
RNA-Sequencing Muscle Plasticity to Resistance Exercise Training and Disuse in Youth and Older Age
 ,
,
Abstract
:1. Introduction
2. Transcriptional Responses to RET

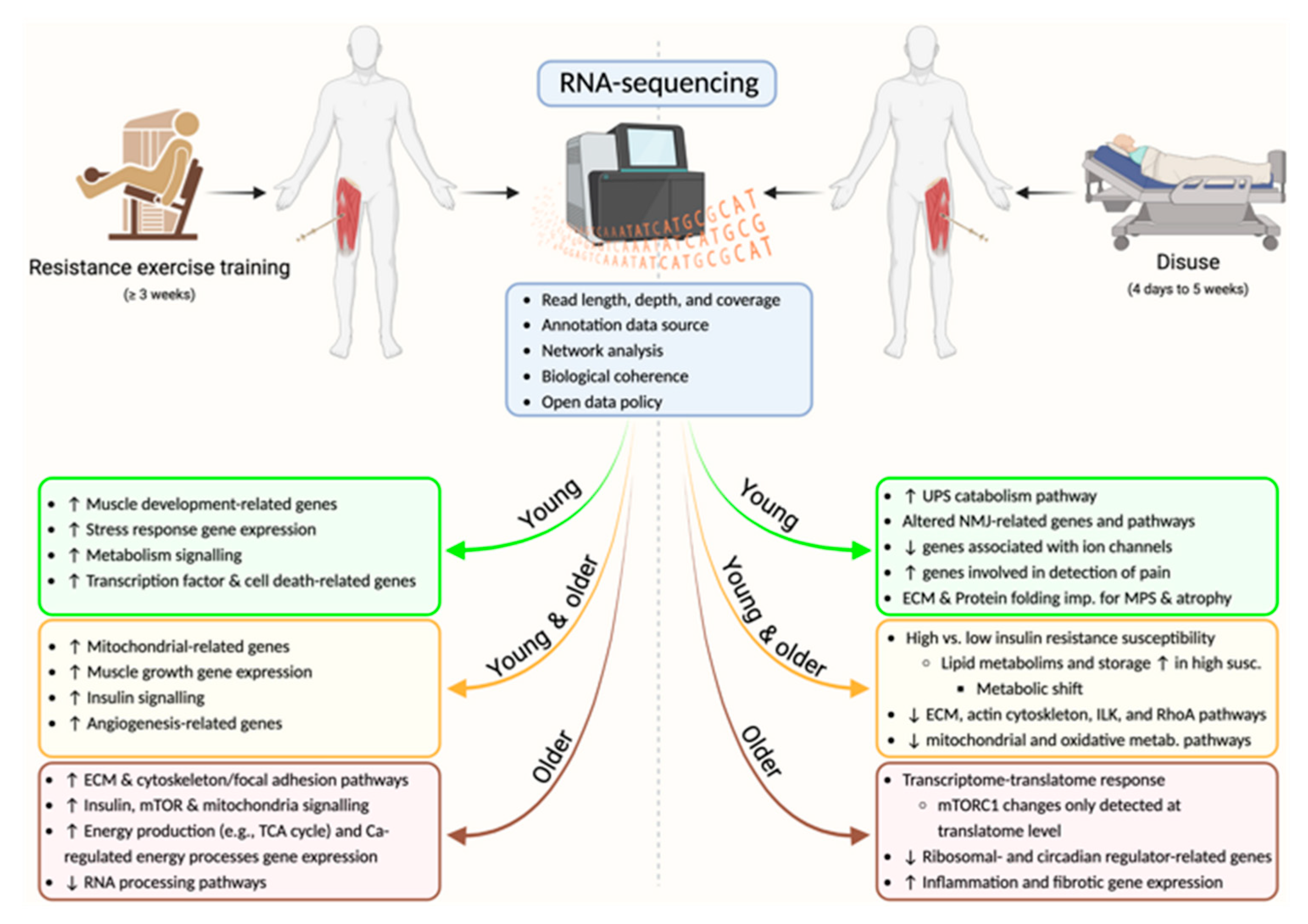{kind=link}
| Reference | Population | Study Design | Transcriptomic Responses | Phenotypic Adaptations |
|---|---|---|---|---|
| RET | ||||
| Robinson et al. [11] | Young adults (n = 11, 5 M/6 F, 23.7 ± 3.5 yr). Older adults (70.3 ± 3.9 yr, n = 9, 5 M/4 F). | 12 weeks RET. (another group: 12 weeks high intensity interval training) (another group: 12 weeks combined aerobic and resistance exercise). | Regulation of mitochondrial, muscle growth and insulin-related genes (albeit to a lesser extent than high intensity interval training). Upregulation of genes pertaining to angiogenesis and regulation of angiogenesis. | Significant improvements in fat free mass, muscle strength and insulin sensitivity in both age groups. No change in mitochondrial respiration in either age group. |
| Lim et al. [33] | Young males (n = 21, 23.7 ± 2.5 yr). | 10 weeks RET. | Up-regulation in genes related to muscle development, stress response, metabolism, tran-scription factor and cell death (albiet to a lesser extent than acute RE). | Increase in muscle strength and fibre cross-sectional area [34]. |
| Chapman et al. [35] | RET-trained males (n = 7, 42.1 ± 5.8 yr). Age-matched untrained females (n = 8) and males (n = 7). | +15 yr RET experience | Upregulation in genes related to cellular respiration pathways compared to untrained controls. Downregulation in pathways associated with the negative regulation of cell proliferation compared untrained controls. | Greater muscle fibre cross sectional area in RET trained adults (vs. untrained controls). |
| Kulkarni et al. [36] | Older males and females with placebo (n = 48, ≥65 yr). Older males and females with metformin (n = 46, ≥65 yr). | 14 weeks RET. | Increased expression of genes related to extracellular matrix remodelling pathways (compared to baseline). Downregulation in genes related to RNA processing pathways (compared to baseline). | Increased lean body mass, thigh muscle mass and muscle strength [37]. |
| Lavin et al. [38] | Older adults (n = 31, 18 F/13 M, 70 ± 4 yr) | 14 weeks RET. | Two modules of genes significantly and positively related to the change in mid-thigh muscle cross-sectional area, of which the hubs were related to immune and inflammatory processes, specifically: defence response to virus, regulation of leukocyte activation, positive regulation of defence response, positive regulation of cytokine production, and negative regulation of immune system processes (part of “prediction analysis”). | Decreased percentage body fat, increased mid-thigh muscle cross-sectional area and thigh muscle mass. |
| Bolotta et al. [39] | Life-long exercise trained older adults (n = 9, 65–80 yr). Sedentary older adults (n = 5, 70–76 yr). | Life-long RET (n = 4) or aerobically exercise trained (n = 5). | Upregulation in genes related to insulin signalling, energy production (e.g., TCA cycle), mTOR signalling, mitochondria, calcium-regulated energy processes and the cytoskeleton/focal adhesions (compared to sedentary controls). | Fast type fibres were larger in RET versus aerobically trained adults. |
| Disuse | ||||
| Willis et al. [40] | Healthy young (22 yr) males (n = 8). | Four-day unilateral lower limb immobilisation. | Downregulation of mitochondrial and myogenesis. Upregulation of ribosome biogenesis, UPS catabolism, and ribonucleoprotein complex organization/mRNA processing. | Decreased muscle mass (−1.7%) and MPS (−16.2%), with high inter-individual variability. Associations between gene networks phenotypic changes. |
| Sarto et al. [41] | Active young (22 yr) male adults (n = 12). Focus on NMJ. | Ten-day unilateral lower limb suspension (ULLS) followed by a 21-day readaptation program. | Upregulation of ACh receptor subunits genes. Downregulation of Homer proteins genes. Changes in expression of other genes (e.g., neuregulings, neurotrophins, ErbBs, Wnts) indicative of NMJ molecular instability. Downregulation of ion channels gene set. Most ULLS-induced transcrioptional changes were restored after the readapatation program. | Deacreased muscle volume (−4.5%). NMJ transmission stability was unchanged after ULLS. Increased motor unit potential complexity and decreased motor unit firing rates after ULLS. Most ULLS-induced phenotypic changes were restored after the readapatation program. |
| McFarland et al. [42] | Healthy men (n = 22) and women (n = 3) (20–54 yrs) randomized in two groups. | Five-week −6° head down tilt bed rest. Two parallel groups: bed rest only (n = 9) and bed rest with exercise (n = 16). | Downregulation of virtually all aspects of mitochondrial activity. Upregulation of ligands with a key role in pain. The exercise countermeasure normalized most of the genes related to mitochondrial activity. | Reported in other manuscript [43]. Decreased muscle volume (quadriceps; −9%). Exercise during bed rest reduced the muscle atrophy (quadriceps; −5%) |
| Mahmassani et al. [44] | Healthy older and younger male (n = 13) and female (n = 13) adults (mean age; ~52 yrs). | Five-day bed rest. Participants were categorized into high or low susceptibility for insuling resistance after bed rest. | Gene ontologies (GO) that changed in both high and low susceptibility groups: muscle contraction, muscle filament sliding, mitonchondrial ATP synthesis. GOs altered only in high-susceptibility group: lipid metabolic processes, lipid storage, protein homotetramerization. | Both high and low susceptibility groups become insulin-resistant after bed rest but the “High” group had 49% lower insulin sensitivity after bed rest, versus 15% in the “Low” group. |
| Mahmassani et al. [45] | Healthy young (23 yr; n = 9) and older (68 yr, n = 18) participants (13 men and 14 women). | Five-day bed rest. | Common pathways altered in both young and older: Acting cytoskeleton signaling, ILK and RhoA signaling, Mitochondrial dysfunction and calcium signaling. Increased inflammation and fibrotic gene expression in older group only. 51 genes changed in young but not older; after bed rest, the expression of these genes in young nearly matched that in older participants. | Leg lean mass decreased 3.4% in the older group, but did not change in the young group (similar results for total lean mass and myofiber CSA). Leg strength decreased after bed rest in both groups. |
| Standley et al. [46] | Healthy older (between 60–79 yrs) men (n = 11) and women (n = 10). | Ten-day bed rest. Two groups; bed rest only (n = 9), and bed rest with nutritional supplementation (n = 12). | Downregulation of genes related to mitochondria, ribosomes, and oxidative metabolism. Upregulation of genes involved in extracellular matrix, focal adhesion, and collagen. The nutritional supplementation offset some of these changes. | CSA of type IIa fibers decreased in the bed rest group. |
| Mahmassani et al. [47] | Healthy older adults (~72 yrs; n = 8, 6 females and 2 males). | Two-week reduced activity period (from 11,000 steps/day to 2200 steps/day). RNA-seq of muscle and ribosomal profiling. | Altered response for several transcripts (e.g., PFKFB3, GADD45A, NMRK2) in response to leucine stimulation. Uncoupled translation for mTORC1 pathway. Reduction in genes related with ribosomal proteins and alteration of circadian regulators | Unchanged leg lean mass. Tendency for reduced Type I fiber size. Glucose tolerance and insulin sensitivity did not change. |
3. Transcriptional Responses to Disuse
4. Analytical Considerations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ivy, J.L.; Katz, A.L.; Cutler, C.L.; Sherman, W.M.; Coyle, E.F. Muscle glycogen synthesis after exercise: Effect of time of carbohydrate ingestion. J. Appl. Physiol. 1988, 64, 1480–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Burguera, B.; Jensen, M.D. Kinetics of intramuscular triglyceride fatty acids in exercising humans. J. Appl. Physiol. 2000, 89, 2057–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, R.R. The underappreciated role of muscle in health and disease. Am. J. Clin. Nutr. 2006, 84, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Wall, B.T.; Dirks, M.L.; Snijders, T.; Senden, J.M.; Dolmans, J.; van Loon, L.J. Substantial skeletal muscle loss occurs during only 5 days of disuse. Acta Physiol. 2014, 210, 600–611. [Google Scholar] [CrossRef]
- Alkner, B.A.; Tesch, P.A. Knee extensor and plantar flexor muscle size and function following 90 days of bed rest with or without resistance exercise. Eur. J. Appl. Physiol. 2004, 93, 294–305. [Google Scholar] [CrossRef]
- Berg, H.E.; Larsson, L.; Tesch, P.A. Lower limb skeletal muscle function after 6 wk of bed rest. J. Appl. Physiol. 1997, 82, 182–188. [Google Scholar] [CrossRef]
- Haus, J.M.; Carrithers, J.A.; Carroll, C.C.; Tesch, P.A.; Trappe, T.A. Contractile and connective tissue protein content of human skeletal muscle: Effects of 35 and 90 days of simulated microgravity and exercise countermeasures. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2007, 293, R1722–R1727. [Google Scholar] [CrossRef] [Green Version]
- Brook, M.S.; Wilkinson, D.J.; Mitchell, W.K.; Lund, J.N.; Szewczyk, N.J.; Greenhaff, P.L.; Smith, K.; Atherton, P.J. Skeletal muscle hypertrophy adaptations predominate in the early stages of resistance exercise training, matching deuterium oxide-derived measures of muscle protein synthesis and mechanistic target of rapamycin complex 1 signaling. FASEB J. 2015, 29, 4485–4496. [Google Scholar] [CrossRef] [Green Version]
- Phillips, B.E.; Williams, J.P.; Greenhaff, P.L.; Smith, K.; Atherton, P.J. Physiological adaptations to resistance exercise as a function of age. JCI Insight 2017, 2, e95581. [Google Scholar] [CrossRef] [Green Version]
- Brook, M.S.; Wilkinson, D.J.; Mitchell, W.K.; Lund, J.N.; Phillips, B.E.; Szewczyk, N.J.; Greenhaff, P.L.; Smith, K.; Atherton, P.J. Synchronous deficits in cumulative muscle protein synthesis and ribosomal biogenesis underlie age-related anabolic resistance to exercise in humans. J. Physiol. 2016, 594, 7399–7417. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.M.; Dasari, S.; Konopka, A.R.; Johnson, M.L.; Manjunatha, S.; Esponda, R.R.; Carter, R.E.; Lanza, I.R.; Nair, K.S. Enhanced Protein Translation Underlies Improved Metabolic and Physical Adaptations to Different Exercise Training Modes in Young and Old Humans. Cell Metab. 2017, 25, 581–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, C.; Reidy, P.T.; Bhattarai, N.; Sidossis, L.S.; Rasmussen, B.B. Resistance Exercise Training Alters Mitochondrial Function in Human Skeletal Muscle. Med. Sci. Sports Exerc. 2015, 47, 1922–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hvid, L.; Suetta, C.; Nielsen, J.; Jensen, M.; Frandsen, U.; Ørtenblad, N.; Kjaer, M.; Aagaard, P. Aging impairs the recovery in mechanical muscle function following 4days of disuse. Exp. Gerontol. 2014, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Timmons, J.A. Variability in training-induced skeletal muscle adaptation. J. Appl. Physiol. 2011, 110, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Gonzalo, R.; Tesch, P.A.; Lundberg, T.R.; Alkner, B.A.; Rullman, E.; Gustafsson, T. Three months of bed rest induce a residual transcriptomic signature resilient to resistance exercise countermeasures. FASEB J. 2020, 34, 7958–7969. [Google Scholar] [CrossRef] [Green Version]
- Rullman, E.; Fernandez-Gonzalo, R.; Mekjavić, I.B.; Gustafsson, T.; Eiken, O. MEF2 as upstream regulator of the transcriptome signature in human skeletal muscle during unloading. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2018, 315, R799–R809. [Google Scholar] [CrossRef] [Green Version]
- Deane, C.S.; Willis, C.R.G.; Phillips, B.E.; Atherton, P.J.; Harries, L.W.; Ames, R.M.; Szewczyk, N.J.; Etheridge, T. Transcriptomic meta-analysis of disuse muscle atrophy vs. resistance exercise-induced hypertrophy in young and older humans. J. Cachexia Sarcopenia Muscle 2021, 12, 629–645. [Google Scholar] [CrossRef]
- Mahoney, D.J.; Tarnopolsky, M.A. Understanding skeletal muscle adaptation to exercise training in humans: Contributions from microarray studies. Phys. Med. Rehabil. Clin. N. Am. 2005, 16, 859–873. [Google Scholar] [CrossRef]
- van Dam, S.; Vosa, U.; van der Graaf, A.; Franke, L.; de Magalhaes, J.P. Gene co-expression analysis for functional classification and gene-disease predictions. Brief. Bioinform. 2018, 19, 575–592. [Google Scholar] [CrossRef] [PubMed]
- Willis, C.R.G.; Ames, R.M.; Deane, C.S.; Phillips, B.E.; Boereboom, C.L.; Abdulla, H.; Bukhari, S.S.I.; Lund, J.N.; Williams, J.P.; Wilkinson, D.J.; et al. Network analysis of human muscle adaptation to aging and contraction. Aging 2020, 12, 740–755. [Google Scholar] [CrossRef] [PubMed]
- Song, W.M.; Zhang, B. Multiscale Embedded Gene Co-expression Network Analysis. PLoS Comput. Biol. 2015, 11, e1004574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokes, T.; Timmons, J.A.; Crossland, H.; Tripp, T.R.; Murphy, K.; McGlory, C.; Mitchell, C.J.; Oikawa, S.Y.; Morton, R.W.; Phillips, B.E.; et al. Molecular Transducers of Human Skeletal Muscle Remodeling under Different Loading States. Cell Rep. 2020, 32, 107980. [Google Scholar] [CrossRef]
- Giorgi, F.M.; Del Fabbro, C.; Licausi, F. Comparative study of RNA-seq- and microarray-derived coexpression networks in Arabidopsis thaliana. Bioinformatics 2013, 29, 717–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, M.S.; Van Vleet, T.R.; Ciurlionis, R.; Buck, W.R.; Mittelstadt, S.W.; Blomme, E.A.G.; Liguori, M.J. Comparison of RNA-Seq and Microarray Gene Expression Platforms for the Toxicogenomic Evaluation of Liver From Short-Term Rat Toxicity Studies. Front. Genet 2018, 9, 636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, B.T.; Landry, J.R. RNA-Seq-quantitative measurement of expression through massively parallel RNA-sequencing. Methods 2009, 48, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yu, Y.; Hertwig, F.; Thierry-Mieg, J.; Zhang, W.; Thierry-Mieg, D.; Wang, J.; Furlanello, C.; Devanarayan, V.; Cheng, J.; et al. Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol. 2015, 16, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, M.F.; Tycksen, E.D.; Sandell, L.J.; Brophy, R.H. Advantages of RNA-seq compared to RNA microarrays for transcriptome profiling of anterior cruciate ligament tears. J. Orthop. Res. 2018, 36, 484–497. [Google Scholar] [CrossRef] [Green Version]
- Tumasian, R.A., 3rd; Harish, A.; Kundu, G.; Yang, J.H.; Ubaida-Mohien, C.; Gonzalez-Freire, M.; Kaileh, M.; Zukley, L.M.; Chia, C.W.; Lyashkov, A.; et al. Skeletal muscle transcriptome in healthy aging. Nat. Commun. 2021, 12, 2014. [Google Scholar] [CrossRef]
- Lindholm, M.E.; Huss, M.; Solnestam, B.W.; Kjellqvist, S.; Lundeberg, J.; Sundberg, C.J. The human skeletal muscle transcriptome: Sex differences, alternative splicing, and tissue homogeneity assessed with RNA sequencing. FASEB J. 2014, 28, 4571–4581. [Google Scholar] [CrossRef]
- Deane, C.S.; Ames, R.M.; Phillips, B.E.; Weedon, M.N.; Willis, C.R.G.; Boereboom, C.; Abdulla, H.; Bukhari, S.S.I.; Lund, J.N.; Williams, J.P.; et al. The acute transcriptional response to resistance exercise: Impact of age and contraction mode. Aging 2019, 11, 2111–2126. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, J.M.; D’Lugos, A.C.; Naymik, M.A.; Siniard, A.L.; Wolfe, A.J.; Curtis, D.R.; Huentelman, M.J.; Carroll, C.C. Transcriptome response of human skeletal muscle to divergent exercise stimuli. J. Appl. Physiol. 2018, 124, 1529–1540. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.; Shimizu, J.; Kawano, F.; Kim, H.J.; Kim, C.K. Adaptive responses of histone modifications to resistance exercise in human skeletal muscle. PLoS ONE 2020, 15, e0231321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.; Kim, H.J.; Morton, R.W.; Harris, R.; Phillips, S.M.; Jeong, T.S.; Kim, C.K. Resistance Exercise-induced Changes in Muscle Phenotype Are Load Dependent. Med. Sci. Sports Exerc. 2019, 51, 2578–2585. [Google Scholar] [CrossRef]
- Chapman, M.A.; Arif, M.; Emanuelsson, E.B.; Reitzner, S.M.; Lindholm, M.E.; Mardinoglu, A.; Sundberg, C.J. Skeletal Muscle Transcriptomic Comparison between Long-Term Trained and Untrained Men and Women. Cell Rep. 2020, 31, 107808. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.S.; Peck, B.D.; Walton, R.G.; Kern, P.A.; Mar, J.C.; Windham, S.T.; Bamman, M.M.; Barzilai, N.; Peterson, C.A. Metformin alters skeletal muscle transcriptome adaptations to resistance training in older adults. Aging 2020, 12, 19852–19866. [Google Scholar] [CrossRef]
- Walton, R.G.; Dungan, C.M.; Long, D.E.; Tuggle, S.C.; Kosmac, K.; Peck, B.D.; Bush, H.M.; Villasante Tezanos, A.G.; McGwin, G.; Windham, S.T.; et al. Metformin blunts muscle hypertrophy in response to progressive resistance exercise training in older adults: A randomized, double-blind, placebo-controlled, multicenter trial: The MASTERS trial. Aging Cell 2019, 18, e13039. [Google Scholar] [CrossRef] [Green Version]
- Lavin, K.M.; Bell, M.B.; McAdam, J.S.; Peck, B.D.; Walton, R.G.; Windham, S.T.; Tuggle, S.C.; Long, D.E.; Kern, P.A.; Peterson, C.A.; et al. Muscle transcriptional networks linked to resistance exercise training hypertrophic response heterogeneity. Physiol. Genom. 2021, 53, 206–221. [Google Scholar] [CrossRef]
- Bolotta, A.; Filardo, G.; Abruzzo, P.M.; Astolfi, A.; De Sanctis, P.; Di Martino, A.; Hofer, C.; Indio, V.; Kern, H.; Lofler, S.; et al. Skeletal Muscle Gene Expression in Long-Term Endurance and Resistance Trained Elderly. Int. J. Mol. Sci. 2020, 21, 3988. [Google Scholar] [CrossRef]
- Willis, C.R.G.; Gallagher, I.J.; Wilkinson, D.J.; Brook, M.S.; Bass, J.J.; Phillips, B.E.; Smith, K.; Etheridge, T.; Stokes, T.; McGlory, C.; et al. Transcriptomic links to muscle mass loss and declines in cumulative muscle protein synthesis during short-term disuse in healthy younger humans. FASEB J. 2021, 35, e21830. [Google Scholar] [CrossRef]
- Sarto, F.; Stashuk, D.W.; Franchi, M.V.; Monti, E.; Zampieri, S.; Valli, G.; Sirago, G.; Candia, J.; Hartnell, L.M.; Paganini, M.; et al. Effects of short-term unloading and active recovery on human motor unit properties, neuromuscular junction transmission and transcriptomic profile. J. Physiol. 2022, 600, 4731–4751. [Google Scholar] [CrossRef] [PubMed]
- McFarland, A.J.; Ray, P.R.; Bhai, S.; Levine, B.D.; Price, T.J. RNA sequencing on muscle biopsy from a 5-week bed rest study reveals the effect of exercise and potential interactions with dorsal root ganglion neurons. Physiol. Rep. 2022, 10, e15176. [Google Scholar] [CrossRef] [PubMed]
- Krainski, F.; Hastings, J.L.; Heinicke, K.; Romain, N.; Pacini, E.L.; Snell, P.G.; Wyrick, P.; Palmer, M.D.; Haller, R.G.; Levine, B.D. The effect of rowing ergometry and resistive exercise on skeletal muscle structure and function during bed rest. J. Appl. Physiol. 2014, 116, 1569–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmassani, Z.S.; Reidy, P.T.; McKenzie, A.I.; Stubben, C.; Howard, M.T.; Drummond, M.J. Disuse-induced insulin resistance susceptibility coincides with a dysregulated skeletal muscle metabolic transcriptome. J. Appl. Physiol. 2019, 126, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Mahmassani, Z.S.; Reidy, P.T.; McKenzie, A.I.; Stubben, C.; Howard, M.T.; Drummond, M.J. Age-dependent skeletal muscle transcriptome response to bed rest-induced atrophy. J. Appl. Physiol. 2019, 126, 894–902. [Google Scholar] [CrossRef]
- Standley, R.A.; Distefano, G.; Trevino, M.B.; Chen, E.; Narain, N.R.; Greenwood, B.; Kondakci, G.; Tolstikov, V.V.; Kiebish, M.A.; Yu, G.; et al. Skeletal Muscle Energetics and Mitochondrial Function Are Impaired Following 10 Days of Bed Rest in Older Adults. J. Gerontol. Ser. A 2020, 75, 1744–1753. [Google Scholar] [CrossRef] [Green Version]
- Mahmassani, Z.S.; McKenzie, A.I.; Petrocelli, J.J.; de Hart, N.M.; Fix, D.K.; Kelly, J.J.; Baird, L.M.; Howard, M.T.; Drummond, M.J. Reduced Physical Activity Alters the Leucine-Stimulated Translatome in Aged Skeletal Muscle. J. Gerontol. Ser. A 2021, 76, 2112–2121. [Google Scholar] [CrossRef]
- Kumar, V.; Selby, A.; Rankin, D.; Patel, R.; Atherton, P.; Hildebrandt, W.; Williams, J.; Smith, K.; Seynnes, O.; Hiscock, N.; et al. Age-related differences in the dose-response relationship of muscle protein synthesis to resistance exercise in young and old men. J. Physiol. 2009, 587, 211–217. [Google Scholar] [CrossRef]
- Raue, U.; Trappe, T.A.; Estrem, S.T.; Qian, H.R.; Helvering, L.M.; Smith, R.C.; Trappe, S. Transcriptome signature of resistance exercise adaptations: Mixed muscle and fiber type specific profiles in young and old adults. J. Appl. Physiol. 2012, 112, 1625–1636. [Google Scholar] [CrossRef] [Green Version]
- Deane, C.S.; Phillips, B.E.; Willis, C.R.G.; Wilkinson, D.J.; Smith, K.; Higashitani, N.; Williams, J.P.; Szewczyk, N.J.; Atherton, P.J.; Higashitani, A.; et al. Proteomic features of skeletal muscle adaptation to resistance exercise training as a function of age. Geroscience 2022. [Google Scholar] [CrossRef]
- Suetta, C.; Frandsen, U.; Jensen, L.; Jensen, M.M.; Jespersen, J.G.; Hvid, L.G.; Bayer, M.; Petersson, S.J.; Schrøder, H.D.; Andersen, J.L.; et al. Aging affects the transcriptional regulation of human skeletal muscle disuse atrophy. PLoS ONE 2012, 7, e51238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjær, M. Role of Extracellular Matrix in Adaptation of Tendon and Skeletal Muscle to Mechanical Loading. Physiol. Rev. 2004, 84, 649–698. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.A.; Carland, C.R.; Guo, Y.; Bernstein, S.I. Getting Folded: Chaperone Proteins in Muscle Development, Maintenance and Disease. Anat. Rec. 2014, 297, 1637–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inns, T.B.; Bass, J.J.; Hardy, E.J.O.; Wilkinson, D.J.; Stashuk, D.W.; Atherton, P.J.; Phillips, B.E.; Piasecki, M. Motor unit dysregulation following 15 days of unilateral lower limb immobilisation. J. Physiol. 2022, 600, 4753–4769. [Google Scholar] [CrossRef]
- Desaphy, J.F.; Pierno, S.; Léoty, C.; George, A.L.; De Luca, A.; Camerino, D.C. Skeletal muscle disuse induces fibre type-dependent enhancement of Na+ channel expression. Brain J. Neurol. 2001, 124, 1100–1113. [Google Scholar] [CrossRef] [Green Version]
- Neubauer, O.; Sabapathy, S.; Ashton, K.J.; Desbrow, B.; Peake, J.M.; Lazarus, R.; Wessner, B.; Cameron-Smith, D.; Wagner, K.-H.; Haseler, L.J.; et al. Time course-dependent changes in the transcriptome of human skeletal muscle during recovery from endurance exercise: From inflammation to adaptive remodeling. J. Appl. Physiol. 2014, 116, 274–287. [Google Scholar] [CrossRef]
- Pillon, N.J.; Smith, J.A.B.; Alm, P.S.; Chibalin, A.V.; Alhusen, J.; Arner, E.; Carninci, P.; Fritz, T.; Otten, J.; Olsson, T.; et al. Distinctive exercise-induced inflammatory response and exerkine induction in skeletal muscle of people with type 2 diabetes. Sci. Adv. 2022, 8, eabo3192. [Google Scholar] [CrossRef]
- Fernandez-Gonzalo, R.; Irimia, J.M.; Cusso, R.; Gustafsson, T.; Linné, A.; Tesch, P.A. Flywheel Resistance Exercise to Maintain Muscle Oxidative Potential During Unloading. Aviat. Space Environ. Med. 2014, 85, 694–699. [Google Scholar] [CrossRef]
- Irimia, J.M.; Guerrero, M.; Rodriguez-Miguelez, P.; Cadefau, J.A.; Tesch, P.A.; Cussó, R.; Fernandez-Gonzalo, R. Metabolic adaptations in skeletal muscle after 84 days of bed rest with and without concurrent flywheel resistance exercise. J. Appl. Physiol. 2017, 122, 96–103. [Google Scholar] [CrossRef] [Green Version]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.S.; Weissman, J.S. Genome-Wide Analysis in Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef]
- Illumina. Considerations for RNA-Seq read length and coverage. Available online: https://emea.support.illumina.com/bulletins/2017/04/considerations-for-rna-seq-read-length-and-coverage-.html (accessed on 4 October 2022).
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szczesniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Zhang, B. A comprehensive evaluation of ensembl, RefSeq, and UCSC annotations in the context of RNA-seq read mapping and gene quantification. BMC Genom. 2015, 16, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldham, M. Transcriptomics: From Differential Expression to Coexpression. In The OMICs: Applications in Neuroscience; Oxford University Press: Oxford, UK, 2014. [Google Scholar]
- Oldham, M.C.; Konopka, G.; Iwamoto, K.; Langfelder, P.; Kato, T.; Horvath, S.; Geschwind, D.H. Functional organization of the transcriptome in human brain. Nat. Neurosci. 2008, 11, 1271–1282. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, S.A.; Gold, E.S.; Aderem, A. A systems biology approach to understanding atherosclerosis. EMBO Mol. Med. 2010, 2, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Joanisse, S.; Lim, C.; McKendry, J.; McLeod, J.C.; Stokes, T.; Phillips, S.M. Recent advances in understanding resistance exercise training-induced skeletal muscle hypertrophy in humans. F1000Research 2020, 9, 141. [Google Scholar] [CrossRef]
- Langfelder, P.; Zhang, B.; Horvath, S. Defining clusters from a hierarchical cluster tree: The Dynamic Tree Cut package for R. Bioinformatics 2008, 24, 719–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA Package FAQ. Available online: https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/faq.html (accessed on 4 October 2022).
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Mubeen, S.; Hoyt, C.T.; Gemund, A.; Hofmann-Apitius, M.; Frohlich, H.; Domingo-Fernandez, D. The Impact of Pathway Database Choice on Statistical Enrichment Analysis and Predictive Modeling. Front. Genet. 2019, 10, 1203. [Google Scholar] [CrossRef]
- Timmons, J.A.; Szkop, K.J.; Gallagher, I.J. Multiple sources of bias confound functional enrichment analysis of global-omics data. Genome Biol. 2015, 16, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijesooriya, K.; Jadaan, S.A.; Perera, K.L.; Kaur, T.; Ziemann, M. Urgent need for consistent standards in functional enrichment analysis. PLoS Comput Biol 2022, 18, e1009935. [Google Scholar] [CrossRef]
- Soneson, C.; Delorenzi, M. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinform. 2013, 14, 91. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillon, N.J.; Gabriel, B.M.; Dollet, L.; Smith, J.A.B.; Sardon Puig, L.; Botella, J.; Bishop, D.J.; Krook, A.; Zierath, J.R. Transcriptomic profiling of skeletal muscle adaptations to exercise and inactivity. Nat. Commun. 2020, 11, 470. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.J.; Hu, P.; Batt, J.; Santos, C.C. Microarray Meta-Analysis and Cross-Platform Normalization: Integrative Genomics for Robust Biomarker Discovery. Microarrays 2015, 4, 389–406. [Google Scholar] [CrossRef] [Green Version]
- Rung, J.; Brazma, A. Reuse of public genome-wide gene expression data. Nat. Rev. Genet 2013, 14, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.C.; Lin, H.M.; Sibille, E.; Tseng, G.C. Meta-analysis methods for combining multiple expression profiles: Comparisons, statistical characterization and an application guideline. BMC Bioinform. 2013, 14, 368. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/A (accessed on 4 October 2022).
- 9Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, 1–6. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Patro, R.; Mount, S.M.; Kingsford, C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat. Biotechnol. 2014, 32, 462–464. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]

| Tool | Highlighted Use | Reference |
|---|---|---|
| FastQC | Quality check of raw sequence reads | [92] |
| Cutadapt | Trimming and/or filtering of raw sequence reads | [93] |
| SOAPnuke | [94] | |
| Trimmomatic | [95] | |
| Histat2 | Splice-aware genome alignment | [96] |
| STAR | [97] | |
| TopHat2 | [98] | |
| featureCounts | Genomic feature counting of aligned reads | [99] |
| htseq-count | [100] | |
| Kallisto | Pseudoalignment and quantification of transcript abundance | [101] |
| Sailfish | [102] | |
| Salmon | [103] | |
| tximport | Infer gene counts from transcript-level estimates | [104] |
| DESeq2 | Differential gene expression analysis | [105] |
| EdgeR | [106] | |
| Limma | [107] | |
| IPA | Facilitates (among other things) knowledge-based network inference | [71] |
| NetworkAnalyst | [73] | |
| STRING | [72] | |
| WGCNA | Facilitates data-driven network analysis | [108] |
| clusterProfiler | Overrepresentation analysis and/or gene set enrichment analysis | [81] |
| DAVID | [82] | |
| Enrichr | [85] | |
| g:Profiler | [86] | |
| Broad Institute GSEA software | [84] | |
| WebGestalt | [83] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez-Gonzalo, R.; Willis, C.R.G.; Etheridge, T.; Deane, C.S. RNA-Sequencing Muscle Plasticity to Resistance Exercise Training and Disuse in Youth and Older Age. Physiologia 2022, 2, 164-179. https://doi.org/10.3390/physiologia2040014
Fernandez-Gonzalo R, Willis CRG, Etheridge T, Deane CS. RNA-Sequencing Muscle Plasticity to Resistance Exercise Training and Disuse in Youth and Older Age. Physiologia. 2022; 2(4):164-179. https://doi.org/10.3390/physiologia2040014
Chicago/Turabian StyleFernandez-Gonzalo, Rodrigo, Craig R. G. Willis, Timothy Etheridge, and Colleen S. Deane. 2022. "RNA-Sequencing Muscle Plasticity to Resistance Exercise Training and Disuse in Youth and Older Age" Physiologia 2, no. 4: 164-179. https://doi.org/10.3390/physiologia2040014