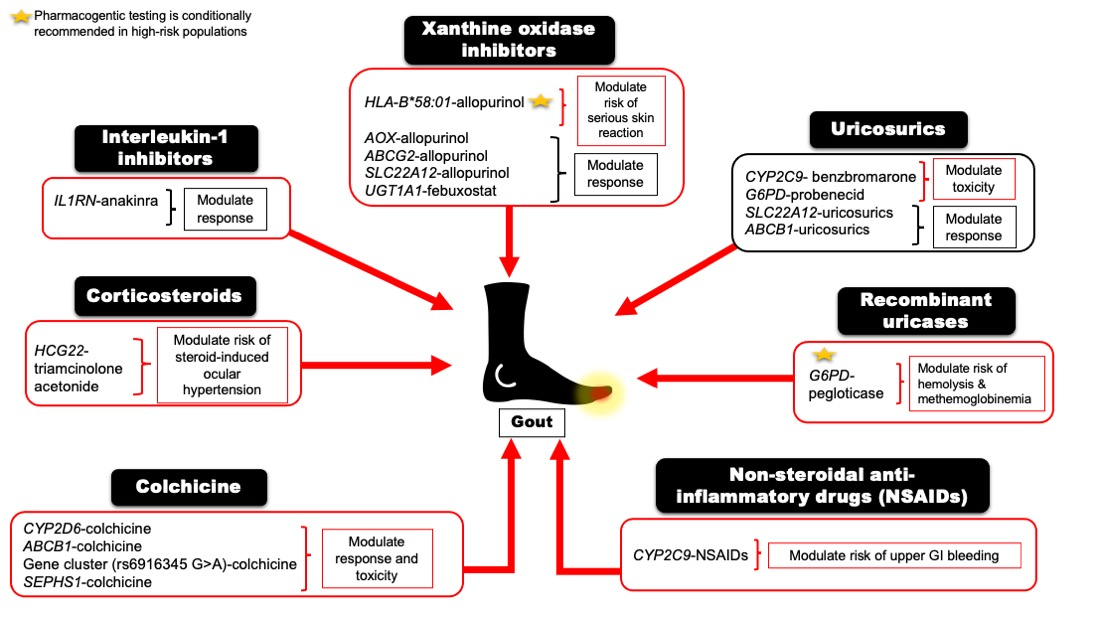
:
1. Background
2. Methods
3. Epidemiology of Hyperuricemia and Gout
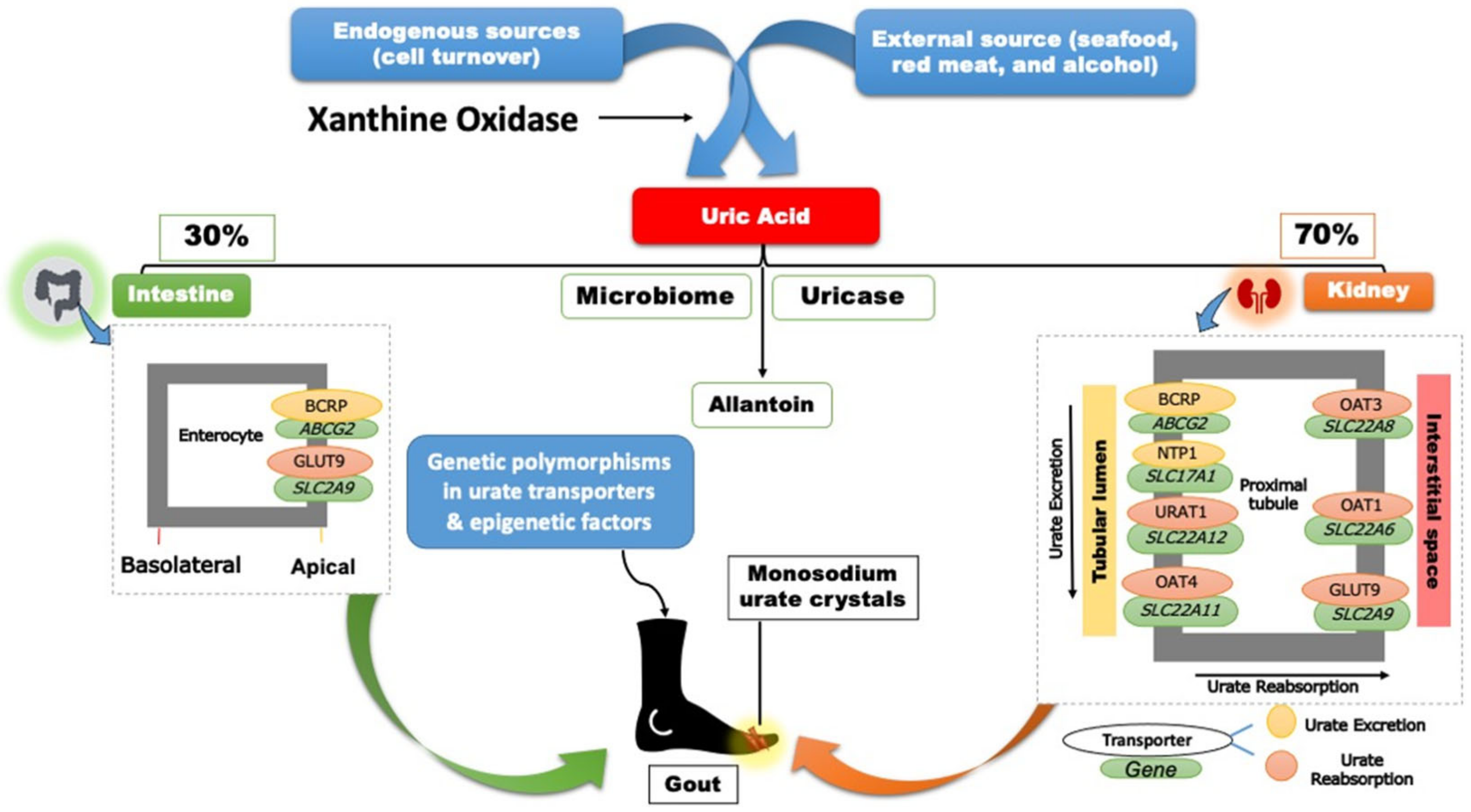
4. Genetics of Hyperuricemia and Gout
5. Gout Management Pharmacotherapy
6. Allopurinol
7. Febuxostat
8. Uricosurics
9. Recombinant Uricases
10. Non-Steroidal Anti-Inflammatory Drugs
11. Colchicine
12. Corticosteroids
13. Interleukin-1 Inhibitors
14. Pharmacogenetic Challenges and Opportunities in Gout Management
15. Conclusions
16. Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roden, D.M.; McLeod, H.L.; Relling, M.V.; Williams, M.S.; Mensah, G.A.; Peterson, J.F.; van Driest, S.L. Pharmacogenomics. Lancet 2019, 394, 521–532. [Google Scholar] [CrossRef]
- Briesacher, B.A.; Andrade, S.E.; Fouayzi, H.; Chan, K.A. Comparison of Drug Adherence Rates Among Patients with Seven Different Medical Conditions. Pharmacotherapy 2008, 28, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Scheepers, L.E.J.M.; van Onna, M.; Stehouwer, C.D.A.; Singh, J.A.; Arts, I.C.W.; Boonen, A. Medication adherence among patients with gout: A systematic review and meta-analysis. Semin. Arthritis Rheum. 2018, 47, 689–702. [Google Scholar] [CrossRef] [PubMed]
- United Nations. Population. Available online: https://www.un.org/en/sections/issues-depth/population/ (accessed on 14 December 2020).
- Fitzgerald, J.D.; Dalbeth, N.; Mikuls, T.; Brignardello-Petersen, R.; Guyatt, G.; Abeles, A.M.; Gelber, A.C.; Harrold, L.R.; Khanna, D.; King, C.; et al. 2020 American College of Rheumatology Guideline for the Management of Gout. Arthritis Rheumatol. 2020, 72, 879–895. [Google Scholar] [CrossRef]
- Krishnan, E.; Lienesch, D.; Kwoh, C.K. Gout in ambulatory care settings in the United States. J. Rheumatol. 2008, 35, 498–501. [Google Scholar]
- Butler, F.; Alghubayshi, A.; Roman, Y. The Epidemiology and Genetics of Hyperuricemia and Gout across Major Racial Groups: A Literature Review and Population Genetics Secondary Database Analysis. J. Pers. Med. 2021, 11, 231. [Google Scholar] [CrossRef]
- Rosenblatt, G.; Decker, J.L.; Healey, L.A., Jr. Gout in hospitalized Filipinos in Hawaii. Pac. Med. Surg. 1966, 74, 312–313. Available online: https://pubmed.ncbi.nlm.nih.gov/5979498/ (accessed on 24 February 2022).
- Murdoch, R.; Barry, M.J.; Choi, H.K.; Hernandez, D.; Johnsen, B.; Labrador, M.; Reid, S.; Singh, J.A.; Terkeltaub, R.; Mellado, J.V.; et al. Original research: Gout, Hyperuricaemia and Crystal-Associated Disease Network (G-CAN) common language definition of gout. RMD Open 2021, 7, e001623. [Google Scholar] [CrossRef]
- Roman, Y.; Tiirikainen, M.; Prom-Wormley, E. The prevalence of the gout-associated polymorphism rs2231142 G > T in ABCG2 in a pregnant female Filipino cohort. Clin. Rheumatol. 2020, 39, 2387–2392. [Google Scholar] [CrossRef]
- Alghubayshi, A.; Edelman, A.; Alrajeh, K.; Roman, Y. Genetic assessment of hyperuricemia and gout in Asian, Native Hawaiian, and Pacific Islander subgroups of pregnant women: Biospecimens repository cross-sectional study. BMC Rheumatol. 2022, 6, 1. [Google Scholar] [CrossRef]
- Kenny, J.-E.S.; Goldfarb, D.S. Update on the Pathophysiology and Management of Uric Acid Renal Stones. Curr. Rheumatol. Rep. 2010, 12, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Huang, J.; Wu, S.; Ji, Q.; Guo, X.; Huang, Y. The Association between the Serum Uric Acid Level and Hypertension in Middle-Aged and Elderly Adults. Cardiovasc. Ther. 2021, 2021, 4626062. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Andres-Hernando, A.; Kuwabara, M. Uric acid and hypertension. Hypertens. Res. 2020, 43, 832–834. [Google Scholar] [CrossRef]
- Hu, X.; Rong, S.; Wang, Q.; Sun, T.; Bao, W.; Chen, L.; Liu, L. Association between plasma uric acid and insulin resistance in type 2 diabetes: A Mendelian randomization analysis. Diabetes Res. Clin. Pract. 2021, 171, 108542. [Google Scholar] [CrossRef] [PubMed]
- Hisatome, I.; Li, P.; Miake, J.; Taufiq, F.; Mahati, E.; Maharani, N.; Utami, S.B.; Kuwabara, M.; Bahrudin, U.; Ninomiya, H. Uric Acid as a Risk Factor for Chronic Kidney Disease and Cardiovascular Disease—Japanese Guideline on the Management of Asymptomatic Hyperuricemia. Circ. J. 2021, 85, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.F.; Rudan, I.; Hastie, N.D.; Campbell, H. A “complexity” of urate transporters. Kidney Int. 2010, 78, 446–452. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Kottgen, A.; Dehghan, A.; Smith, A.V.; Glazer, N.L.; Chen, M.H.; Chasman, D.I.; Aspelund, T.; Eiriksdottir, G.; Harris, T.B.; et al. Multiple Genetic Loci Influence Serum Urate Levels and Their Relationship with Gout and Cardiovascular Disease Risk Factors. Circ. Cardiovasc. Genet. 2010, 3, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Kolz, M.; Johnson, T.; Sanna, S.; Teumer, A.; Vitart, V.; Perola, M.; Mangino, M.; Albrecht, E.; Wallace, C.; Farrall, M.; et al. Meta-Analysis of 28,141 Individuals Identifies Common Variants within Five New Loci That Influence Uric Acid Concentrations. PLoS Genet. 2009, 5, e1000504. [Google Scholar] [CrossRef] [Green Version]
- Kottgen, A.; Albrecht, E.; Teumer, A.; Vitart, V.; Krumsiek, J.; Hundertmark, C.; Pistis, G.; Ruggiero, D.; O’Seaghdha, C.M.; Haller, T.; et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat. Genet. 2013, 45, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Tin, A.; Marten, J.; Kuhns, V.L.H.; Li, Y.; Wuttke, M.; Kirsten, H.; Sieber, K.B.; Qiu, C.; Gorski, M.; German Chronic Kidney Disease Study; et al. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat. Genet. 2019, 51, 1459–1474. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Anzai, N.; Jutabha, P.; Amonpatumrat-Takahashi, S.; Sakurai, H. Recent advances in renal urate transport: Characterization of candidate transporters indicated by genome-wide association studies. Clin. Exp. Nephrol. 2012, 16, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Woodward, O.M.; Köttgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Köttgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caulfield, M.J.; Munroe, P.B.; O’Neill, D.; Witkowska, K.; Charchar, F.; Doblado, M.; Evans, S.; Eyheramendy, S.; Onipinla, A.; Howard, P.; et al. SLC2A9 Is a High-Capacity Urate Transporter in Humans. PLoS Med. 2008, 5, 1509–1523. [Google Scholar] [CrossRef]
- Vitart, V.; Rudan, I.; Hayward, C.; Gray, N.; Floyd, J.; Palmer, C.; Knott, S.A.; Kolcic, I.; Polasek, O.; Graessler, J.; et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat. Genet. 2008, 40, 437–442. [Google Scholar] [CrossRef]
- Hung, S.-I.; Chung, W.-H.; Liou, L.-B.; Chu, C.-C.; Lin, M.; Huang, H.-P.; Lin, Y.-L.; Lan, J.-L.; Yang, L.-C.; Hong, H.-S.; et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc. Natl. Acad. Sci. USA 2005, 102, 4134–4139. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Stamp, L.K.; Caudle, K.E.; Hershfield, M.; McDonagh, E.M.; Callaghan, J.T.; Tassaneeyakul, W.; Mushiroda, T.; Kamatani, N.; Goldspiel, B.R.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for human leukocyte antigen B (HLA-B) genotype and allopurinol dosing: 2015 update. Clin. Pharmacol. Ther. 2016, 99, 36–37. [Google Scholar] [CrossRef] [Green Version]
- Carroll, M.B.; Smith, D.M.; Shaak, T.L. Genomic sequencing of uric acid metabolizing and clearing genes in relationship to xanthine oxidase inhibitor dose. Rheumatol. Int. 2017, 37, 445–453. [Google Scholar] [CrossRef]
- Vora, B.; Brackman, D.J.; Zou, L.; Garcia-Cremades, M.; Sirota, M.; Savic, R.M.; Giacomini, K.M. Oxypurinol pharmacokinetics and pharmacodynamics in healthy volunteers: Influence of BCRP Q141K polymorphism and patient characteristics. Clin. Transl. Sci. 2021, 14, 1431–1443. [Google Scholar] [CrossRef]
- Iwanaga, T. Involvement of Uric Acid Transporter in Increased Renal Clearance of the Xanthine Oxidase Inhibitor Oxypurinol Induced by a Uricosuric Agent, Benzbromarone. Drug Metab. Dispos. 2005, 33, 1791–1795. [Google Scholar] [CrossRef] [Green Version]
- Roman, Y.M.; Culhane-Pera, K.A.; Menk, J.; Straka, R.J. Assessment of genetic polymorphisms associated with hyperuricemia or gout in the Hmong. Pers. Med. 2016, 13, 429–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichida, K.; Hosoyamada, M.; Hisatome, I.; Enomoto, A.; Hikita, M.; Endou, H.; Hosoya, T. Clinical and Molecular Analysis of Patients with Renal Hypouricemia in Japan-Influence of URAT1 Gene on Urinary Urate Excretion. J. Am. Soc. Nephrol. 2004, 15, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Hamada, T.; Ichida, K.; Hosoyamada, M.; Mizuta, E.; Yanagihara, K.; Sonoyama, K.; Sugihara, S.; Igawa, O.; Hosoya, T.; Ohtahara, A.; et al. Uricosuric Action of Losartan via the Inhibition of Urate Transporter 1 (URAT 1) in Hypertensive Patients. Am. J. Hypertens. 2008, 21, 1157–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beringer, P.M.; Kriengkauykiat, J.; Zhang, X.; Hidayat, L.; Liu, S.; Louie, S.; Synold, T.; Burckart, G.J.; Rao, P.A.; Shapiro, B.; et al. Lack of Effect of P-glycoprotein Inhibition on Renal Clearance of Dicloxacillin in Patients with Cystic Fibrosis. Pharmacotherapy 2008, 28, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.K.; Todd, D.; Tso, S.C. Drug-induced haemolysis in glucose-6-phosphate dehydrogenase deficiency. Br. Med. J. 1976, 2, 1227. [Google Scholar] [CrossRef] [Green Version]
- Uchida, S.; Shimada, K.; Misaka, S.; Imai, H.; Katoh, Y.; Inui, N.; Takeuchi, K.; Ishizaki, T.; Yamada, S.; Ohashi, K.; et al. Benzbromarone Pharmacokinetics and Pharmacodynamics in Different Cytochrome P450 2C9 Genotypes. Drug Metab. Pharmacokinet. 2010, 25, 605–610. [Google Scholar] [CrossRef]
- Dalbeth, N.; Stamp, L.K.; Merriman, T.R. The genetics of gout: Towards personalised medicine? BMC Med. 2017, 15, 108. [Google Scholar] [CrossRef] [Green Version]
- Relling, M.V.; McDonagh, E.M.; Chang, T.; Caudle, K.E.; McLeod, H.L.; Haidar, C.E.; Klein, T.; Luzzatto, L. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for Rasburicase Therapy in the Context of G6PD Deficiency Genotype. Clin. Pharmacol. Ther. 2014, 96, 169–174. [Google Scholar] [CrossRef]
- Theken, K.N.; Lee, C.R.; Gong, L.; Caudle, K.E.; Formea, C.M.; Gaedigk, A.; Klein, T.E.; Agúndez, J.A.; Grosser, T. Clinical Pharmacogenetics Implementation Consortium Guideline (CPIC) for CYP2C9 and Nonsteroidal Anti-Inflammatory Drugs. Clin. Pharmacol. Ther. 2020, 108, 191–200. [Google Scholar] [CrossRef]
- Figueiras, A.; Estany-Gestal, A.; Aguirre, C.; Ruiz, B.; Vidal, X.; Carvajal, A.; Salado, I.; Salgado-Barreira, A.; Rodella, L.; Moretti, U.; et al. CYP2C9 variants as a risk modifier of NSAID-related gastrointestinal bleeding: A case-control study. Pharmacogenet. Genom. 2016, 26, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Yalcıntepe, S.; Ozdemır, O.; Sılan, C.; Ozen, F.; Uludag, A.; Candan, F.; Sılan, F. The CYP4502D6 *4 and *6 alleles are the molecular genetic markers for drug response: Implications in colchicine non-responder FMF patients. Eur. J. Drug Metab. Pharmacokinet. 2016, 41, 281–286. [Google Scholar] [CrossRef]
- Bezalel, Y.; Gershoni-Baruch, R.; Dagan, E.; Lidar, M.; Livneh, A. The 3435T polymorphism in the ABCB1 gene and colchicine unre-sponsiveness in familial Mediterranean fever. Clin. Exp. Rheumatol. 2009, 7, S103–S104. Available online: https://www.clinexprheumatol.org/article.asp?a=3689 (accessed on 24 February 2022).
- Babaoglu, M.O.; Yasar, U.; Tufan, A.; Akdogan, A.; Calguneri, M.; Kiraz, S.; Bozkurt, A. Association of the 3435C > T polymorphism of the drug transporter gene ABCB1 with colchicine response in patients with familial Mediterranean fever. FASEB J. 2007, 21, A414–A415. [Google Scholar] [CrossRef]
- Dubé, M.-P.; Legault, M.-A.; Lemaçon, A.; Perreault, L.-P.L.; Fouodjio, R.; Waters, D.D.; Kouz, S.; Pinto, F.J.; Maggioni, A.P.; Diaz, R.; et al. Pharmacogenomics of the Efficacy and Safety of Colchicine in COLCOT. Circ. Genom. Precis. Med. 2021, 14, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Patel, N.; Edlund, C.K.; Hartiala, J.; Hazelett, D.J.; Itakura, T.; Wu, P.-C.; Avery, R.L.; Davis, J.L.; Flynn, H.W.; et al. Identification of a Novel Mucin Gene HCG22 Associated with Steroid-Induced Ocular Hypertension. Investig. Opthalmol. Vis. Sci. 2015, 56, 2737–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardeo, M.; Rossi, M.N.; Marafon, D.P.; Sacco, E.; Bracaglia, C.; Passarelli, C.; Caiello, I.; Marucci, G.; Insalaco, A.; Perrone, C.; et al. Early Treatment and IL1RN Single-Nucleotide Polymorphisms Affect Response to Anakinra in Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2021, 73, 1053–1061. [Google Scholar] [CrossRef]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Avena-Woods, C.; Hilas, O. Febuxostat (Uloric), A New Treatment Option for Gout. Pharm. Ther. 2010, 35, 82. Available online: https://pmc/articles/PMC2827920/ (accessed on 15 December 2021).
- Nakamura, M.; Fujita, K.; Toyoda, Y.; Takada, T.; Hasegawa, H.; Ichida, K. Investigation of the transport of xanthine dehydrogenase inhibitors by the urate transporter ABCG2. Drug Metab. Pharmacokinet. 2018, 33, 77–81. [Google Scholar] [CrossRef]
- Stamp, L.K.; Chapman, P.T. Allopurinol hypersensitivity: Pathogenesis and prevention. Best Pract. Res. Clin. Rheumatol. 2020, 34, 101501. [Google Scholar] [CrossRef]
- Stamp, L.K.; Taylor, W.J.; Jones, P.B.; Dockerty, J.L.; Drake, J.; Frampton, C.; Dalbeth, N. Starting dose is a risk factor for allopurinol hypersensitivity syndrome: A proposed safe starting dose of allopurinol. Arthritis Rheum. 2012, 64, 2529–2536. [Google Scholar] [CrossRef] [PubMed]
- Hande, K.R.; Noone, R.M.; Stone, W.J. Severe allopurinol toxicity: Description and guidelines for prevention in patients with renal insufficiency. Am. J. Med. 1984, 76, 47–56. [Google Scholar] [CrossRef]
- Lu, N.; Rai, S.K.; Terkeltaub, R.; Kim, S.; Menendez, M.E.; Choi, H.K. Racial disparities in the risk of Stevens-Johnson Syndrome and toxic epidermal necrolysis as urate-lowering drug adverse events in the United States. Semin. Arthritis Rheum. 2016, 46, 253–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Food & Drug Administration. Table of Pharmacogenetic Associations. FDA. Available online: https://www.fda.gov/medical-devices/precision-medicine/table-pharmacogenetic-associations (accessed on 16 December 2020).
- Hershfield, M.S.; Callaghan, J.T.; Tassaneeyakul, W.; Mushiroda, T.; Thorn, C.F.; Klein, T.E.; Lee, M.T.M. Clinical Pharmacogenetics Implementation Consortium Guidelines for Human Leukocyte Antigen-B Genotype and Allopurinol Dosing. Clin. Pharmacol. Ther. 2013, 93, 153–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, C.C.; Yee, S.W.; Liang, X.; Hoffmann, T.J.; Kvale, M.N.; Banda, Y.; Jorgenson, E.; Schaefer, C.; Risch, N.; Giacomini, K.M. Genome-wide association study identifies ABCG2 (BCRP) as an allopurinol transporter and a determinant of drug response. Clin. Pharmacol. Ther. 2015, 97, 518–525. [Google Scholar] [CrossRef] [Green Version]
- Roman, Y.M.; Culhane-Pera, K.A.; Lo, M.C.; Yang, S.; Yang, J.; Lo, M.; Straka, R.J. The Impact of Rs505802 for Slc22a12 on Oxipurinol and Uric Acid Disposition in Hmong Patients on Allopurinol from the Genetics of Hyperuricemia Therapy in Hmong (Gouth) Study. Clin. Pharmacol. Ther. 2017, 101, S48. [Google Scholar]
- Anzai, N.; Ichida, K.; Jutabha, P.; Kimura, T.; Babu, E.; Jin, C.J.; Srivastava, S.; Kitamura, K.; Hisatome, I.; Endou, H.; et al. Plasma Urate Level Is Directly Regulated by a Voltage-driven Urate Efflux Transporter URATv1 (SLC2A9) in Humans. J. Biol. Chem. 2008, 283, 26834–26838. [Google Scholar] [CrossRef] [Green Version]
- Veenstra, F.; Wanten, S.A.C.; Verhoef, L.M.; Stal, M.T.; Kwok, W.-Y.; Hoogen, F.H.J.V.D.; Flendrie, M.; van Herwaarden, N. Sex differences in response to allopurinol and benzbromarone in gout: A retrospective cohort study. Rheumatol. Adv. Pract. 2021, 5, rkab002. [Google Scholar] [CrossRef]
- Drug Approval Package: Uloric (Febuxostat) Tablets NDA 21856. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/021856s000toc.cfm (accessed on 15 December 2021).
- FDA. Highlights of Prescribing Information. Available online: www.fda.gov/medwatch (accessed on 15 December 2021).
- White, W.B.; Saag, K.G.; Becker, M.A.; Borer, J.S.; Gorelick, P.B.; Whelton, A.; Hunt, B.; Castillo, M.; Gunawardhana, L.; CARES Investigators. Cardiovascular Safety of Febuxostat or Allopurinol in Patients with Gout. N. Engl. J. Med. 2018, 378, 1200–1210. [Google Scholar] [CrossRef]
- Zhang, M.; Solomon, D.H.; Desai, R.J.; Kang, E.H.; Liu, J.; Neogi, T.; Kim, S.C. Assessment of Cardiovascular Risk in Older Patients with Gout Initiating Febuxostat Versus Allopurinol. Circulation 2018, 138, 1116–1126. [Google Scholar] [CrossRef]
- Foody, J.A.; Turpin, R.S.; Tidwell, B.A.; Lawrence, D.; Schulman, K.L. Major Cardiovascular Events in Patients with Gout and Associated Cardiovascular Disease or Heart Failure and Chronic Kidney Disease Initiating a Xanthine Oxidase Inhibitor. Am. Health Drug Benefits 2017, 10, 393–401. Available online: https://pmc/articles/PMC5726059/ (accessed on 15 December 2021).
- Barbarino, J.M.; Haidar, C.E.; Klein, T.E.; Altman, R.B. PharmGKB summary: Very important pharmacogene information for UGT1A1. Pharmcogenet. Genom. 2014, 24, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, M.H.; Simkin, P.A. Uricosuric drugs: The once and future therapy for hyperuricemia? Curr. Opin. Rheumatol. 2014, 26, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Pharmacogenomics Knowledge Base (PharmGKB). ABCB1—Overview. Available online: https://www.pharmgkb.org/gene/PA267 (accessed on 16 December 2020).
- Pharmacogenomics Knowledge Base (PharmGKB). Drug Label Information and Legend. Available online: https://www.pharmgkb.org/page/drugLabelLegend#pgx-level (accessed on 16 December 2020).
- Lee, M.-H.H.; Graham, G.G.; Williams, K.M.; Day, R.O. A Benefit-Risk Assessment of Benzbromarone in the Treatment of Gout. Was its withdrawal from the market in the best interest of patients? Drug Saf. 2008, 31, 643–665. [Google Scholar] [CrossRef]
- Kang, E.H.; Park, E.H.; Shin, A.; Song, J.S.; Kim, S.C. Cardiovascular risk associated with allopurinol vs. benzbromarone in patients with gout. Eur. Heart J. 2021, 42, 4578–4588. [Google Scholar] [CrossRef]
- Azevedo, V.F.; Kos, I.A.; Vargas-Santos, A.B.; da Rocha Castelar Pinheiro, G.; dos Santos Paiva, E. Benzbromarone in the treatment of gout. Adv. Rheumatol. 2019, 59, 37. [Google Scholar] [CrossRef] [Green Version]
- rs1057910 (SNP)—Population Genetics—Homo sapiens—Ensembl Genome Browser 105. Available online: https://useast.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=10:94980796-94981796;v=rs1057910;vdb=variation;vf=166334188 (accessed on 22 March 2022).
- McDonagh, E.M.; Thorn, C.F.; Callaghan, J.T.; Altman, R.; Klein, T.E. PharmGKB summary: Uric acid-lowering drugs pathway, pharmacodynamics. Pharmacogenet. Genom. 2014, 24, 464–476. [Google Scholar] [CrossRef] [Green Version]
- Pui, C.-H. Rasburicase: A potent uricolytic agent. Expert Opin. Pharmacother. 2002, 3, 433–442. [Google Scholar] [CrossRef]
- Pui, C.-H.; Relling, M.; Lascombes, F.; Harrison, P.; Struxiano, A.; Mondesir, J.-M.; Ribeiro, R.; Sandlund, J.; Rivera, G.; Evans, W.; et al. Urate oxidase in prevention and treatment of hyperuricemia associated with lymphoid malignancies. Leukemia 1997, 11, 1813–1816. [Google Scholar] [CrossRef] [Green Version]
- Pharmacogenomics Knowledge Base (PharmGKB). G6PD—Clinical Guideline Annotations. Available online: https://www.pharmgkb.org/gene/PA28469/guidelineAnnotation (accessed on 16 December 2020).
- Beutler, E.; Gaetani, G.; Der Kaloustian, V.; Luzzatto, L.; Sodeinde, O. Hexose-6-phosphate Dehydrogenase Deficiency. Bull. World Health Organ. 1989, 67, 601–611. [Google Scholar] [CrossRef]
- Nkhoma, E.T.; Poole, C.; Vannappagari, V.; Hall, S.A.; Beutler, E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: A systematic review and meta-analysis. Blood Cells Mol. Dis. 2009, 42, 267–278. [Google Scholar] [CrossRef]
- Tiitinen, S.; Nissilä, M.; Ruutsalo, H.M.; Isomäki, H. Effect of nonsteroidal anti-inflammatory drugs on the renal excretion of uric acid. Clin. Rheumatol. 1983, 2, 233–236. [Google Scholar] [CrossRef]
- Zhou, X.-Y.; Lu, X.-R.; Li, Y.-H.; Ma, Y.-Q.; Zhao, S.-W.; Wang, F.; Xu, R.-A.; Hu, G.-X.; Cai, J.-P. Identification and Enzymatic Activity Evaluation of a Novel CYP2C9 Allelic Variant Discovered in a Patient. Front. Pharmacol. 2021, 12, 619339. [Google Scholar] [CrossRef]
- Mukai, Y.; Senda, A.; Toda, T.; Eliasson, E.; Rane, A.; Inotsume, N. The Role of CYP2C8 and CYP2C9 Genotypes in Losartan-Dependent Inhibition of Paclitaxel Metabolism in Human Liver Microsomes. Basic Clin. Pharmacol. Toxicol. 2016, 118, 408–414. [Google Scholar] [CrossRef] [Green Version]
- Daly, A.K.; Rettie, A.E.; Fowler, D.M.; Miners, J.O. Pharmacogenomics of CYP2C9: Functional and Clinical Considerations. J. Pers. Med. 2018, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Sekino, K.; Kubota, T.; Okada, Y.; Yamada, Y.; Yamamoto, K.; Horiuchi, R.; Kimura, K.; Iga, T. Effect of the single CYP2C9*3 allele on pharmacokinetics and pharmacodynamics of losartan in healthy Japanese subjects. Eur. J. Clin. Pharmacol. 2003, 59, 589–592. [Google Scholar] [CrossRef]
- Lee, C.; Pieper, J.A.; Hinderliter, A.L.; Frye, R.F.; Blaisdell, J.A.; Goldstein, J.A. Tolbutamide, Flurbiprofen, and Losartan as Probes of CYP2C9 Activity in Humans. J. Clin. Pharmacol. 2003, 43, 84–91. [Google Scholar] [CrossRef]
- PharmVar-CYP2C9. Available online: https://www.pharmvar.org/gene/CYP2C9 (accessed on 15 January 2022).
- Van Booven, D.; Marsh, S.; McLeod, H.; Whirl-Carrillo, M.; Sangkuhl, K.; Klein, T.E.; Altman, R.B. Cytochrome P450 2C9-CYP2C9. Pharmacogenet. Genom. 2010, 20, 277–281. [Google Scholar] [CrossRef]
- Pharmacogenomics Knowledge Base (PharmGKB). Very Important Pharmacogene: CYP2C9. Available online: https://www.pharmgkb.org/vip/PA166169913 (accessed on 17 December 2020).
- Dorado, P.; Cavaco, I.; Cáceres, M.C.; Piedade, R.; Ribeiro, V.; Llerena, A. Relationship between CYP2C8 genotypes and diclofenac 5-hydroxylation in healthy Spanish volunteers. Eur. J. Clin. Pharmacol. 2008, 64, 967–970. [Google Scholar] [CrossRef]
- Krasniqi, V.; Dimovski, A.; Domjanović, I.K.; Bilić, I.; Božina, N. How polymorphisms of the cytochrome P450 genes affect ibuprofen and diclofenac metabolism and toxicity. Arh. Hig. Rada Toksikol. 2016, 67, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Schlesinger, N. Treatment of Acute Gout. Rheum. Dis. Clin. N. Am. 2014, 40, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Slobodnick, A.; Shah, B.; Krasnokutsky, S.; Pillinger, M.H. Update on colchicine, 2017. Rheumatology 2018, 57 (Suppl. 1), i4–i11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slobodnick, A.; Shah, B.; Pillinger, M.H.; Krasnokutsky, S. Colchicine: Old and New. Am. J. Med. 2015, 128, 461–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, S.; Yang, K.C.K.; Atkins, K.; Dalbeth, N.; Robinson, P.C. Adverse events during oral colchicine use: A systematic review and meta-analysis of randomised controlled trials. Arthritis Res. Ther. 2020, 22, 28. [Google Scholar] [CrossRef] [Green Version]
- PharmVar. Available online: https://www.pharmvar.org/gene/CYP2D6 (accessed on 15 January 2022).
- Brown, J.T.; Bishop, J.R.; Sangkuhl, K.; Nurmi, E.L.; Mueller, D.J.; Dinh, J.C.; Gaedigk, A.; Klein, T.E.; Caudle, K.E.; McCracken, J.T.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline for Cytochrome P450 (CYP) 2D6 Genotype and Atomoxetine Therapy. Clin. Pharmacol. Ther. 2019, 106, 94–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohar, E.; Gafni, J.; Pras, M.; Heller, H. Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am. J. Med. 1967, 43, 227–253. [Google Scholar] [CrossRef]
- Drenth, J.P.H.; Van Der Meer, J.W. Hereditary Periodic Fever. N. Engl. J. Med. 2001, 345, 1748–1757. [Google Scholar] [CrossRef]
- Ozen, F.; Silan, C.; Uludag, A.; Candan, F.; Silan, F.; Özdemir, S.; Atik, S.; Ozdemir, O. Association between ABCB1 (MDR1) Gene 3435 C > T Polymorphism and Colchicine Unresponsiveness of FMF Patients. Ren. Fail. 2011, 33, 899–903. [Google Scholar] [CrossRef] [Green Version]
- Marzolini, C.; Paus, E.; Buclin, T.; Kim, R.B. Polymorphisms in human MDR1 (P-glycoprotein): Recent advances and clinical relevance. Clin. Pharmacol. Ther. 2004, 75, 13–33. [Google Scholar] [CrossRef]
- De Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Jostins, L.; Rice, D.L.; Gutierrez-Achury, J.; Ji, S.-G.; et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef] [Green Version]
- SEPHS1 Selenophosphate Synthetase 1 [Homo sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/22929 (accessed on 4 February 2022).
- HUGO Gene Nomenclature Committee. Gene Symbol Report. Available online: https://www.genenames.org/data/gene-symbol-report/#!/hgnc_id/HGNC:19685 (accessed on 4 February 2022).
- Gentschew, L.; Bishop, K.S.; Han, D.Y.; Morgan, A.R.; Fraser, A.G.; Lam, W.J.; Karunasinghe, N.; Campbell, B.; Ferguson, L.R. Selenium, Selenoprotein Genes and Crohn’s Disease in a Case-Control Population from Auckland, New Zealand. Nutrients 2012, 4, 1247–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Q.-Q.; Xie, W.-Y.; Tang, Y.-J.; Zhang, J.; Liu, J. Genetic variation in the glucocorticoid pathway involved in interindividual differences in the glucocorticoid treatment. Pharmacogenomics 2017, 18, 293–316. [Google Scholar] [CrossRef] [PubMed]
- Corticosteroids. Available online: https://www.pharmgkb.org/chemical/PA10832/clinicalAnnotation (accessed on 21 December 2021).
- Janssen, C.A.; Voshaar, M.A.H.O.; Vonkeman, H.E.; Jansen, T.L.T.A.; Janssen, M.; Kok, M.; Radovits, B.; Van Durme, C.; Baan, H.; Laar, M.A.F.J.V.D. Anakinra for the treatment of acute gout flares: A randomized, double-blind, placebo-controlled, active-comparator, non-inferiority trial. Rheumatology 2019, 58, 1344–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, Y.M. Race and precision medicine: Is it time for an upgrade? Pharm. J. 2019, 19, 1–4. [Google Scholar] [CrossRef]
- Roman, Y. The United States 2020 Census data: Implications for precision medicine and the research landscape. Pers. Med. 2021, 19, 5–8. [Google Scholar] [CrossRef]
- Roman, Y.M.; Lor, K.; Xiong, T.; Culhane-Pera, K.; Straka, R.J. Gout prevalence in the Hmong: A prime example of health disparity and the role of community-based genetic research. Pers. Med. 2021, 18, 311–327. [Google Scholar] [CrossRef]
- Coronado, G.; Chio-Lauri, J.; Cruz, R.D.; Roman, Y.M. Health Disparities of Cardiometabolic Disorders Among Filipino Americans: Implications for Health Equity and Community-Based Genetic Research. J. Racial Ethn. Health Disparities 2021. [Google Scholar] [CrossRef]
- Roman, Y.M.; Dixon, D.L.; Salgado, T.M.; Price, E.T.; Zimmerman, K.M.; Sargent, L.; Slattum, P.W. Challenges in pharmacotherapy for older adults: A framework for pharmacogenomics implementation. Pharmacogenomics 2020, 21, 627–635. [Google Scholar] [CrossRef]
- Anderson, A.N.; Chan, A.R.; Roman, Y.M. Pharmacogenomics and clinical cultural competency: Pathway to overcome the limitations of race. Pharmacogenomics 2022, 23, 363–370. [Google Scholar] [CrossRef]
- Ke, C.-H.; Chung, W.-H.; Wen, Y.-H.; Huang, Y.-B.; Chuang, H.-Y.; Tain, Y.-L.; Wang, Y.-C.L.; Wu, C.-C.; Hsu, C.-N. Cost-effectiveness Analysis for Genotyping before Allopurinol Treatment to Prevent Severe Cutaneous Adverse Drug Reactions. J. Rheumatol. 2017, 44, 835–843. [Google Scholar] [CrossRef] [Green Version]
- Park, D.-J.; Kang, J.-H.; Lee, J.-W.; Lee, K.-E.; Wen, L.; Kim, T.-J.; Park, Y.-W.; Park, S.-H.; Lee, S.-S. Cost-Effectiveness Analysis of HLA-B5801 Genotyping in the Treatment of Gout Patients with Chronic Renal Insufficiency in Korea. Arthritis Care Res. 2015, 67, 280–287. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Drug | Mapped Genes | Effect | Clinical Outcomes | CPIC Guideline Level of Evidence a | References |
|---|---|---|---|---|---|
| Xanthine oxidase inhibitors (XO) | |||||
| Allopurinol or Oxypurinol | HLA-B | Safety | HLA-B*58:01 allele significantly increases the risk of allopurinol-induced serious cutaneous reaction | A | [5,28] |
| AOX | Response | rs3731722 A>G is associated with a better response to the standard dose of allopurinol (300 mg/day) vs. non-carriers | NA | [29] | |
| ABCG2 | Response/PK | rs2231142 C>A (Q141K) is associated with poor response to allopurinol | NA | [30] | |
| SLC22A12 | Response/PK | rs505802 C>T may influence the response to allopurinol and the PK of oxypurinol as they are substrates for the URAT1 | NA | [11,31,32] | |
| Febuxostat | UGT1A1 | Response/PK | rs34650714 C>T is associated with lower doses of febuxostat | NA | [29] |
| Uricosuric Agents | |||||
| Probenecid | SLC22A12 | Response | Homozygous or heterozygous for the mutant allele (G774A) have impaired response to loading tests of probenecid | NA | [33,34] |
| ABCB1 | PK | rs1045642 C>T could influence the PK effect of probenecid as an inhibitor when co-administered with Beta-lactam | NA | [35] | |
| G6PD | Safety | Possible hematologic adverse reactions in G6PD deficient patients | B | [36] | |
| Benzbromarone | CYP2C9 | Safety | Carriers of the no-function allele (CYP2C9*3) have reduced metabolic activity leading to prolonged exposure to benzbromarone relative to normal metabolizers | NA | [37,38] |
| Recombinant Uricase | |||||
| Pegloticase | G6PD | Safety | Risk of hemolysis or methemoglobinemia in G6PD deficient patients | B | [39] |
| Non-steroidal anti-inflammatory drugs (NSAIDs) | |||||
| Ibuprofen, celecoxib, and other NSAIDs | CYP2C9 | Safety/PK | Increased risk of NSAID-related GI bleeding in no-function allele (*3) carriers relative to normal function, as well as reduced metabolism and prolonged exposure to ibuprofen and celecoxib in CYP2C9 poor metabolizers | A (ibuprofen and celecoxib); C (indomethacin, diclofenac, naproxen) | [40,41] |
| Anti-inflammatory | |||||
| Colchicine | CYP2D6 | Response | Diminished response to colchicine in CYP2D6*4 variant carriers | NA | [42] |
| ABCB1 | Inconsistent evidence wherein one study indicates good response in the T allele carriers of the SNP rs10455642 C>T, while another study suggests no response with the T allele | NA | [43,44] | ||
| SEPHS1 | Safety | The risk allele G of rs74795203 A>G significantly increases the risk of gastrointestinal adverse events by 2.5-fold with using colchicine | NA | [45] | |
| KIF13A, RNU6-793Pb | Safety | The risk allele A of rs6916345 G>A (intergenic) was significantly associated with a ~2-fold increased risk of gastrointestinal adverse events with colchicine compared with the G allele | NA | [45] | |
| Corticosteroids | |||||
| Injectable triamcinolone acetonide | HCG22 | Safety | The G and T alleles of rs3873352 C>G and rs2523864 C>T, respectively, increase the risk of steroid-induced ocular hypertension | NA | [46] |
| IL-1 inhibitor | |||||
| Anakinra | IL1RN | Response | SNP cluster in strong linkage disequilibrium associated with poor response to anakinra | NA | [47] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alrajeh, K.Y.; Roman, Y.M. Pharmacogenetic Perspective for Optimal Gout Management. Future Pharmacol. 2022, 2, 135-152. https://doi.org/10.3390/futurepharmacol2020011
Alrajeh KY, Roman YM. Pharmacogenetic Perspective for Optimal Gout Management. Future Pharmacology. 2022; 2(2):135-152. https://doi.org/10.3390/futurepharmacol2020011
Chicago/Turabian StyleAlrajeh, Khalifa Y., and Youssef M. Roman. 2022. "Pharmacogenetic Perspective for Optimal Gout Management" Future Pharmacology 2, no. 2: 135-152. https://doi.org/10.3390/futurepharmacol2020011