
Mitochondrial Transplantation: Is It a Feasible Therapy to Prevent the Cardiorenal Side Effects of Cisplatin?

 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Acute Kidney Injury
1.1. Platinum Based Drugs in Cancer
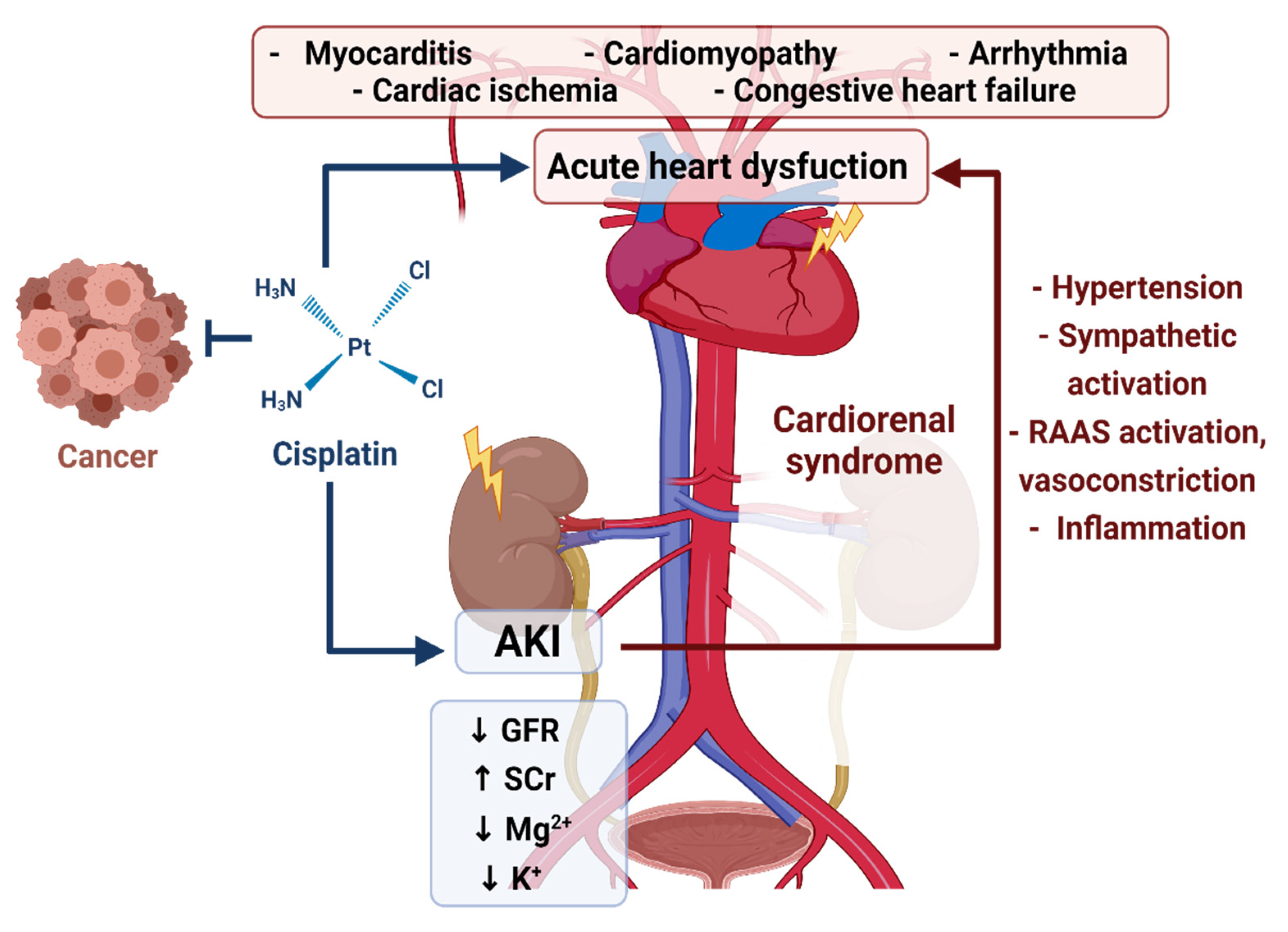
1.2. Cisplatin Induced AKI
1.3. Cisplatin-Induced AKI and the Cardiorenal Association
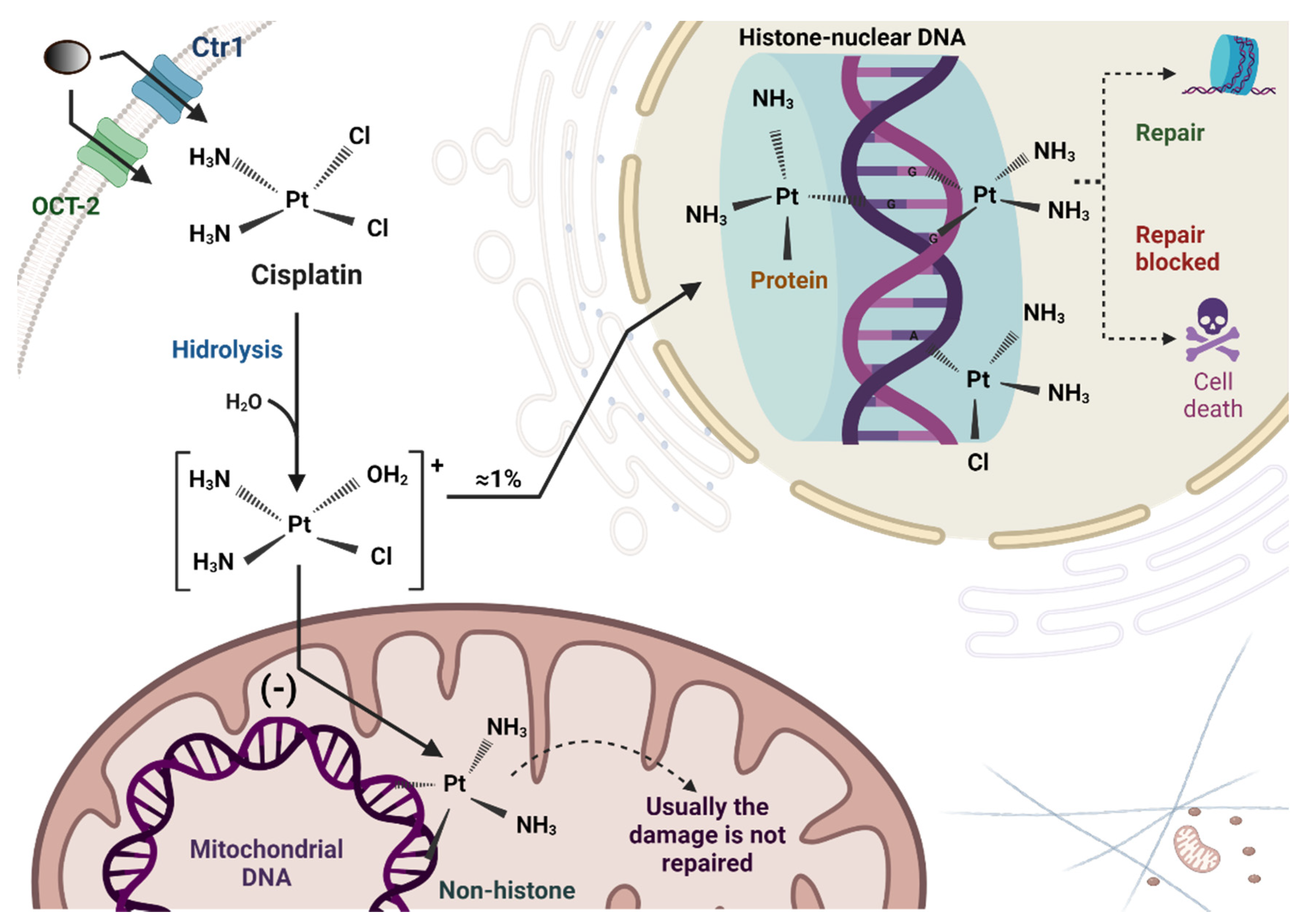
2. Cisplatin Uptake and Biotransformation in Tubular Cells
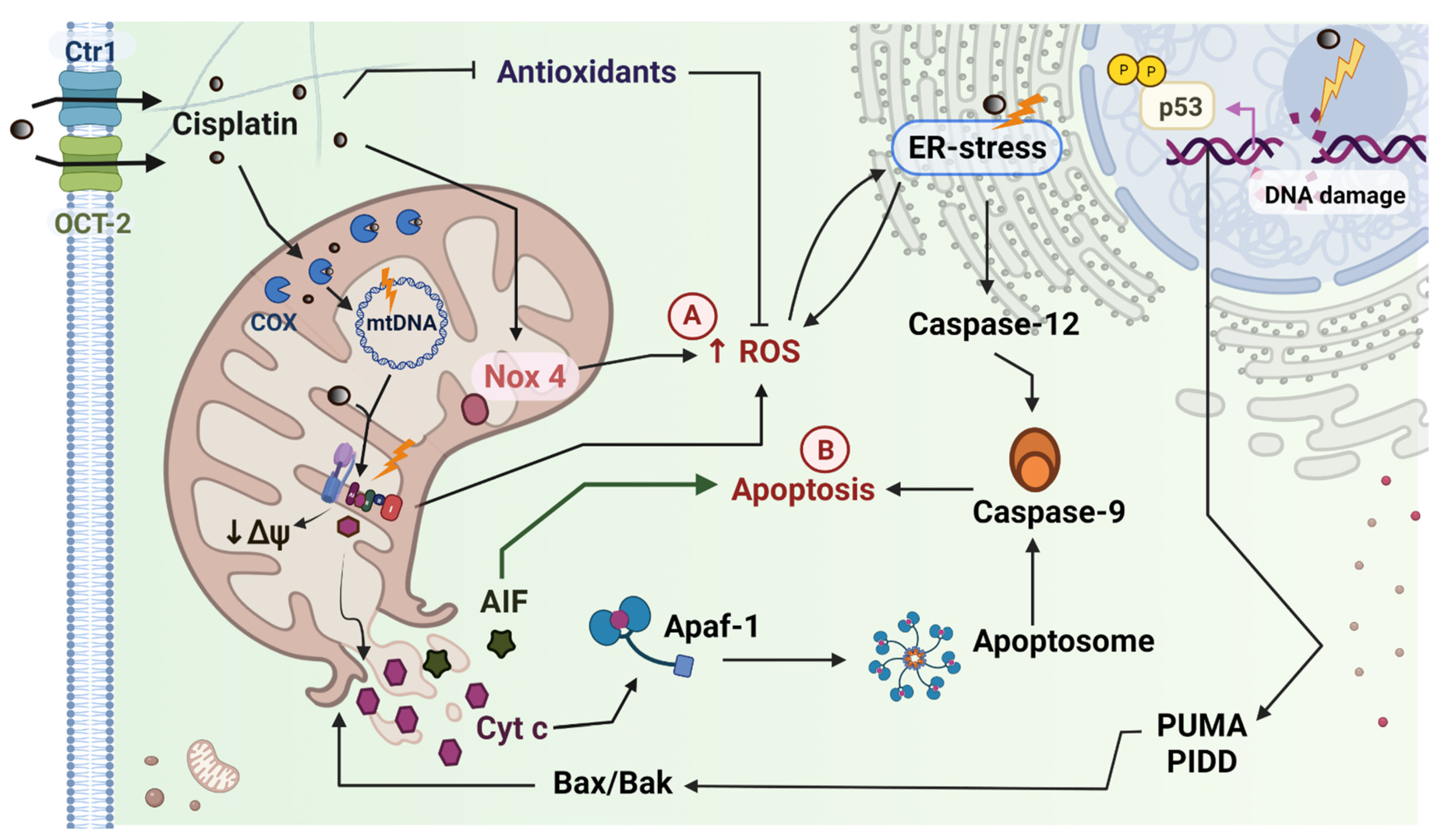
2.1. Cisplatin Induced Mitochondrial Dysfunction in the Kidney
2.2. Cisplatin Induced Mitochondrial Dysfunction in the Heart
3. Therapeutic Approaches to Mitigate the Nephrotoxic Effects of Cisplatin
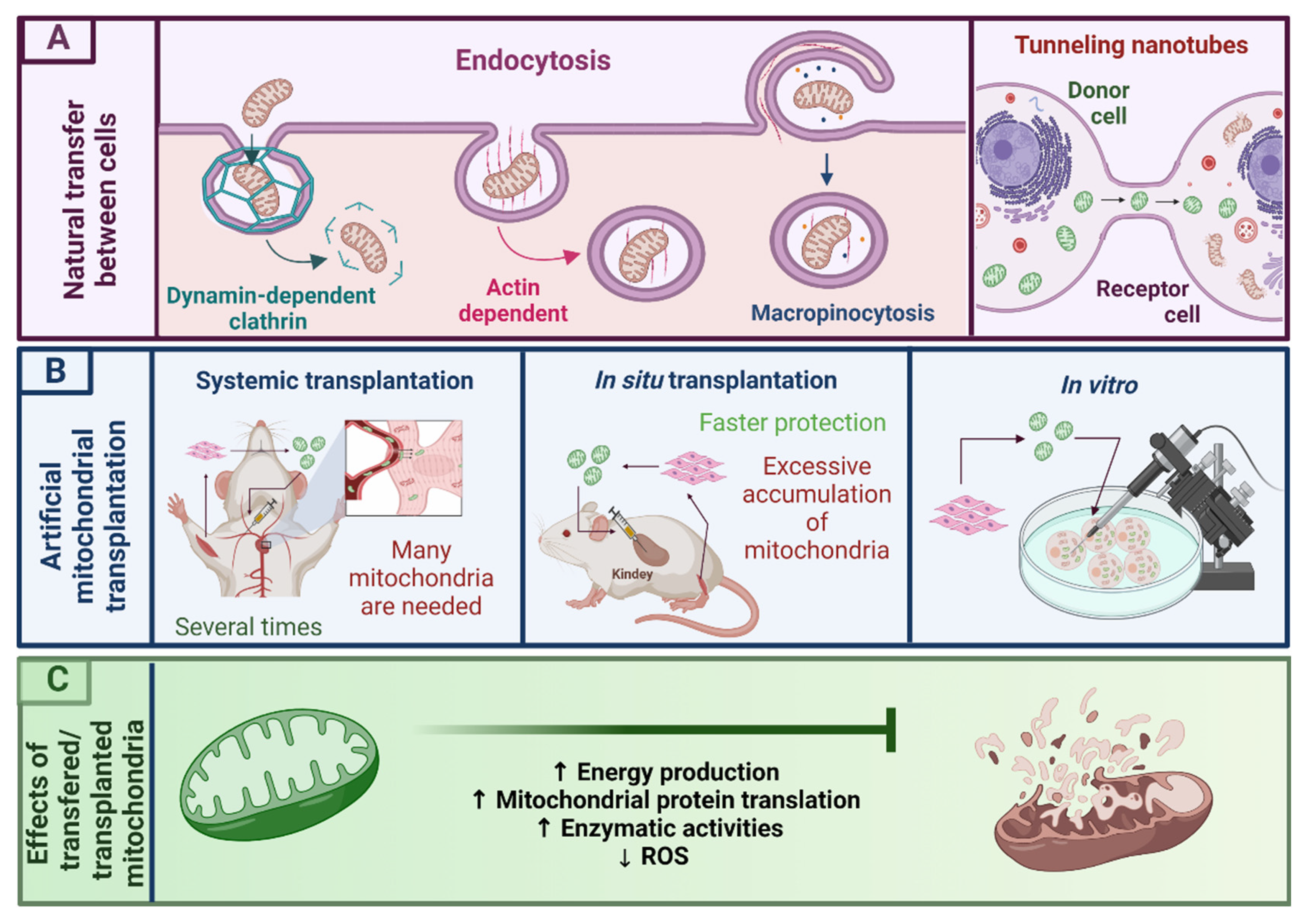
3.1. Mitochondrial Transplantation as a Novel Therapy for Mitochondrial Dysfunction
3.2. Mitochondrial Isolation
3.3. MT in Pathology Induced Mitochondrial Dysfunction
- (a)
- Liver
- (b)
- Kidney
- (c)
- Heart
3.4. MT in Drug Induced Mitochondrial Dysfunction
- (a)
- Liver
- (b)
- Kidney
- (c)
- Heart
4. MT as a Possible Therapy against Cisplatin-Induced Cardiorenal Alterations
5. Concluding Remarks and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hoste, E.A.J.; Kellum, J.A.; Selby, N.M.; Zarbock, A.; Palevsky, P.M.; Bagshaw, S.M.; Goldstein, S.L.; Cerdá, J.; Chawla, L.S. Global Epidemiology and Outcomes of Acute Kidney Injury. Nat. Rev. Nephrol. 2018, 14, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Ikizler, T.A.; Parikh, C.R.; Himmelfarb, J.; Chinchilli, V.M.; Liu, K.D.; Coca, S.G.; Garg, A.X.; Hsu, C.; Siew, E.D.; Wurfel, M.M.; et al. A Prospective Cohort Study of Acute Kidney Injury and Kidney Outcomes, Cardiovascular Events, and Death. Kidney Int. 2021, 99, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Kane-Gill, S.L.; Goldstein, S.L. Drug-Induced Acute Kidney Injury. Crit. Care Clin. 2015, 31, 675–684. [Google Scholar] [CrossRef]
- Mehta, R.L.; Pascual, M.T.; Soroko, S.; Savage, B.R.; Himmelfarb, J.; Ikizler, T.A.; Paganini, E.P.; Chertow, G.M. Spectrum of Acute Renal Failure in the Intensive Care Unit: The PICARD Experience. Kidney Int. 2004, 66, 1613–1621. [Google Scholar] [CrossRef] [Green Version]
- Faught, L.N.; Greff, M.J.E.; Rieder, M.J.; Koren, G. Drug-Induced Acute Kidney Injury in Children: Drug-Induced AKI in Children. Br. J. Clin. Pharmacol. 2015, 80, 901–909. [Google Scholar] [CrossRef] [Green Version]
- Uchino, S. The Epidemiology of Acute Renal Failure in the World. Curr. Opin. Crit. Care 2006, 12, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Sales, G.T.M.; Foresto, R.D. Drug-Induced Nephrotoxicity. Rev. Assoc. Méd. Bras. 2020, 66 (Suppl. S1), s82–s90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Fu, Y.; Liu, Z.; Shu, S.; Wang, Y.; Tang, C.; Cai, J.; Dong, Z. NAM Protects against Cisplatin-Induced Acute Kidney Injury by Suppressing the PARP1/P53 Pathway. Toxicol. Appl. Pharmacol. 2021, 418, 115492. [Google Scholar] [CrossRef]
- Kwiatkowska, E.; Domański, L.; Dziedziejko, V.; Kajdy, A.; Stefańska, K.; Kwiatkowski, S. The Mechanism of Drug Nephrotoxicity and the Methods for Preventing Kidney Damage. Int. J. Mol. Sci. 2021, 22, 6109. [Google Scholar] [CrossRef]
- Zhou, J.; Kang, Y.; Chen, L.; Wang, H.; Liu, J.; Zeng, S.; Yu, L. The Drug-Resistance Mechanisms of Five Platinum-Based Antitumor Agents. Front. Pharmacol. 2020, 11, 343. [Google Scholar] [CrossRef] [Green Version]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The Side Effects of Platinum-Based Chemotherapy Drugs: A Review for Chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Buechel, M.; Herzog, T.J.; Westin, S.N.; Coleman, R.L.; Monk, B.J.; Moore, K.N. Treatment of Patients with Recurrent Epithelial Ovarian Cancer for Whom Platinum Is Still an Option. Ann. Oncol. 2019, 30, 721–732. [Google Scholar] [CrossRef]
- Liu, Y.L.; Zhou, Q.; Iasonos, A.; Emengo, V.N.; Friedman, C.; Konner, J.A.; O’Cearbhaill, R.E.; Aghajanian, C.; Zamarin, D. Subsequent Therapies and Survival after Immunotherapy in Recurrent Ovarian Cancer. Gynecol. Oncol. 2019, 155, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wu, M.; Wu, D.; Wang, L.; Li, H.; Lin, Z.; Li, J. Calculating the Dose of Cisplatin That Is Actually Utilized in Hyperthermic Intraperitoneal Chemotherapy among Ovarian Cancer Patients. J. Ovarian Res. 2021, 14, 9. [Google Scholar] [CrossRef]
- Tsao, L.R.; Young, F.D.; Otani, I.M.; Castells, M.C. Hypersensitivity Reactions to Platinum Agents and Taxanes. Clin. Rev. Allergy Immunol. 2021, 1–17. (In Press) [Google Scholar] [CrossRef]
- Funakoshi, Y.; Fujiwara, Y.; Kiyota, N.; Mukohara, T.; Shimada, T.; Toyoda, M.; Imamura, Y.; Chayahara, N.; Tomioka, H.; Umezu, M.; et al. Validity of New Methods to Evaluate Renal Function in Cancer Patients Treated with Cisplatin. Cancer Chemother. Pharmacol. 2016, 77, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Tixier, F.; Ranchon, F.; Iltis, A.; Vantard, N.; Schwiertz, V.; Bachy, E.; Bouafia-Sauvy, F.; Sarkozy, C.; Tournamille, J.F.; Gyan, E.; et al. Comparative Toxicities of 3 Platinum-Containing Chemotherapy Regimens in Relapsed/Refractory Lymphoma Patients: Platinum-Containing Chemotherapy Regimens. Hematol. Oncol. 2017, 35, 584–590. [Google Scholar] [CrossRef]
- Sato, K.; Watanabe, S.; Ohtsubo, A.; Shoji, S.; Ishikawa, D.; Tanaka, T.; Nozaki, K.; Kondo, R.; Okajima, M.; Miura, S.; et al. Nephrotoxicity of Cisplatin Combination Chemotherapy in Thoracic Malignancy Patients with CKD Risk Factors. BMC Cancer 2016, 16, 222. [Google Scholar] [CrossRef] [Green Version]
- Latcha, S.; Jaimes, E.A.; Patil, S.; Glezerman, I.G.; Mehta, S.; Flombaum, C.D. Long–Term Renal Outcomes after Cisplatin Treatment. Clin. J. Am. Soc. Nephrol. 2016, 11, 1173–1179. [Google Scholar] [CrossRef]
- Shi, M.; McMillan, K.L.; Wu, J.; Gillings, N.; Flores, B.; Moe, O.W.; Hu, M.C. Cisplatin Nephrotoxicity as a Model of Chronic Kidney Disease. Lab. Investig. 2018, 98, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- Małyszko, J.; Kozłowska, K.; Kozłowski, L.; Małyszko, J. Nephrotoxicity of Anticancer Treatment. Nephrol. Dial. Transplant. 2016, 32, 924–936. [Google Scholar] [CrossRef]
- Pabla, N.; Dong, Z. Cisplatin Nephrotoxicity: Mechanisms and Renoprotective Strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [Green Version]
- Gudsoorkar, P.S.; Thakar, C.V. Acute Kidney Injury, Heart Failure, and Health Outcomes. Cardiol. Clin. 2019, 37, 297–305. [Google Scholar] [CrossRef]
- Gómez-Sierra, T.; Eugenio-Pérez, D.; Sánchez-Chinchillas, A.; Pedraza-Chaverri, J. Role of Food-Derived Antioxidants against Cisplatin Induced-Nephrotoxicity. Food Chem. Toxicol. 2018, 120, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Perazella, M.A. Onco-Nephrology: Renal Toxicities of Chemotherapeutic Agents. Clin. J. Am. Soc. Nephrol. 2012, 7, 1713–1721. [Google Scholar] [CrossRef]
- McSweeney, K.R.; Gadanec, L.K.; Qaradakhi, T.; Ali, B.A.; Zulli, A.; Apostolopoulos, V. Mechanisms of Cisplatin-Induced Acute Kidney Injury: Pathological Mechanisms, Pharmacological Interventions, and Genetic Mitigations. Cancers 2021, 13, 1572. [Google Scholar] [CrossRef]
- Kramann, R.; Kusaba, T.; Humphreys, B.D. Who Regenerates the Kidney Tubule? Nephrol. Dial. Transplant. 2015, 30, 903–910. [Google Scholar] [CrossRef] [Green Version]
- Oh, G.-S.; Kim, H.-J.; Shen, A.; Lee, S.B.; Khadka, D.; Pandit, A.; So, H.-S. Cisplatin-Induced Kidney Dysfunction and Perspectives on Improving Treatment Strategies. Electrolytes Blood Press. 2014, 12, 55. [Google Scholar] [CrossRef] [Green Version]
- Rangaswami, J.; Bhalla, V.; Blair, J.E.A.; Chang, T.I.; Costa, S.; Lentine, K.L.; Lerma, E.V.; Mezue, K.; Molitch, M.; Mullens, W.; et al. Cardiorenal Syndrome: Classification, Pathophysiology, Diagnosis, and Treatment Strategies: A Scientific Statement From the American Heart Association. Circulation 2019, 139, e840–e878. [Google Scholar] [CrossRef] [PubMed]
- McCullough, P.A. Cardiorenal Syndromes: Pathophysiology to Prevention. Int. J. Nephrol. 2011, 2011, 762590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubattu, S.; Mennuni, S.; Testa, M.; Mennuni, M.; Pierelli, G.; Pagliaro, B.; Gabriele, E.; Coluccia, R.; Autore, C.; Volpe, M. Pathogenesis of Chronic Cardiorenal Syndrome: Is There a Role for Oxidative Stress? Int. J. Mol. Sci. 2013, 14, 23011–23032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soranno, D.E.; Kirkbride-Romeo, L.; Wennersten, S.A.; Ding, K.; Cavasin, M.A.; Baker, P.; Altmann, C.; Bagchi, R.A.; Haefner, K.R.; Steinkühler, C.; et al. Acute Kidney Injury Results in Long-Term Diastolic Dysfunction That Is Prevented by Histone Deacetylase Inhibition. JACC Basic Transl. Sci. 2021, 6, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Patanè, S. Cardiotoxicity: Cisplatin and Long-Term Cancer Survivors. Int. J. Cardiol. 2014, 175, 201–202. [Google Scholar] [CrossRef]
- El-Hawwary, A.A.; Omar, N.M. The Influence of Ginger Administration on Cisplatin-Induced Cardiotoxicity in Rat: Light and Electron Microscopic Study. Acta Histochem. 2019, 121, 553–562. [Google Scholar] [CrossRef]
- Hanchate, L.P. Cisplatin Induced Acute Myocardial Infarction and Dyslipidemia. J. Clin. Diagn. Res. 2017, 11, OD05–OD07. [Google Scholar] [CrossRef]
- Dugbartey, G.J.; Peppone, L.J.; de Graaf, I.A.M. An Integrative View of Cisplatin-Induced Renal and Cardiac Toxicities: Molecular Mechanisms, Current Treatment Challenges and Potential Protective Measures. Toxicology 2016, 371, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjerring, A.W.; Fosså, S.D.; Haugnes, H.S.; Nome, R.; Stokke, T.M.; Haugaa, K.H.; Kiserud, C.E.; Edvardsen, T.; Sarvari, S.I. The Cardiac Impact of Cisplatin-Based Chemotherapy in Survivors of Testicular Cancer: A 30-Year Follow-Up. Eur. Heart J.-Cardiovasc. Imaging 2021, 22, 443–450. [Google Scholar] [CrossRef]
- Steingart, R. Mechanisms of Late Cardiovascular Toxicity from Cancer Chemotherapy. J. Clin. Oncol. 2005, 23, 9051–9052. [Google Scholar] [CrossRef]
- Steingart, R.M.; Yadav, N.; Manrique, C.; Carver, J.R.; Liu, J. Cancer Survivorship: Cardiotoxic Therapy in the Adult Cancer Patient; Cardiac Outcomes With Recommendations for Patient Management. Semin. Oncol. 2013, 40, 690–708. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Bakhaat, G.A.; Tammam, H.G.; Mohamed, R.M.; El-Naggar, S.A. Cardioprotective Effect of Green Tea Extract and Vitamin E on Cisplatin-Induced Cardiotoxicity in Mice: Toxicological, Histological and Immunohistochemical Studies. Biomed. Pharmacother. 2019, 113, 108731. [Google Scholar] [CrossRef]
- Cui, Y.; Li, C.; Zeng, C.; Li, J.; Zhu, Z.; Chen, W.; Huang, A.; Qi, X. Tongmai Yangxin Pills Anti-Oxidative Stress Alleviates Cisplatin-Induced Cardiotoxicity: Network Pharmacology Analysis and Experimental Evidence. Biomed. Pharmacother. 2018, 108, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Bahadır, A. Protective Effects of Curcumin and Beta-Carotene on Cisplatin-Induced Cardiotoxicity: An Experimental Rat Model. Anatol. J. Cardiol. 2018, 19, 213–221. [Google Scholar] [CrossRef]
- Saleh, D.O.; Mansour, D.F.; Mostafa, R.E. Rosuvastatin and Simvastatin Attenuate Cisplatin-Induced Cardiotoxicity via Disruption of Endoplasmic Reticulum Stress-Mediated Apoptotic Death in Rats: Targeting ER-Chaperone GRP78 and Calpain-1 Pathways. Toxicol. Rep. 2020, 7, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Chirino, Y.I.; Pedraza-Chaverri, J. Role of Oxidative and Nitrosative Stress in Cisplatin-Induced Nephrotoxicity. Exp. Toxicol. Pathol. 2009, 61, 223–242. [Google Scholar] [CrossRef]
- Mapuskar, K.A.; Wen, H.; Holanda, D.G.; Rastogi, P.; Steinbach, E.; Han, R.; Coleman, M.C.; Attanasio, M.; Riley, D.P.; Spitz, D.R.; et al. Persistent Increase in Mitochondrial Superoxide Mediates Cisplatin-Induced Chronic Kidney Disease. Redox Biol. 2019, 20, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin Induces a Mitochondrial-ROS Response That Contributes to Cytotoxicity Depending on Mitochondrial Redox Status and Bioenergetic Functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin Nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef] [Green Version]
- Mandic, A.; Hansson, J.; Linder, S.; Shoshan, M.C. Cisplatin Induces Endoplasmic Reticulum Stress and Nucleus-Independent Apoptotic Signaling. J. Biol. Chem. 2003, 278, 9100–9106. [Google Scholar] [CrossRef] [Green Version]
- Qian, W.; Nishikawa, M.; Haque, A.M.; Hirose, M.; Mashimo, M.; Sato, E.; Inoue, M. Mitochondrial Density Determines the Cellular Sensitivity to Cisplatin-Induced Cell Death. Am. J. Physiol.-Cell Physiol. 2005, 289, C1466–C1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghadasali, R.; Azarnia, M.; Hajinasrollah, M.; Arghani, H.; Nassiri, S.M.; Molazem, M.; Vosough, A.; Mohitmafi, S.; Najarasl, M.; Ajdari, Z.; et al. Intra-Renal Arterial Injection of Autologous Bone Marrow Mesenchymal Stromal Cells Ameliorates Cisplatin-Induced Acute Kidney Injury in a Rhesus Macaque Mulatta Monkey Model. Cytotherapy 2014, 16, 734–749. [Google Scholar] [CrossRef]
- Harrach, S.; Ciarimboli, G. Role of Transporters in the Distribution of Platinum-Based Drugs. Front. Pharmacol. 2015, 6, 85. [Google Scholar] [CrossRef] [Green Version]
- Freitas-Lima, L.C.; Budu, A.; Arruda, A.C.; Perilhão, M.S.; Barrera-Chimal, J.; Araujo, R.C.; Estrela, G.R. PPAR-α Deletion Attenuates Cisplatin Nephrotoxicity by Modulating Renal Organic Transporters MATE-1 and OCT-2. Int. J. Mol. Sci. 2020, 21, 7416. [Google Scholar] [CrossRef]
- Townsend, D.M.; Deng, M.; Zhang, L.; Lapus, M.G.; Hanigan, M.H. Metabolism of Cisplatin to a Nephrotoxin in Proximal Tubule Cells. J. Am. Soc. Nephrol. 2003, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Townsend, D.M.; Tew, K.D.; He, L.; King, J.B.; Hanigan, M.H. Role of Glutathione S-Transferase Pi in Cisplatin-Induced Nephrotoxicity. Biomed. Pharmacother. 2009, 63, 79–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichinomiya, M.; Shimada, A.; Ohta, N.; Inouchi, E.; Ogihara, K.; Naya, Y.; Nagane, M.; Morita, T.; Satoh, M. Demonstration of Mitochondrial Damage and Mitophagy in Cisplatin-Mediated Nephrotoxicity. Tohoku J. Exp. Med. 2018, 246, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega-Domínguez, B.; Aparicio-Trejo, O.E.; García-Arroyo, F.E.; León-Contreras, J.C.; Tapia, E.; Molina-Jijón, E.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Barrera-Oviedo, D.; Pedraza-Chaverri, J. Curcumin Prevents Cisplatin-Induced Renal Alterations in Mitochondrial Bioenergetics and Dynamic. Food Chem. Toxicol. 2017, 107, 373–385. [Google Scholar] [CrossRef]
- Yan, M.; Tang, C.; Ma, Z.; Huang, S.; Dong, Z. DNA Damage Response in Nephrotoxic and Ischemic Kidney Injury. Toxicol. Appl. Pharmacol. 2016, 313, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Bernal-Barquero, C.E.; Vazquez-Zapien, G.J.; Mata-Miranda, M.M. Review of Alterations in Gene Expression and Apoptotic Pathways Caused in Nephrotoxicity Induced by Cisplatin. Nefrología 2019, 39, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, Y.; Inagi, R. Mitochondria: A Therapeutic Target in Acute Kidney Injury. Nephrol. Dial. Transplant. 2016, 31, 1062–1069. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, C.; Cai, J.; Chen, G.; Zhang, D.; Zhang, Z.; Dong, Z. PINK1/Parkin-Mediated Mitophagy Is Activated in Cisplatin Nephrotoxicity to Protect against Kidney Injury. Cell Death Dis. 2018, 9, 1113. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Ciofi-Baffoni, S.; Hadjiloi, T.; Martinelli, M.; Palumaa, P. Mitochondrial Copper(I) Transfer from Cox17 to Sco1 Is Coupled to Electron Transfer. Proc. Natl. Acad. Sci. USA 2008, 105, 6803–6808. [Google Scholar] [CrossRef] [Green Version]
- Ong, J.X.; Le, H.V.; Lee, V.E.Y.; Ang, W.H. A Cisplatin-Selective Fluorescent Probe for Real-Time Monitoring of Mitochondrial Platinum Accumulation in Living Cells. Angew. Chem. Int. Ed. 2021, 60, 9264–9269. [Google Scholar] [CrossRef]
- Meyer, J.N.; Hartman, J.H.; Mello, D.F. Mitochondrial Toxicity. Toxicol. Sci. 2018, 162, 15–23. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, N.A.G.; Carvalho Rodrigues, M.A.; Martins, N.M.; dos Santos, A.C. Cisplatin-Induced Nephrotoxicity and Targets of Nephroprotection: An Update. Arch. Toxicol. 2012, 86, 1233–1250. [Google Scholar] [CrossRef]
- Ueki, M.; Ueno, M.; Morishita, J.; Maekawa, N. Curcumin Ameliorates Cisplatin-Induced Nephrotoxicity by Inhibiting Renal Inflammation in Mice. J. Biosci. Bioeng. 2013, 115, 547–551. [Google Scholar] [CrossRef]
- Demkow, U.; Stelmaszczyk-Emmel, A. Cardiotoxicity of Cisplatin-Based Chemotherapy in Advanced Non-Small Cell Lung Cancer Patients. Respir. Physiol. Neurobiol. 2013, 187, 64–67. [Google Scholar] [CrossRef] [PubMed]
- van der Bliek, A.M.; Sedensky, M.M.; Morgan, P.G. Cell Biology of the Mitochondrion. Genetics 2017, 207, 843–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, H.; John, M.C.G. Free Radical in Biology and Medicine, 5th ed.; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative Stress, Aging, and Diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Gyurászová, M.; Gurecká, R.; Bábíčková, J.; Tóthová, Ľ. Oxidative Stress in the Pathophysiology of Kidney Disease: Implications for Noninvasive Monitoring and Identification of Biomarkers. Oxid. Med. Cell. Longev. 2020, 2020, 5478708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briones-Herrera, A.; Avila-Rojas, S.H.; Aparicio-Trejo, O.E.; Cristóbal, M.; León-Contreras, J.C.; Hernández-Pando, R.; Pinzón, E.; Pedraza-Chaverri, J.; Sánchez-Lozada, L.G.; Tapia, E. Sulforaphane Prevents Maleic Acid-Induced Nephropathy by Modulating Renal Hemodynamics, Mitochondrial Bioenergetics and Oxidative Stress. Food Chem. Toxicol. 2018, 115, 185–197. [Google Scholar] [CrossRef]
- Pan, H.; Chen, J.; Shen, K.; Wang, X.; Wang, P.; Fu, G.; Meng, H.; Wang, Y.; Jin, B. Mitochondrial Modulation by Epigallocatechin 3-Gallate Ameliorates Cisplatin Induced Renal Injury through Decreasing Oxidative/Nitrative Stress, Inflammation and NF-KB in Mice. PLoS ONE 2015, 10, e0124775. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, Y.; Li, B.; Ding, P.; Jin, D.; Hou, S.; Cai, X.; Sheng, X. The Effect of Monotropein on Alleviating Cisplatin-Induced Acute Kidney Injury by Inhibiting Oxidative Damage, Inflammation and Apoptosis. Biomed. Pharmacother. 2020, 129, 110408. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of Reactive Oxygen Species Generation by Mitochondria Oxidizing Different Substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, C.L.; Treberg, J.R.; Perevoshchikova, I.V.; Orr, A.L.; Brand, M.D. Native Rates of Superoxide Production from Multiple Sites in Isolated Mitochondria Measured Using Endogenous Reporters. Free Radic. Biol. Med. 2012, 53, 1807–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugasundaram, K.; Nayak, B.K.; Friedrichs, W.E.; Kaushik, D.; Rodriguez, R.; Block, K. NOX4 Functions as a Mitochondrial Energetic Sensor Coupling Cancer Metabolic Reprogramming to Drug Resistance. Nat. Commun. 2017, 8, 997. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Fermín, L.M.; Avila-Rojas, S.H.; Aparicio-Trejo, O.E.; Tapia, E.; Rivero, I.; Pedraza-Chaverri, J. The Protective Effect of Alpha-Mangostin against Cisplatin-Induced Cell Death in LLC-PK1 Cells Is Associated to Mitochondrial Function Preservation. Antioxidants 2019, 8, 133. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Yuan, Y.; Yuan, L.; Li, L.; Liu, F.; Liu, J.; Chen, Y.; Lu, Y.; Cheng, J. Activation of TFEB-Mediated Autophagy by Trehalose Attenuates Mitochondrial Dysfunction in Cisplatin-Induced Acute Kidney Injury. Theranostics 2020, 10, 5829–5844. [Google Scholar] [CrossRef] [PubMed]
- Perico, L.; Morigi, M.; Rota, C.; Breno, M.; Mele, C.; Noris, M.; Introna, M.; Capelli, C.; Longaretti, L.; Rottoli, D.; et al. Human Mesenchymal Stromal Cells Transplanted into Mice Stimulate Renal Tubular Cells and Enhance Mitochondrial Function. Nat. Commun. 2017, 8, 983. [Google Scholar] [CrossRef]
- Javadov, S.; Jang, S.; Agostini, B. Crosstalk between Mitogen-Activated Protein Kinases and Mitochondria in Cardiac Diseases: Therapeutic Perspectives. Pharmacol. Ther. 2014, 144, 202–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gollihue, J.L.; Rabchevsky, A.G. Prospects for Therapeutic Mitochondrial Transplantation. Mitochondrion 2017, 35, 70–79. [Google Scholar] [CrossRef]
- Meng, X.-M.; Ren, G.-L.; Gao, L.; Yang, Q.; Li, H.-D.; Wu, W.-F.; Huang, C.; Zhang, L.; Lv, X.; Li, J. NADPH Oxidase 4 Promotes Cisplatin-Induced Acute Kidney Injury via ROS-Mediated Programmed Cell Death and Inflammation. Lab. Investig. 2018, 98, 63–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.-N.; Yang, Q.; Yang, C.; Cai, Y.-T.; Xing, T.; Gao, L.; Wang, F.; Chen, X.; Liu, X.-Q.; He, X.-Y.; et al. Smad3 Promotes AKI Sensitivity in Diabetic Mice via Interaction with P53 and Induction of NOX4-Dependent ROS Production. Redox Biol. 2020, 32, 101479. [Google Scholar] [CrossRef]
- El-Awady, E.-S.E.; Moustafa, Y.M.; Abo-Elmatty, D.M.; Radwan, A. Cisplatin-Induced Cardiotoxicity: Mechanisms and Cardioprotective Strategies. Eur. J. Pharmacol. 2011, 650, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Meng, L.; Wu, S.; Li, Y.; Zhong, Y.; Xu, F.; Zhou, X.; Li, G.; Xu, G.; Peng, K. LIGHT Deficiency Aggravates Cisplatin-Induced Acute Kidney Injury by Upregulating Mitochondrial Apoptosis. Int. Immunopharmacol. 2020, 89, 106999. [Google Scholar] [CrossRef] [PubMed]
- Achkar, I.W.; Abdulrahman, N.; Al-Sulaiti, H.; Joseph, J.M.; Uddin, S.; Mraiche, F. Cisplatin Based Therapy: The Role of the Mitogen Activated Protein Kinase Signaling Pathway. J. Transl. Med. 2018, 16, 96. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, H.; Roushandeh, A.M.; Rostami, M.K.; Razavi-Toosi, M.T.; Shokrgozar, M.A.; Jahanian-Najafabadi, A.; Kuwahara, Y.; Roudkenar, M.H. Mitochondrial Transplantation Ameliorates Ischemia/Reperfusion-Induced Kidney Injury in Rat. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2020, 1866, 165809. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Kaushal, V.; Shah, S.V.; Kaushal, G.P. Autophagy Is Associated with Apoptosis in Cisplatin Injury to Renal Tubular Epithelial Cells. Am. J. Physiol.-Ren. Physiol. 2008, 294, F777–F787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konari, N.; Nagaishi, K.; Kikuchi, S.; Fujimiya, M. Mitochondria Transfer from Mesenchymal Stem Cells Structurally and Functionally Repairs Renal Proximal Tubular Epithelial Cells in Diabetic Nephropathy in Vivo. Sci. Rep. 2019, 9, 5184. [Google Scholar] [CrossRef] [Green Version]
- Luan, Z.; Wei, Y.; Huo, X.; Sun, X.; Zhang, C.; Ming, W.; Luo, Z.; Du, C.; Li, Y.; Xu, H.; et al. Pregnane X Receptor (PXR) Protects against Cisplatin-Induced Acute Kidney Injury in Mice. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2021, 1867, 165996. [Google Scholar] [CrossRef] [PubMed]
- McCully, J.D.; Levitsky, S.; Nido, P.J.; Cowan, D.B. Mitochondrial Transplantation for Therapeutic Use. Clin. Transl. Med. 2016, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Roushandeh, A.M.; Kuwahara, Y.; Roudkenar, M.H. Mitochondrial Transplantation as a Potential and Novel Master Key for Treatment of Various Incurable Diseases. Cytotechnology 2019, 71, 647–663. [Google Scholar] [CrossRef]
- Mietsch, M.; Hinkel, R. “Empowering” Cardiac Cells via Stem Cell Derived Mitochondrial Transplantation- Does Age Matter? Int. J. Mol. Sci. 2021, 22, 1824. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.N. Targeting the Mitochondria in Heart Failure. JACC Basic Transl. Sci. 2020, 5, 88–106. [Google Scholar] [CrossRef]
- Paliwal, S.; Chaudhuri, R.; Agrawal, A.; Mohanty, S. Regenerative Abilities of Mesenchymal Stem Cells through Mitochondrial Transfer. J. Biomed. Sci. 2018, 25, 31. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial Substrate Metabolism in the Normal and Failing Heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Shaker, M.E.; Shaaban, A.A.; El-Shafey, M.M.; El-Mesery, M.E. The Selective C-Met Inhibitor Capmatinib Offsets Cisplatin-Nephrotoxicity and Doxorubicin-Cardiotoxicity and Improves Their Anticancer Efficacies. Toxicol. Appl. Pharmacol. 2020, 398, 115018. [Google Scholar] [CrossRef]
- Kanzaki, Y.; Terasaki, F.; Okabe, M.; Otsuka, K.; Katashima, T.; Fujita, S.; Ito, T.; Kitaura, Y. Giant Mitochondria in the Myocardium of a Patient With Mitochondrial Cardiomyopathy: Transmission and 3-Dimensional Scanning Electron Microscopy. Circulation 2010, 121, 831–832. [Google Scholar] [CrossRef]
- Huang, X.; Sun, L.; Ji, S.; Zhao, T.; Zhang, W.; Xu, J.; Zhang, J.; Wang, Y.; Wang, X.; Franzini-Armstrong, C.; et al. Kissing and Nanotunneling Mediate Intermitochondrial Communication in the Heart. Proc. Natl. Acad. Sci. USA 2013, 110, 2846–2851. [Google Scholar] [CrossRef] [Green Version]
- Gollihue, J.L.; Patel, S.P.; Mashburn, C.; Eldahan, K.C.; Sullivan, P.G.; Rabchevsky, A.G. Optimization of Mitochondrial Isolation Techniques for Intraspinal Transplantation Procedures. J. Neurosci. Methods 2017, 287, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukelmoune, N.; Chiu, G.S.; Kavelaars, A.; Heijnen, C.J. Mitochondrial Transfer from Mesenchymal Stem Cells to Neural Stem Cells Protects against the Neurotoxic Effects of Cisplatin. Acta Neuropathol. Commun. 2018, 6, 139. [Google Scholar] [CrossRef]
- Lynch, E.D.; Gu, R.; Pierce, C.; Kil, J. Reduction of Acute Cisplatin Ototoxicity and Nephrotoxicity in Rats by Oral Administration of Allopurinol and Ebselen. Hear. Res. 2005, 201, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Valentovic, M.A.; Ball, J.G.; Mike Brown, J.; Terneus, M.V.; McQuade, E.; Van Meter, S.; Hedrick, H.M.; Roy, A.A.; Williams, T. Resveratrol Attenuates Cisplatin Renal Cortical Cytotoxicity by Modifying Oxidative Stress. Toxicol. In Vitro 2014, 28, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Jung, Y.J.; Lee, J.E.; Lee, A.S.; Kang, K.P.; Lee, S.; Park, S.K.; Han, M.K.; Lee, S.Y.; Ramkumar, K.M.; et al. SIRT1 Activation by Resveratrol Ameliorates Cisplatin-Induced Renal Injury through Deacetylation of P53. Am. J. Physiol.-Ren. Physiol. 2011, 301, F427–F435. [Google Scholar] [CrossRef] [Green Version]
- Badary, O.A.; Abdel-Maksoud, S.; Ahmed, W.A.; Owieda, G.H. Naringenin Attenuates Cisplatin Nephrotoxicity in Rats. Life Sci. 2005, 76, 2125–2135. [Google Scholar] [CrossRef]
- Ramesh, G.; Reeves, W.B. Salicylate Reduces Cisplatin Nephrotoxicity by Inhibition of Tumor Necrosis Factor-α. Kidney Int. 2004, 65, 490–498. [Google Scholar] [CrossRef] [Green Version]
- McCully, J.D.; Cowan, D.B.; Pacak, C.A.; Toumpoulis, I.K.; Dayalan, H.; Levitsky, S. Injection of Isolated Mitochondria during Early Reperfusion for Cardioprotection. Am. J. Physiol.-Heart Circ. Physiol. 2009, 296, H94–H105. [Google Scholar] [CrossRef] [Green Version]
- McCully, J.D.; Cowan, D.B.; Emani, S.M.; del Nido, P.J. Mitochondrial Transplantation: From Animal Models to Clinical Use in Humans. Mitochondrion 2017, 34, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Shi, X.; Zhang, H.; Fu, B. Mitotherapy for Fatty Liver by Intravenous Administration of Exogenous Mitochondria in Male Mice. Front. Pharmacol. 2017, 8, 241. [Google Scholar] [CrossRef] [Green Version]
- Cowan, D.B.; Yao, R.; Akurathi, V.; Snay, E.R.; Thedsanamoorthy, J.K.; Zurakowski, D.; Ericsson, M.; Friehs, I.; Wu, Y.; Levitsky, S.; et al. Intracoronary Delivery of Mitochondria to the Ischemic Heart for Cardioprotection. PLoS ONE 2016, 11, e0160889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; del Nido, P.J.; McCully, J.D. Autologous Mitochondrial Transplantation for Dysfunction after Ischemia-Reperfusion Injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289. [Google Scholar] [CrossRef] [Green Version]
- Ali Pour, P.; Kenney, M.C.; Kheradvar, A. Bioenergetics Consequences of Mitochondrial Transplantation in Cardiomyocytes. J. Am. Heart Assoc. 2020, 9, e014501. [Google Scholar] [CrossRef]
- Court, A.C.; Le-Gatt, A.; Luz-Crawford, P.; Parra, E.; Aliaga-Tobar, V.; Bátiz, L.F.; Contreras, R.A.; Ortúzar, M.I.; Kurte, M.; Elizondo-Vega, R.; et al. Mitochondrial Transfer from MSCs to T Cells Induces Treg Differentiation and Restricts Inflammatory Response. EMBO Rep. 2020, 21, e48052. [Google Scholar] [CrossRef]
- Torralba, D.; Baixauli, F.; Sánchez-Madrid, F. Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front. Cell Dev. Biol. 2016, 4, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Zhao, M.; Fu, C.; Fu, A. Intravenous Administration of Mitochondria for Treating Experimental Parkinson’s Disease. Mitochondrion 2017, 34, 91–100. [Google Scholar] [CrossRef]
- Kitani, T.; Kami, D.; Matoba, S.; Gojo, S. Internalization of Isolated Functional Mitochondria: Involvement of Macropinocytosis. J. Cell. Mol. Med. 2014, 18, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial Transfer between Cells Can Rescue Aerobic Respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, D.; Vacca, R.A.; Moro, L.; Atlante, A. Mitochondria Can Cross Cell Boundaries: An Overview of the Biological Relevance, Pathophysiological Implications and Therapeutic Perspectives of Intercellular Mitochondrial Transfer. Int. J. Mol. Sci. 2021, 22, 8312. [Google Scholar] [CrossRef] [PubMed]
- Yip, H.-K.; Dubey, N.K.; Lin, K.-C.; Sung, P.-H.; Chiang, J.Y.; Chu, Y.-C.; Huang, C.-R.; Chen, Y.-L.; Deng, Y.-H.; Cheng, H.-C.; et al. Melatonin Rescues Cerebral Ischemic Events through Upregulated Tunneling Nanotube-Mediated Mitochondrial Transfer and Downregulated Mitochondrial Oxidative Stress in Rat Brain. Biomed. Pharmacother. 2021, 139, 111593. [Google Scholar] [CrossRef]
- Pacak, C.A.; Preble, J.M.; Kondo, H.; Seibel, P.; Levitsky, S.; del Nido, P.J.; Cowan, D.B.; McCully, J.D. Actin-Dependent Mitochondrial Internalization in Cardiomyocytes: Evidence for Rescue of Mitochondrial Function. Biol. Open 2015, 4, 622–626. [Google Scholar] [CrossRef] [Green Version]
- Levoux, J.; Prola, A.; Lafuste, P.; Gervais, M.; Chevallier, N.; Koumaiha, Z.; Kefi, K.; Braud, L.; Schmitt, A.; Yacia, A.; et al. Platelets Facilitate the Wound-Healing Capability of Mesenchymal Stem Cells by Mitochondrial Transfer and Metabolic Reprogramming. Cell Metab. 2021, 33, 283–299. [Google Scholar] [CrossRef]
- Cowan, D.B.; Yao, R.; Thedsanamoorthy, J.K.; Zurakowski, D.; del Nido, P.J.; McCully, J.D. Transit and Integration of Extracellular Mitochondria in Human Heart Cells. Sci. Rep. 2017, 7, 17450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preble, J.M.; Pacak, C.A.; Kondo, H.; MacKay, A.A.; Cowan, D.B.; McCully, J.D. Rapid Isolation and Purification of Mitochondria For Transplantation By Tissue Dissociation And Differential Filtration. J. Vis. Exp. 2014, 91, e51682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, H.; Kami, D.; Maeda, R.; Shikuma, A.; Gojo, S. Generation of Somatic Mitochondrial DNA-Replaced Cells for Mitochondrial Dysfunction Treatment. Sci. Rep. 2021, 11, 10897. [Google Scholar] [CrossRef]
- Maeda, H.; Kami, D.; Maeda, R.; Murata, Y.; Jo, J.; Kitani, T.; Tabata, Y.; Matoba, S.; Gojo, S. TAT-dextran–Mediated Mitochondrial Transfer Enhances Recovery from Models of Reperfusion Injury in Cultured Cardiomyocytes. J. Cell. Mol. Med. 2020, 24, 5007–5020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, B.; Saeed, M.Y.; Esch, J.J.; Guariento, A.; Blitzer, D.; Moskowitzova, K.; Ramirez-Barbieri, G.; Orfany, A.; Thedsanamoorthy, J.K.; Cowan, D.B.; et al. A Novel Biological Strategy for Myocardial Protection by Intracoronary Delivery of Mitochondria: Safety and Efficacy. JACC Basic Transl. Sci. 2019, 4, 871–888. [Google Scholar] [CrossRef]
- Hwang, J.W.; Lee, M.J.; Chung, T.N.; Lee, H.A.R.; Lee, J.H.; Choi, S.Y.; Park, Y.J.; Kim, C.H.; Jin, I.; Kim, S.H.; et al. The Immune Modulatory Effects of Mitochondrial Transplantation on Cecal Slurry Model in Rat. Crit. Care 2021, 25, 20. [Google Scholar] [CrossRef]
- Xu, Y.; Yu, Y.; Yang, B.; Hui, J.; Zhang, C.; Fang, H.; Bian, X.; Tao, M.; Lu, Y.; Shang, Z. Extracellular Mitochondrial Components and Effects on Cardiovascular Disease. DNA Cell Biol. 2021, 40, 1–13. [Google Scholar] [CrossRef]
- Masuzawa, A.; Black, K.M.; Pacak, C.A.; Ericsson, M.; Barnett, R.J.; Drumm, C.; Seth, P.; Bloch, D.B.; Levitsky, S.; Cowan, D.B.; et al. Transplantation of Autologously Derived Mitochondria Protects the Heart from Ischemia-Reperfusion Injury. Am. J. Physiol.-Heart Circ. Physiol. 2013, 304, H966–H982. [Google Scholar] [CrossRef]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef]
- Kaza, A.K.; Wamala, I.; Friehs, I.; Kuebler, J.D.; Rathod, R.H.; Berra, I.; Ericsson, M.; Yao, R.; Thedsanamoorthy, J.K.; Zurakowski, D.; et al. Myocardial Rescue with Autologous Mitochondrial Transplantation in a Porcine Model of Ischemia/Reperfusion. J. Thorac. Cardiovasc. Surg. 2017, 153, 934–943. [Google Scholar] [CrossRef] [Green Version]
- Moskowitzova, K.; Shin, B.; Liu, K.; Ramirez-Barbieri, G.; Guariento, A.; Blitzer, D.; Thedsanamoorthy, J.K.; Yao, R.; Snay, E.R.; Inkster, J.A.H.; et al. Mitochondrial Transplantation Prolongs Cold Ischemia Time in Murine Heart Transplantation. J. Heart Lung Transplant. 2019, 38, 92–99. [Google Scholar] [CrossRef]
- Song, X.; Hu, W.; Yu, H.; Wang, H.; Zhao, Y.; Korngold, R.; Zhao, Y. Existence of Circulating Mitochondria in Human and Animal Peripheral Blood. Int. J. Mol. Sci. 2020, 21, 2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulger, O.; Kubat, G.B.; Cicek, Z.; Celik, E.; Atalay, O.; Suvay, S.; Ozler, M. The Effects of Mitochondrial Transplantation in Acetaminophen-Induced Liver Toxicity in Rats. Life Sci. 2021, 279, 119669. [Google Scholar] [CrossRef] [PubMed]
- Kubat, G.B.; Ozler, M.; Ulger, O.; Ekinci, O.; Atalay, O.; Celik, E.; Safali, M.; Budak, M.T. The Effects of Mesenchymal Stem Cell Mitochondrial Transplantation on Doxorubicin-mediated Nephrotoxicity in Rats. J. Biochem. Mol. Toxicol. 2021, 35, e22612. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, Z.; Jiang, D.; Liang, X.; Liao, S.; Zhang, Z.; Yue, W.; Li, X.; Chiu, S.-M.; Chai, Y.-H.; et al. IPSC-MSCs with High Intrinsic MIRO1 and Sensitivity to TNF-α Yield Efficacious Mitochondrial Transfer to Rescue Anthracycline-Induced Cardiomyopathy. Stem Cell Rep. 2016, 7, 749–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.J. Delivery of Exogenous Mitochondria via Centrifugation Enhances Cellular Metabolic Function. Sci. Rep. 2018, 13, 3330. [Google Scholar] [CrossRef]
- Nakamura, Y.; Park, J.-H.; Hayakawa, K. Therapeutic Use of Extracellular Mitochondria in CNS Injury and Disease. Exp. Neurol. 2020, 324, 113114. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Marin, D.; Sisti, G. New Frontiers in IVF: MtDNA and Autologous Germline Mitochondrial Energy Transfer. Reprod. Biol. Endocrinol. 2019, 17, 55. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial Mutations and Mitoepigenetics: Focus on Regulation of Oxidative Stress-Induced Responses in Breast Cancers. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Park, A.; Oh, M.; Lee, S.J.; Oh, K.-J.; Lee, E.-W.; Lee, S.C.; Bae, K.-H.; Han, B.S.; Kim, W.K. Mitochondrial Transplantation as a Novel Therapeutic Strategy for Mitochondrial Diseases. Int. J. Mol. Sci. 2021, 22, 4793. [Google Scholar] [CrossRef]
- Hersant, B.; Sid-Ahmed, M.; Braud, L.; Jourdan, M.; Baba-Amer, Y.; Meningaud, J.-P.; Rodriguez, A.-M. Platelet-Rich Plasma Improves the Wound Healing Potential of Mesenchymal Stem Cells through Paracrine and Metabolism Alterations. Stem Cells Int. 2019, 2019, 1234263. [Google Scholar] [CrossRef]
- Wu, H.-C.; Fan, X.; Hu, C.-H.; Chao, Y.-C.; Liu, C.-S.; Chang, J.-C.; Sen, Y. Comparison of Mitochondrial Transplantation by Using a Stamp-Type Multineedle Injector and Platelet-Rich Plasma Therapy for Hair Aging in Naturally Aging Mice. Biomed. Pharmacother. 2020, 130, 110520. [Google Scholar] [CrossRef]
- Weissig, V.; Boddapati, S.V.; Cheng, S.-M.; D’souza, G.G.M. Liposomes and Liposome-like Vesicles for Drug and DNA Delivery to Mitochondria. J. Liposome Res. 2006, 16, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.-C.; Hoel, F.; Liu, K.-H.; Wei, Y.-H.; Cheng, F.-C.; Kuo, S.-J.; Tronstad, K.J.; Liu, C.-S. Peptide-Mediated Delivery of Donor Mitochondria Improves Mitochondrial Function and Cell Viability in Human Cybrid Cells with the MELAS A3243G Mutation. Sci. Rep. 2017, 7, 10710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH Oxidase-2 Derived Superoxide Drives Mitochondrial Transfer from Bone Marrow Stromal Cells to Leukemic Blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Sahinbegovic, H.; Jelinek, T.; Hrdinka, M.; Bago, J.R.; Turi, M.; Sevcikova, T.; Kurtovic-Kozaric, A.; Hajek, R.; Simicek, M. Intercellular Mitochondrial Transfer in the Tumor Microenvironment. Cancers 2020, 12, 1787. [Google Scholar] [CrossRef]
- Dong, L.-F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal Transfer of Whole Mitochondria Restores Tumorigenic Potential in Mitochondrial DNA-Deficient Cancer Cells. eLife 2017, 6, e22187. [Google Scholar] [CrossRef] [Green Version]
- Elliott, R.L.; Jiang, X.P.; Head, J.F. Mitochondria Organelle Transplantation: Introduction of Normal Epithelial Mitochondria into Human Cancer Cells Inhibits Proliferation and Increases Drug Sensitivity. Breast Cancer Res. Treat. 2012, 136, 347–354. [Google Scholar] [CrossRef]
- Caicedo, A.; Fritz, V.; Brondello, J.-M.; Ayala, M.; Dennemont, I.; Abdellaoui, N.; de Fraipont, F.; Moisan, A.; Prouteau, C.A.; Boukhaddaoui, H.; et al. MitoCeption as a New Tool to Assess the Effects of Mesenchymal Stem/Stromal Cell Mitochondria on Cancer Cell Metabolism and Function. Sci. Rep. 2015, 5, 9073. [Google Scholar] [CrossRef] [Green Version]
- Lohlamoh, W.; Soontornworajit, B.; Rotkrua, P. Anti-Proliferative Effect of Doxorubicin-Loaded AS1411 Aptamer on Colorectal Cancer Cell. Asian Pac. J. Cancer Prev. 2021, 22, 2209–2219. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Cisplatin Dose/Time of Administration | Heart Alterations | Other Alterations | Reference |
|---|---|---|---|---|
| Female 8-week-old Wistar albino rats 190–250 g | Single doses of 5 mg/kg/week, (i.p), two times as once a week 19 days of study | ↑ MDA ↓ SOD, CAT ↑ Proinflammatory cytokines: IL-1β, IL-6, and TNF-α in the cardiac tissue. Muscle fiber damage, cytoplasmic vacuolization, hemorrhage, and interstitial edema. | None | Bahadir et al. [42] |
| Male Wistar rats 200–250 g | 4 mg/kg (i.p), 4 repetitions: day 1, 5, 9, and 13 (Total = 16 mg/kg) 14 days of study | ↑ CK-MB, cTnI Disarrayed cardiac muscle fibers arrangement, severe interstitial edema, vacuolar degeneration of muscle cells, and congestion of blood vessels. ↑ LVSP ↓ LVEDP ↑ Cardiac MDA ↓ SOD, CAT, GHS, and GPx ↑ Apoptosis ↑ Caspase-3 ↑ PARP | Weight loss Hypotension ↑ Nrf-2 ↑ keap1, NQO1, and HO-1 | Y. Cui et al. [41] |
| Male albino rats 180–220 g | Repeated doses of 2 mg/mg/kg/day (i.p) for 1 week 12 days of study | Disturbed architecture of cardiac muscle and separated muscle fibers and extravasation of RBCs. Degenerated and disrupted cardiac muscle fibers. Deposition of collagenous fibers and cardiac muscle fibers with irregular shrunken nuclei, swollen and disrupted mitochondria, and dilated sarcoplasmic reticulum. ↑ CK-MB, LDH | ↑p53 in the cardiomyocytes nuclei. ↑ TNF-α | El-Hawwary & Omar. [34] |
| Male 5-week-old Balb/c mice 20–30 g | Single-dose of 7 mg/kg, (i.p) 31 days of study | ↑ Troponin 1, CPK, CK-MB ↑ MDA ↓ GPx, SOD, catalase ↑ NO Disruption of cardiac muscle fibers, loss of striations, pyknotic nuclei, interrupted cardiac muscle fibers of the myocardium. | ↓ Body weight ↓ Heart weight ↓ Heart index | M.A Ibrahim et al. [40] |
| Male Wistar rats 180–200 g | Single-dose of 10 mg/kg, (i.p) 5 days of study | ECG changes (elongation of QTc duration and increased ST and T wave amplitude). ↑ Heart rate ↑ Troponin I, LDH, CK-MB ↓ GSH, SOD ↑ Caspase-12, Bax ↓ Bcl-2 ↑ Bax/Bcl-2 ratio in heart | ↑ GRP78 ↑ Calpain-1 ↑ aCASP3 | Saleh et al. [43] |
| Model | Cisplatin Dose/Time Administration | Kidney Alterations | Other Alterations | Reference |
|---|---|---|---|---|
| Male Wistar rats (200–250 g) | 5 mg/kg (i.p) 3 days | ↑ SCr and BUN Swelling of the epithelial cells from CPTs. Extensive cytoplasmic vacuolization detached necrotic cells and cellular debris in the tubular lumen. ↓ RCI, ADP/O ratio, ATP synthase activity, mtMP, OPA-1, and Sirt-3. | Mitochondria alterations: ↑ FIS-1, swelling, rupture of cristae, and autophagic bodies in CPTs. Decreased state III and I of the ETS as well as for RCI and ADP/O ratio. State 4 was unchanged. | Ortega-Domínguez et al. [56] |
| Male 10-week-old C57BL/6J mice | 10 mg/kg (i.p) 7 days | ↑ BUN and tubular necrosis ↑ mRNA expression of injury markers: NGAL and KIM-1. Increased DHE oxidation. | ↓ Body weight | Mapuskar et al. [45] |
| Male 8-weeks old C57BL/6N mice | 16 mg/kg (i.p) 4 days | ↑ BUN, SCr, and apoptosis. Histopathological alterations: loss of the brush border, tubular cell loss, and cast formation. | ↑ Autophagy | Zhu et al. [78] |
| BALB/c mice (20–25 g) | 20 mg/kg (i.p) 3 days | ↑ BUN, SCr, MDA, and apoptosis. ↓ GSH, SOD, CAT. Kidney lesions, whitening, and tubular injury. | ↑ mortality, Nrf-2, HO-1, and NQO-1. ↑ TNF-α and IL-1β in the renal tissue. | Zhang et al. [73] |
| Male 8–12 weeks old C57BL/6 mice 22–24 g | 20 mg/kg (i.p) 3 days | ↑ BUN, SCr, KIM-1 mRNA, and proinflammatory cytokines: IL-6, IL-1β, TNF-α, and MCP-1. Tubular dilation, protein casts, and loss of proximal brush border. ↑ kidney size Pale appearance | - | Yang et al. [85] |
| Male 8-week-old C57BL/6 mice | 30 mg/kg (i.p) 2 days | ↑ SCr, BUN, and apoptosis. Severe damage with cast formation, vacuolization, and dilation in renal tubules. | RPTCs treated with 20 µM of cisplatin for 16 h produced apoptotic cells with nuclear condensation and fragmentation. | Wu et al. [8] |
| Male 10-week-old C57BL/6 mice | 20 mg/kg (i.p) 8 days | ↑ BUN, SCr, and KIM-1. ↓ CCR, urinary NAG, and albumin excretion. Acute renal tubule damage with loss of the brush border and extensive necrosis of the proximal tubules. | ↑ cleaved caspase-3, Bax, and p53. ↓ levels of Bcl-2. | Luan et al. [90] |
| Mitochondrial Source | Delivery Method | Injury/Targeted Organs | Therapeutic Outcome | Mitochondrial Concentration Range | Reference |
|---|---|---|---|---|---|
| BM-MSCs | Directly injected under the renal capsule of the left kidney | Diabetic nephropathy induced with streptozotocin (55 mg/kg) in male Sprague-Dawley rats | Improved cellular morphology of PTECs and the structure of the tubular basement membrane and cells with brush border. | Mitochondria obtained from 1 × 106 BM-MSCs | Konari et al. [89] |
| MSCs | Transplanted directly into the cortex of right and left kidneys | Nephrotoxicity induced with 6 mg/kg of doxorubicin in Male Sprague-Dawley rats | cellular OS Regeneration of tubular cells Reverted renal deficits, Bcl-2 caspase-3 levels | Mitochondria obtained from 2 × 107 cells/mL | Kubat et al. [135] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amador-Martínez, I.; Hernández-Cruz, E.Y.; Jiménez-Uribe, A.P.; Sánchez-Lozada, L.G.; Aparicio-Trejo, O.E.; Tapia, E.; Barrera-Chimal, J.; Pedraza-Chaverri, J. Mitochondrial Transplantation: Is It a Feasible Therapy to Prevent the Cardiorenal Side Effects of Cisplatin? Future Pharmacol. 2021, 1, 3-26. https://doi.org/10.3390/futurepharmacol1010002
Amador-Martínez I, Hernández-Cruz EY, Jiménez-Uribe AP, Sánchez-Lozada LG, Aparicio-Trejo OE, Tapia E, Barrera-Chimal J, Pedraza-Chaverri J. Mitochondrial Transplantation: Is It a Feasible Therapy to Prevent the Cardiorenal Side Effects of Cisplatin? Future Pharmacology. 2021; 1(1):3-26. https://doi.org/10.3390/futurepharmacol1010002
Chicago/Turabian StyleAmador-Martínez, Isabel, Estefani Yaquelin Hernández-Cruz, Alexis Paulina Jiménez-Uribe, Laura Gabriela Sánchez-Lozada, Omar Emiliano Aparicio-Trejo, Edilia Tapia, Jonatan Barrera-Chimal, and José Pedraza-Chaverri. 2021. "Mitochondrial Transplantation: Is It a Feasible Therapy to Prevent the Cardiorenal Side Effects of Cisplatin?" Future Pharmacology 1, no. 1: 3-26. https://doi.org/10.3390/futurepharmacol1010002