1. Introduction
Beef cattle are the primary agricultural commodity in Tennessee and account for a multi-billion dollar industry worldwide [
1]. The greatest input costs for the beef industry are feed costs, accounting for 40–70% of the total cost of production [
2,
3]. Given this, finding methods to reduce input costs through improved feed efficiencies stands to provide millions of dollars in savings to industry stakeholders. This is particularly important in heifers and cows, given that they are the members primarily responsible for the propagation of genotypes and phenotypes. In order to select more efficient animals, methods must be identified to detect efficient animals. Current methods for evaluating feed efficiency are often expensive, time-intensive, or require contemporary groups of similar animals. Biomarkers that can be readily measured in the blood or other easily-accessible body fluid may provide a more effective method for producers to evaluate feed efficiency phenotypes. A biomarker may be defined as “any substance, structure, or process that can be measured in the body or its products and influence or predict the incidence of outcome or disease” [
4]. In agriculture, biomarkers may be developed for producers to make management decisions [
5]. In order to accomplish this, biomarkers must be first identified and then tests developed that are economically feasible to producers [
5]. Several methods may be used in order to do this, including microbiomics and metabolomics.
One of the primary contributors to variation in feed efficiency is the rumen microbiome. The rumen microbiota provide the majority of energy precursors required by the host animal as well as many of the required nutrients, such as all water-soluble vitamins [
6,
7]. Several studies have been conducted to examine the relationship between the rumen microbiome and feed efficiency in beef cattle. Myer and colleagues found several bacterial populations that were associated with variation in feed efficiency in beef steers, including Prevotella, the predominant genus within the rumen [
8]. Other studies have supported these data in other beef cattle production classes, including in bulls [
9,
10] and steers [
11], as well as in dairy cattle and other ruminants [
12,
13,
14,
15]. However, few, if any, studies have examined the rumen microbiome and feed efficiency in female beef cattle. Given that these cattle contribute to the propagation of genetics in the herd, understanding feed efficiency in female beef cattle is imperative for improving feed efficiency in the herd.
Although the rumen microbiome is strongly associated with feed efficiency in ruminants, the rumen microbiome is not readily available to producers and those without access to research equipment. Therefore, identifying readily accessible and inexpensive methods for examining the rumen microbiome as well as host physiology that contribute to variations in feed efficiency phenotypes are necessary. Measuring how serum metabolites are associated with the ruminal microbiome and metabolites could serve as a method of identifying more feed-efficient cattle [
5]. Further understanding of the relationships among the rumen microbiome and metabolome, blood metabolome, and cattle feed efficiency is expected to lead to improved selection criteria and overall herd feed efficiency. To date, most studies conducted to understand variation in feed efficient phenotypes have thus far been conducted in beef steers and bulls. Therefore, this study was conducted to establish the relationship among the rumen fluid and serum metabolomes, rumen microbiome, and feed efficiency in female cattle. The hypothesis was that there would be differences in the rumen microbiome and metabolomes of heifers that differed in feed efficiency, leading to the objective to determine potential biomarkers of feed efficiency in heifers.
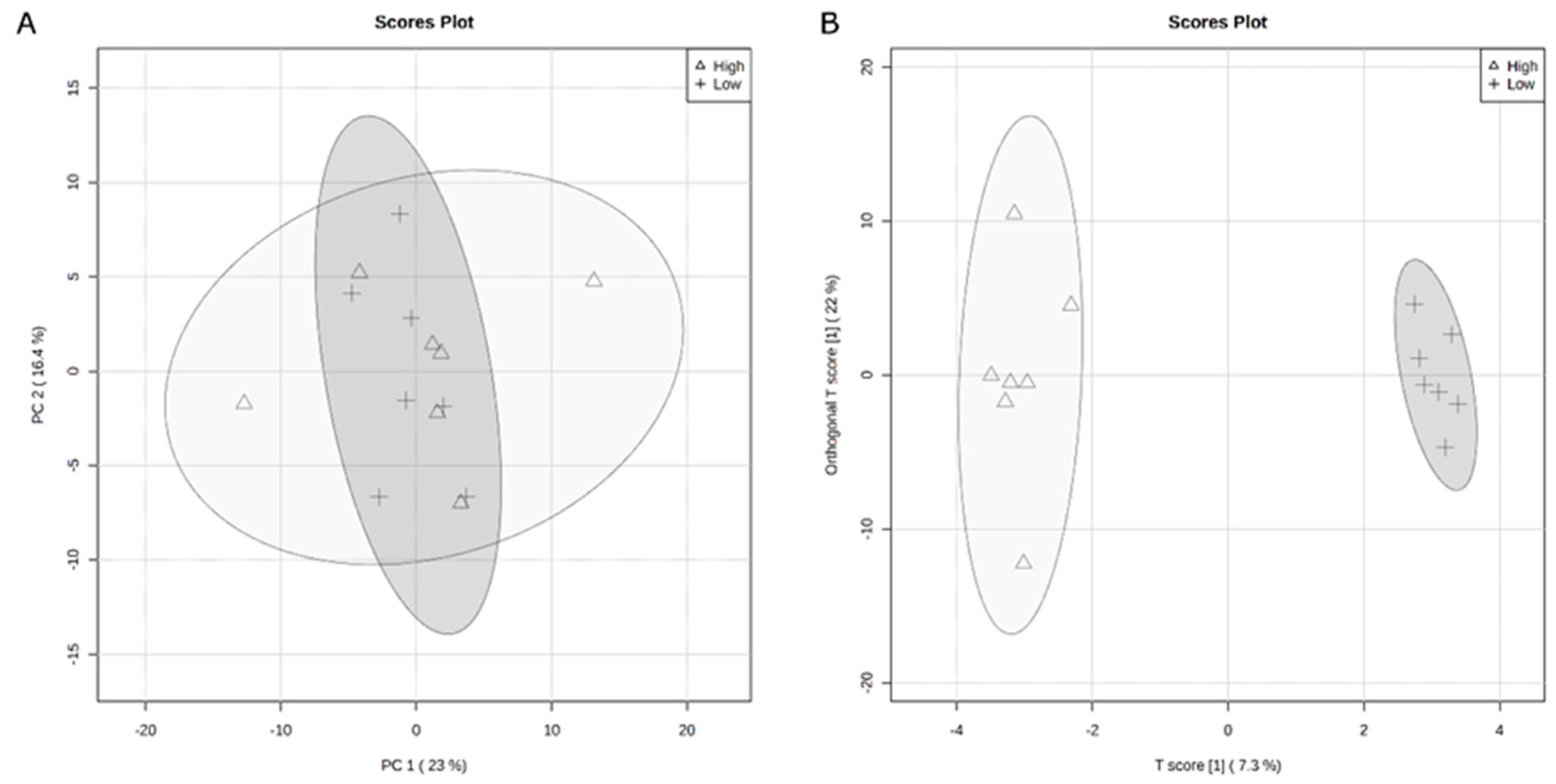
4. Discussion
Globally, beef consumption is rising, as beef provides an excellent source of animal protein [
33]. The United States is one of the largest exporters of beef, exporting more than one million tons of beef annually [
33,
34]. In the United States beef industry, feed costs account for upwards of 70% of the total input costs of production [
3,
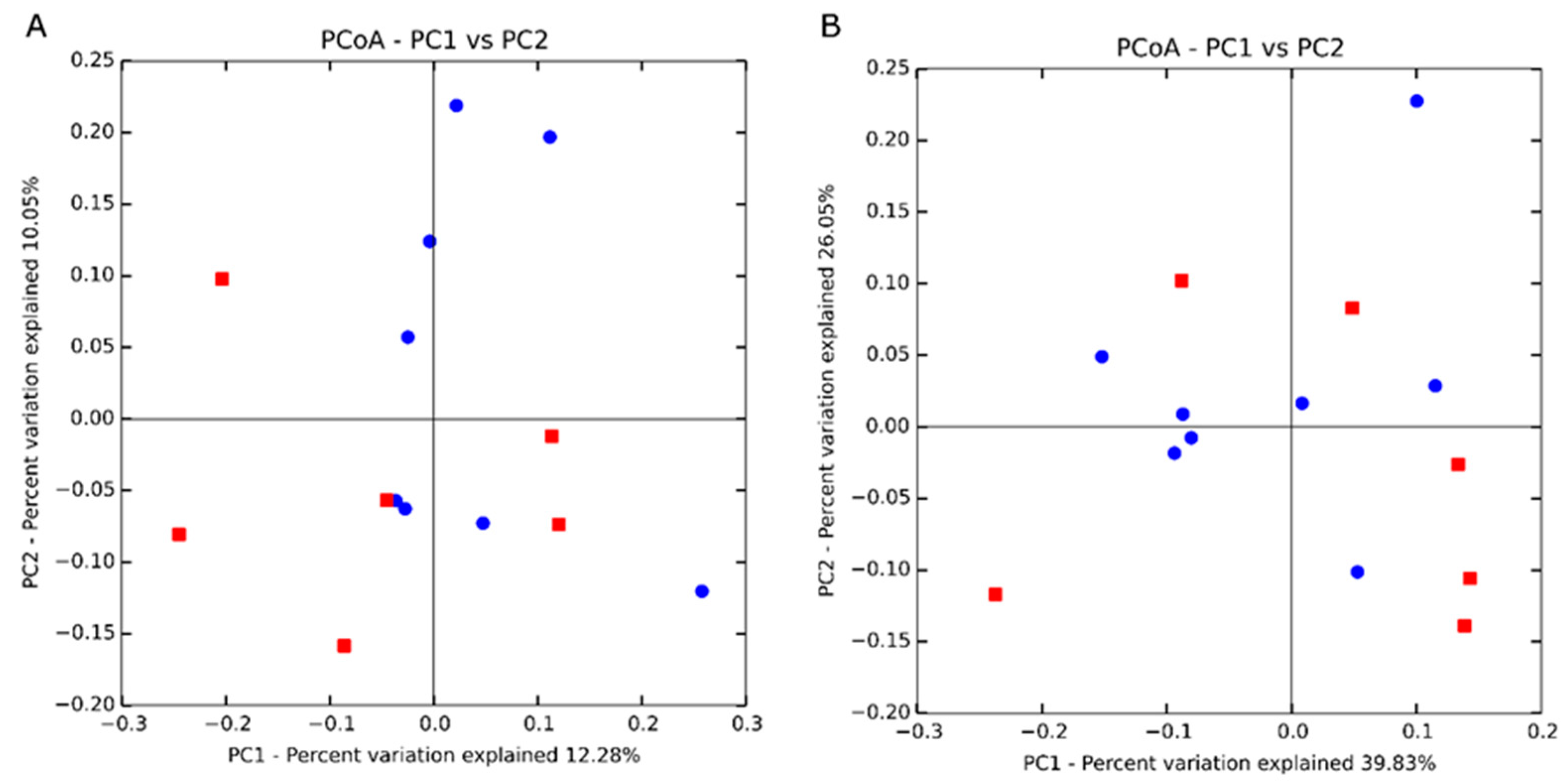
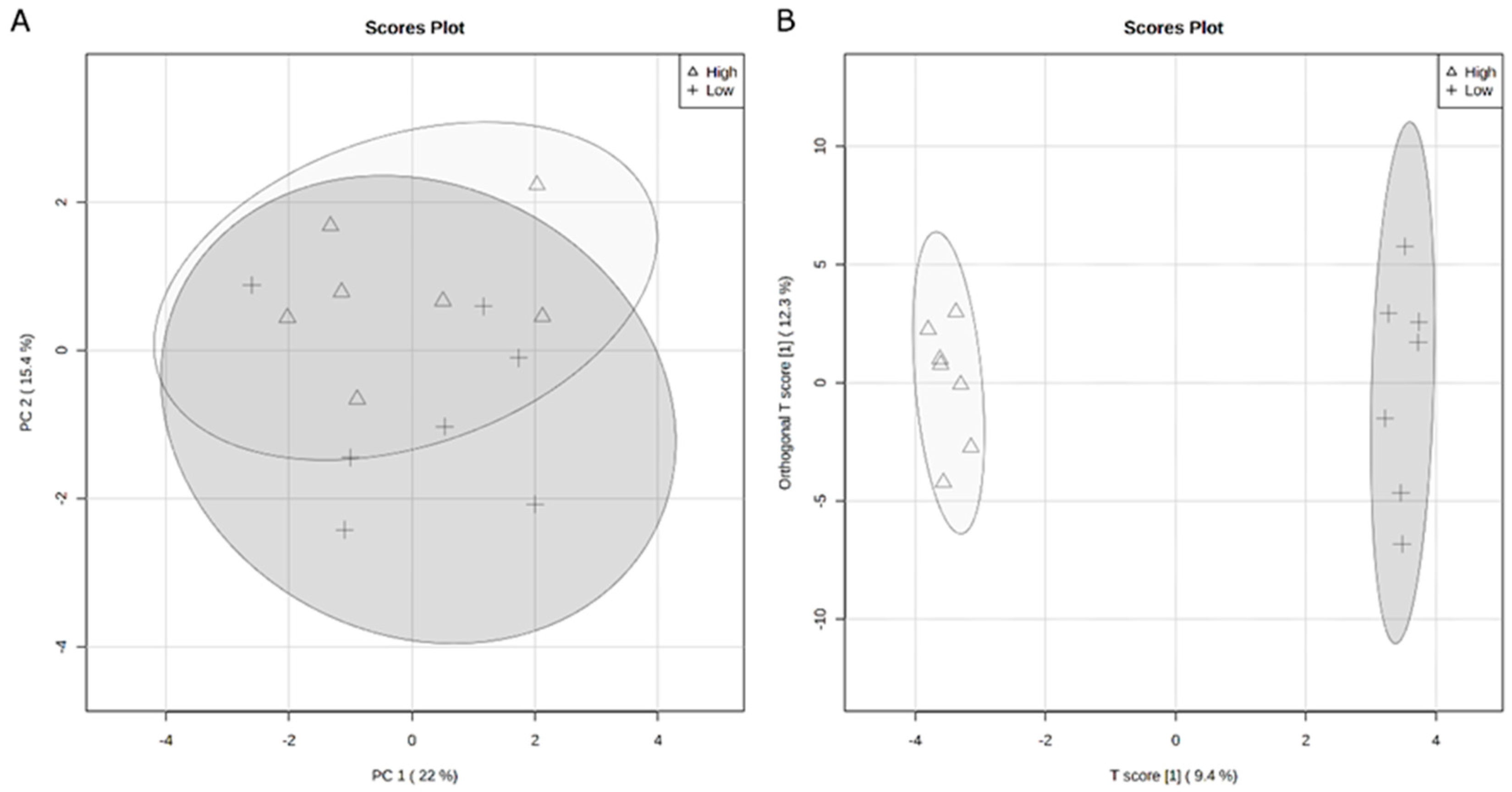
34]. Thus, identifying methods for improving feed efficiency would decrease production costs as well as allow for greater amounts of animal protein available for global consumption. The rumen microbiome contributes to the variation in feed efficiency phenotypes. However, typically, studies focus on terminal beef cattle instead of reproductive members of the herd. Therefore, this study examined the variation in feed efficiency of heifers related to serum and rumen fluid metabolomes as well as rumen bacterial populations.
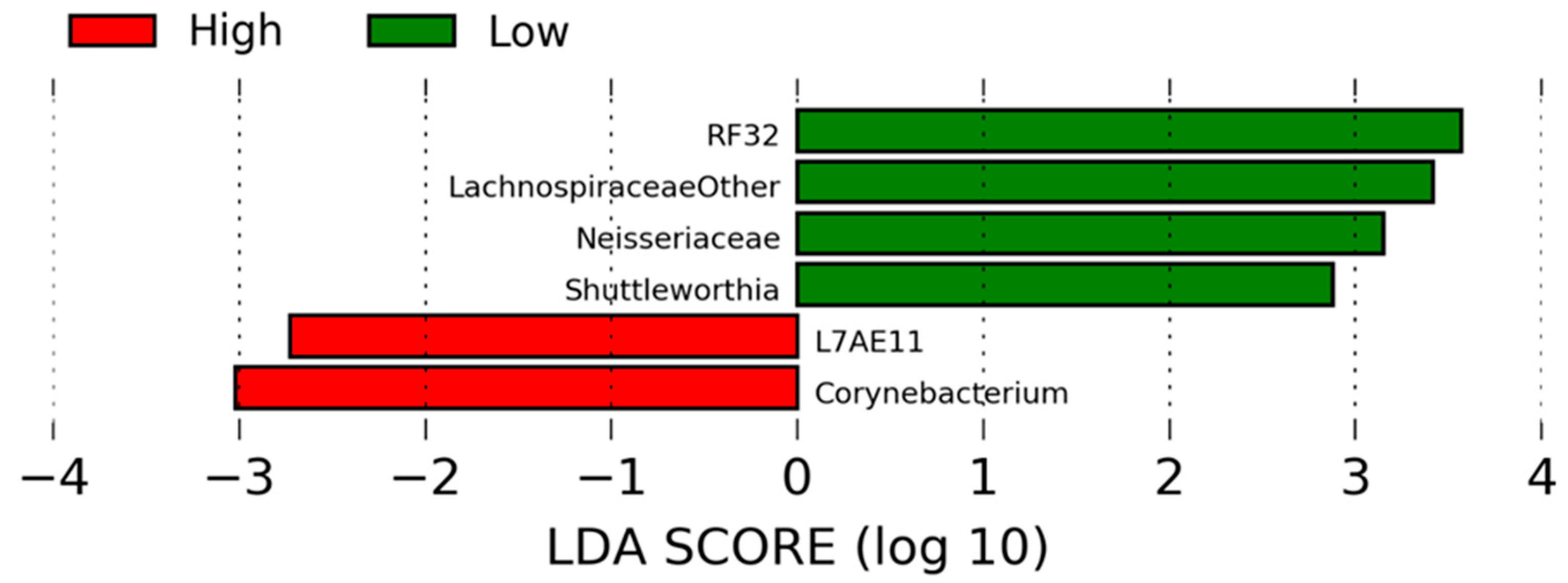
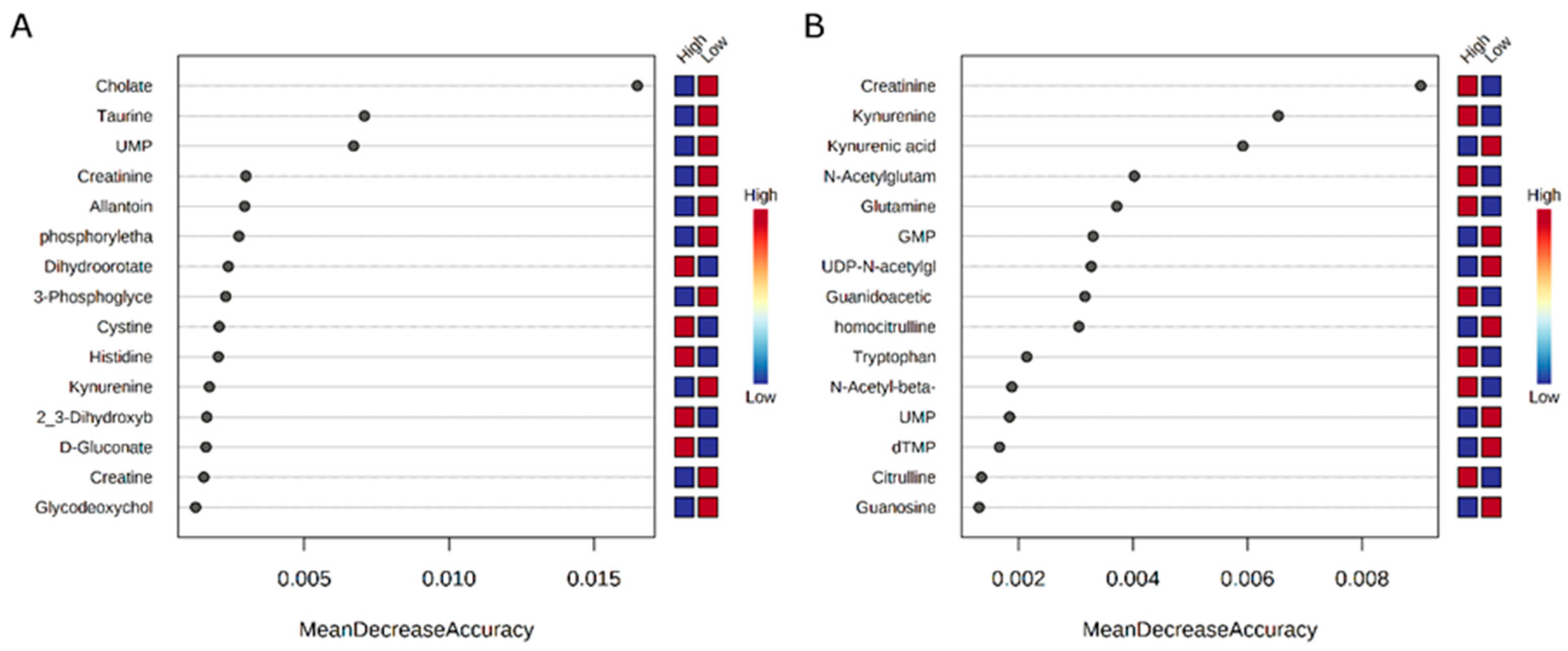
In this study, several relationships were detected between the bacterial communities and host metabolic physiology. A relationship observed among feed efficiency, the rumen microbiome, and rumen metabolome was a positive correlation between asparagine concentrations and
Shuttleworthia. Furthermore, there were greater relative abundances of
Shuttleworthia as well as asparagine in the rumen of low-RFI heifers. Asparagine, though not considered an essential amino acid, is required for proper brain development in mammals as well as several other biological functions [
35]. It can become essential in specific situations, such as in periods of gestation, when animals are consuming low-quality forages, or in diseased states [
36]. In the rumen, asparagine can be rapidly degraded by the rumen microbiota, although it has been demonstrated rates of degradation may differ. Warner examined the rate of degradation of asparagine, glutamine, nicotinamide, and formamide in sheep at various time points [
37]. Warner observed that asparagine was primarily degraded by bacterial populations, but rates of degradation were affected by time or individual animals, possibly suggesting that different bacterial populations may result in variation in rates of asparagine degradation [
37]. The results from Warner, coupled with the results from the present study, illustrate that there are likely differences in asparagine and other amino acid degradation in the rumen as a result of divergent microbial populations. This is of particular interest and importance for ruminant production because microbial utilization of amino acids results in fewer amino acids available to the host ruminant for muscle growth, neurotransmission and development, and other biological activities, as amino acids are catabolized to ammonia which is excreted in the urine as urea [
38].
Furthermore, relationships between bile acid products and bacterial taxa that differed by RFI phenotype were observed. Bacteria that were present in greater relative abundance in high-RFI heifers, including
Corynebacterium and
L7A-E11, were both negatively correlated with taurine, a component for the generation of secondary bile acids. The microbiota found in greater relative abundance in low-RFI heifers, which were positively correlated with metabolites associated with secondary bile acid formation, included
Lachnospiraceae,
Neisseriaceae, and
Shuttleworthia. These bacteria were positively correlated with taurine, glycodeoxycholate, and taurodeoxycholate. The dynamics between bile acids and gut microbiota are interesting and could account for some of the variation observed in feed efficiency phenotypes in cattle. Bile acids are known regulators of gut microbial populations in the mammalian gut as well as host metabolism [
39]. Interestingly, microbially-conjugated bile acids (such as glycodeoxycholate and taurodeoxycholate) rapidly activate insulin regulation pathways via G-protein coupled receptors (GPCR) as well as activate gluconeogenic pathways via FOX01, CEBPα, and HNF4α [
40] in vitro and in monogastric species. Although these specific genes do not appear to be up-or-down-regulated in cattle divergent in feed efficiency, feed efficiency is associated with differential expression in genes related to energy metabolism [
11,
41,
42].
Lachnospiraceae, in particular, is known to produce microbial-derived secondary bile acids [
43,
44].
Lachnospiraceae, as well as the other bacteria found to be associated with differences in RFI and the secondary bile acids, could be altering host gene expression related to metabolism. The relationship between
Lachnospiraceae and RFI in the heifers, as well as with the bile acids, may contribute to variation in feed efficiency phenotypes.
In this study, several microbial taxa of interest were correlated with 2-oxoisovalerate, including those with negative correlations (
p-75-a5 and
Corynebacterium) and those with positive correlations (
Lachnospiraceae [Other] and
Shuttleworthia). Interestingly, those microbes that were negatively correlated with 2-oxoisovalerate were present in greater abundances in high-RFI heifers. Additionally, bacteria positively correlated with 2-oxoisovalerate were observed in greater abundance in the low-RFI heifers. The metabolite 2-oxoisovalerate is involved in several vital metabolic reactions, including valine, leucine, and isoleucine biosynthesis, as well as pantothenate and coenzyme A biosynthesis. Primarily, it is involved in branched-chain amino acid (BCAA) metabolism via the branch chain α-keto acid dehydrogenase (BCKDH) complex as a product of transamination of valine [
45]. The production of α-keto acids, such as 2-oxoisovalerate, results in allosteric inhibition of the BCKDH kinase [
45]. The inhibition of the BCKDH kinase prevents activation of the BCKDH, resulting in conserved BCAA [
45]. Branched-chain amino acids are important for muscle growth, particularly in growing cattle. A study conducted by Li and others examined the effects of BCAA supplementation during nursing on the ADG of growing dairy cattle from three to twenty-eight days after birth [
46]. The authors observed that the steers that received the treatment during the first four weeks of life had sustained greater ADG than the control group starting at two months of age through slaughter at eighteen months of age [
46]. The treatment group had, on average, a 14.8% greater final body weight at slaughter than the control group [
46]. The greater availability of BCAA to the growing animal may provide greater muscle growth potential [
46]. Some bacteria possess the ability to degrade BCAA into byproducts, including 2-oxoisovalerate, that can subsequently be absorbed by the ruminant [
47]. The greater production and absorption of 2-oxoisovalerate could potentially result in some decreased transamination of BCAA, allowing for greater muscle development in low-RFI heifers. This is further supported by other results observed in this study, including the negative correlation of
p-75-a5 with valine and the positive correlation of
Shuttleworthia with 2-oxo-4-methylthiobutanoate, an additional intermediate of BCKDH.
Another metabolic pathway that is associated with RFI, as well as specific microbiota in the rumen, is that involved in the amino acid metabolism of serine and the homocysteine cycle. In the production of serine, 3-phosphoglycerate is converted through a series of enzymatic processes to serine. Serine can, in turn, be converted into glycine, cysteine, and homocysteine. Additionally, serine is a precursor for sphingolipids. In the present study, 3-phosphoglycerate was negatively correlated with
p-75-a5 and
Corynebacterium, both of which were present in greater abundances in high-RFI heifers.
Lachnospiraceae (Other), found in greater abundances in low-RFI heifers, was positively correlated with 3-phosphoglycerate, as well as cysteine and phosphorylethanolamine, a sphingolipid precursor. Rumen bacteria possess the ability to break down cysteine and serine into smaller components [
48], which may make the 3-phosphoglycerate available to the host animal for use, as cattle possess the ability to produce serine and cysteine from 3-phosphoglycerate [
49]. Indeed, Shabat et al. found that amino acid/protein metabolism was upregulated in the rumen of high-RFI cattle, indicating that less feed efficient cattle possess rumen microbiota that may decrease the availability of amino acids or amino acid precursors available to the animal [
11].
Relationships were also observed among glutathione, cysteine, and various bacteria with RFI, in which glutathione and cysteine were positively associated with RFI, as well as bacteria present in greater abundance in high-RFI heifers. The opposite trend was also observed with bacteria present in low-RFI heifers. Glutathione and cysteine are vital antioxidants that reduce the presence of destructive reactive oxygen species (ROS). Low abundances of glutathione and cysteine have been implicated in increased oxidative stress, which is interesting given that greater amounts of glutathione and cysteine were observed in the high-RFI heifers in this study. However, previous studies have demonstrated that antioxidant species may decrease during the depletion of ROS as well as the release of glutathione into media during the loss of intracellular glutathione in vitro [
50]. Kizil and others examined the response of plasma markers of oxidative stress in response to subclinical and clinical mastitis infection [
51]. The authors found that plasma glutathione levels increased with the severity of mastitis infection, indicating increased oxidative stress during mastitis [
51]. The increased levels of glutathione and cysteine in the serum of high-RFI heifers in this study, as well as the negative correlation of various bacteria with cysteine in the rumen of low-RFI heifers, suggest a potential increased oxidative stress in high-RFI. However, the underlying cause of this is yet unknown but warrants further investigation.
Feed efficiency is an incredibly complex phenotypic trait, can be measured using several different methods, and can change based on time of year, production stage, and diet, among others. In ruminants, this is further compounded by the fact that the animal relies substantially on the gut microbiome, primarily the rumen microbiome, as the primary source of energy precursors, vitamins, and other nutrients [
6,
7]. Physiologically, feed efficiency, including RFI, is associated with differences in energy partitioning and methane production [
52]. Researchers have also demonstrated that beef cattle with decreased feed efficiency have altered liver function, the primary site of gluconeogenesis. Alexandre and others examined the liver transcriptome in 16 beef steers differing in feed efficiency using a residual intake and body weight gain (RIG) method [
53]. The authors observed that in the low feed efficient group, cattle had greater fat deposition and as well as increased transcripts for inflammatory responses, the result of which was corroborated using histological methods [
53]. Additionally, the low feed efficient steers demonstrated an upregulation of genes associated with saturated fatty acid synthesis and processes involving lipid metabolism [
53]. The results have been further supported using other methods in beef cattle in further studies [
54,
55,
56,
57,
58]. However, one significant factor that is largely ignored in most studies is the effect of sex on the relationships between physiological differences and feed efficiency. Little is currently known about variation in the metabolome and microbiome in relation to feed efficiency as an effect of sex in cattle. The majority of beef cattle studies regarding feed efficiency are conducted in steers and occasionally bulls, but very few, if any, focus solely on female cattle or examine sex as a factor affecting feed efficiency.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}