
Bovine Rumen Microbiome: Impact of DNA Extraction Methods and Comparison of Non-Invasive Sampling Sites

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Animals and Sample Collection
2.3. DNA Extraction Methodologies
2.4. Amplification and Sequencing of Ribosomal RNA Genes
2.5. Sequence Processing and Analyses
2.6. Statistical Analysis of Data
2.7. Sequence Data Deposition
3. Results
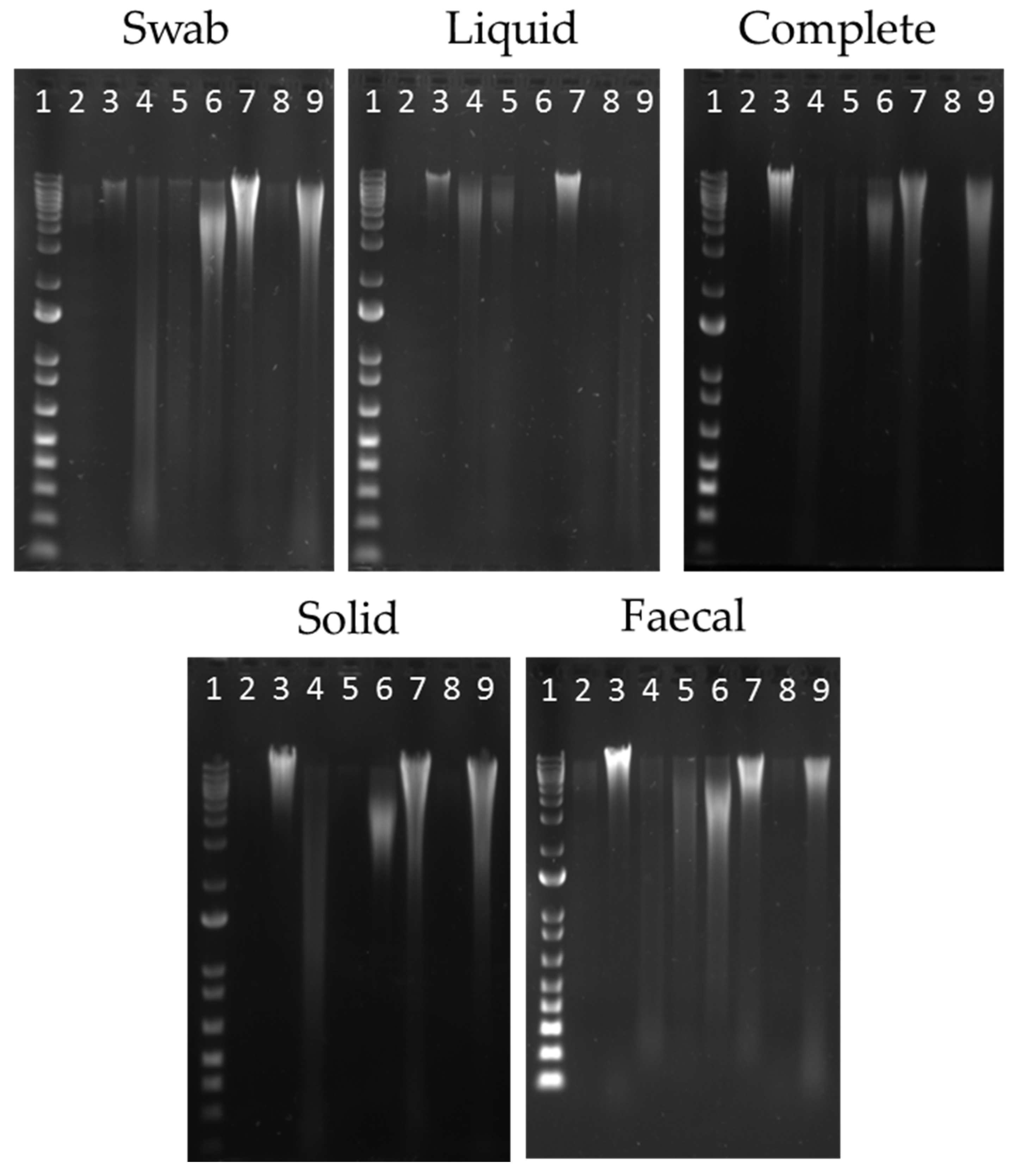
3.1. Evaluation of DNA Extraction Methods
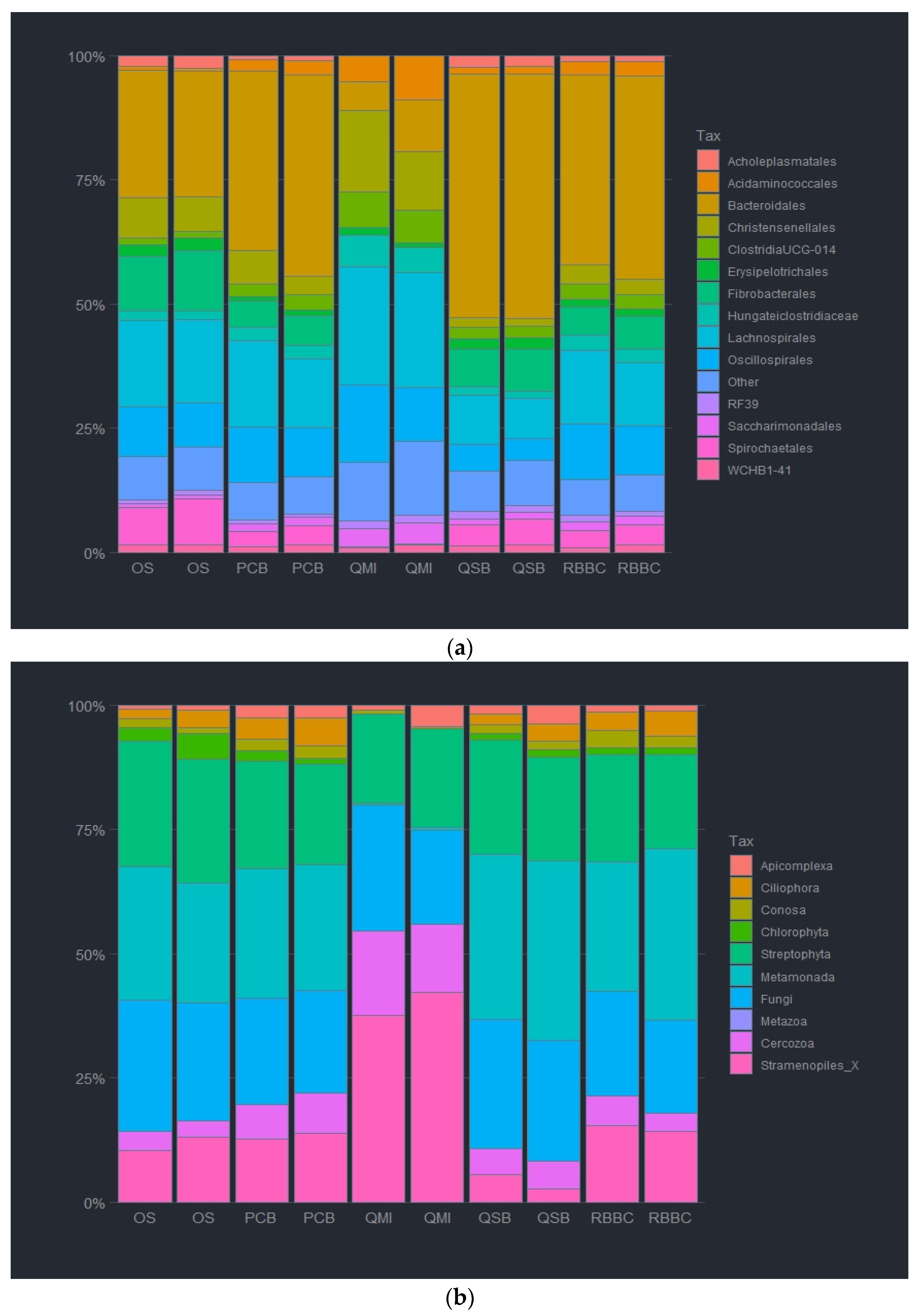
3.2. Impact of DNA Extraction Method on Microbial Relative Abundancies
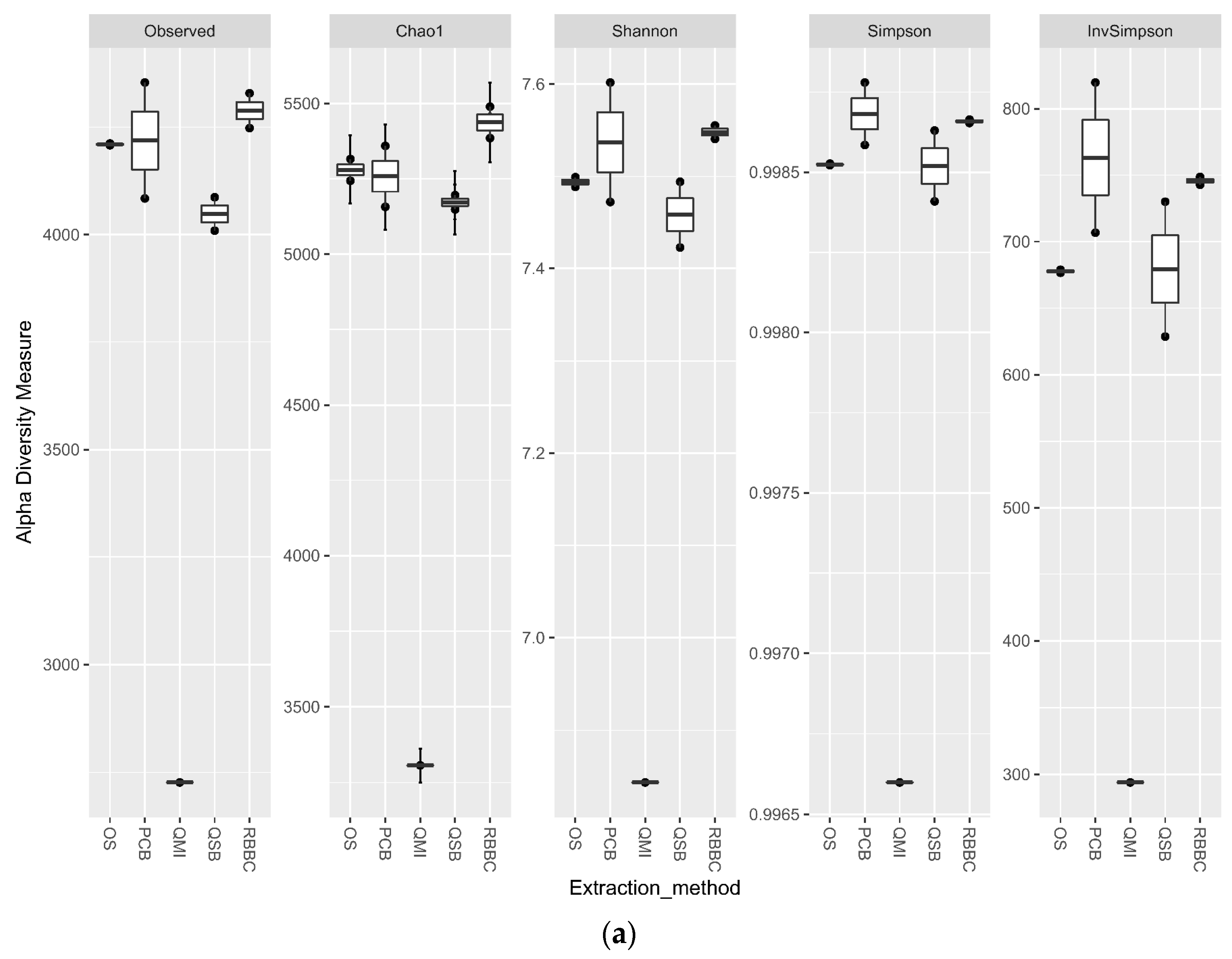
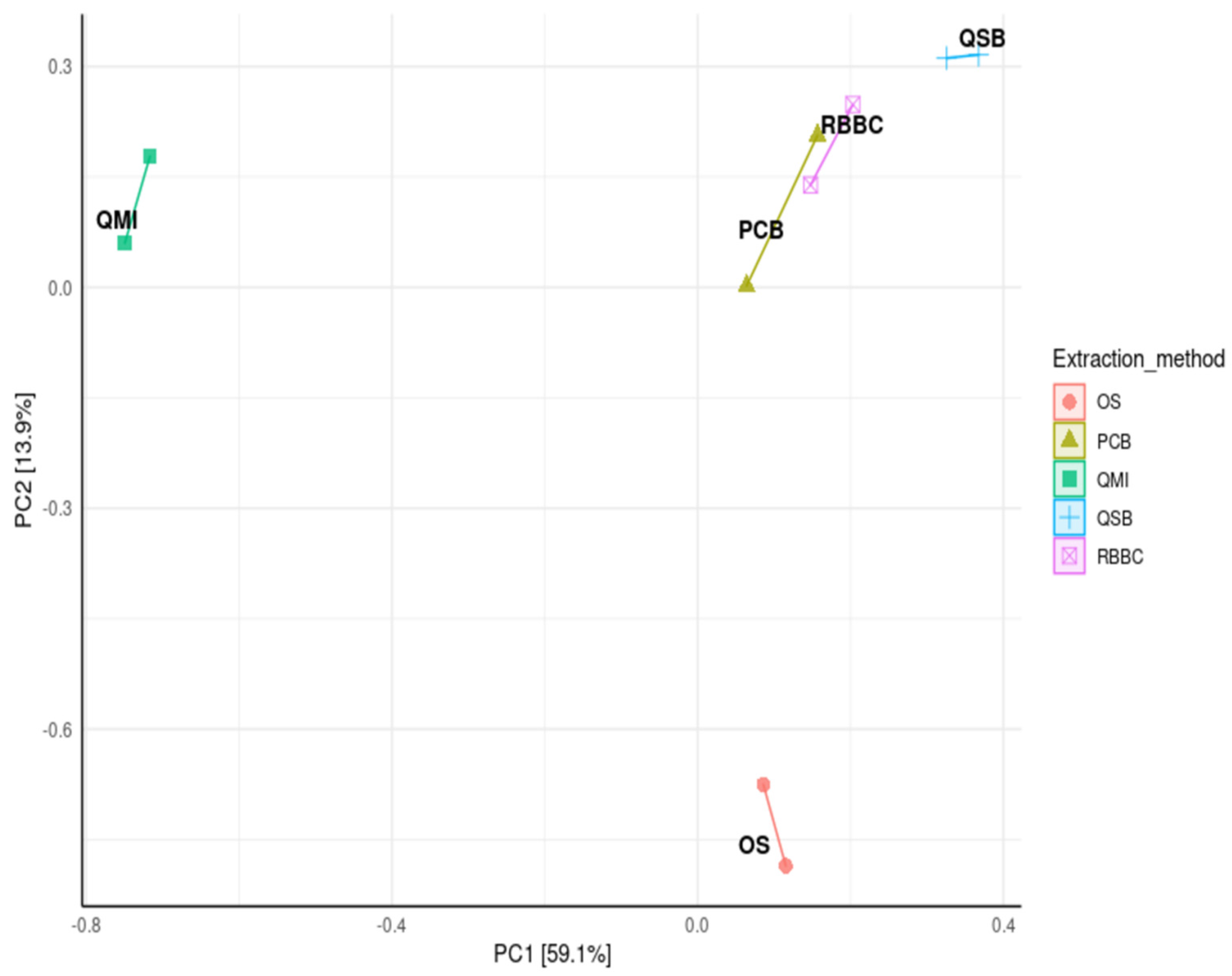
3.3. Impact of DNA Extraction Method on Alpha Diversity and Similarity Analysis
3.4. Impact of Sample Site Selection on Microbial Relative Abundancies
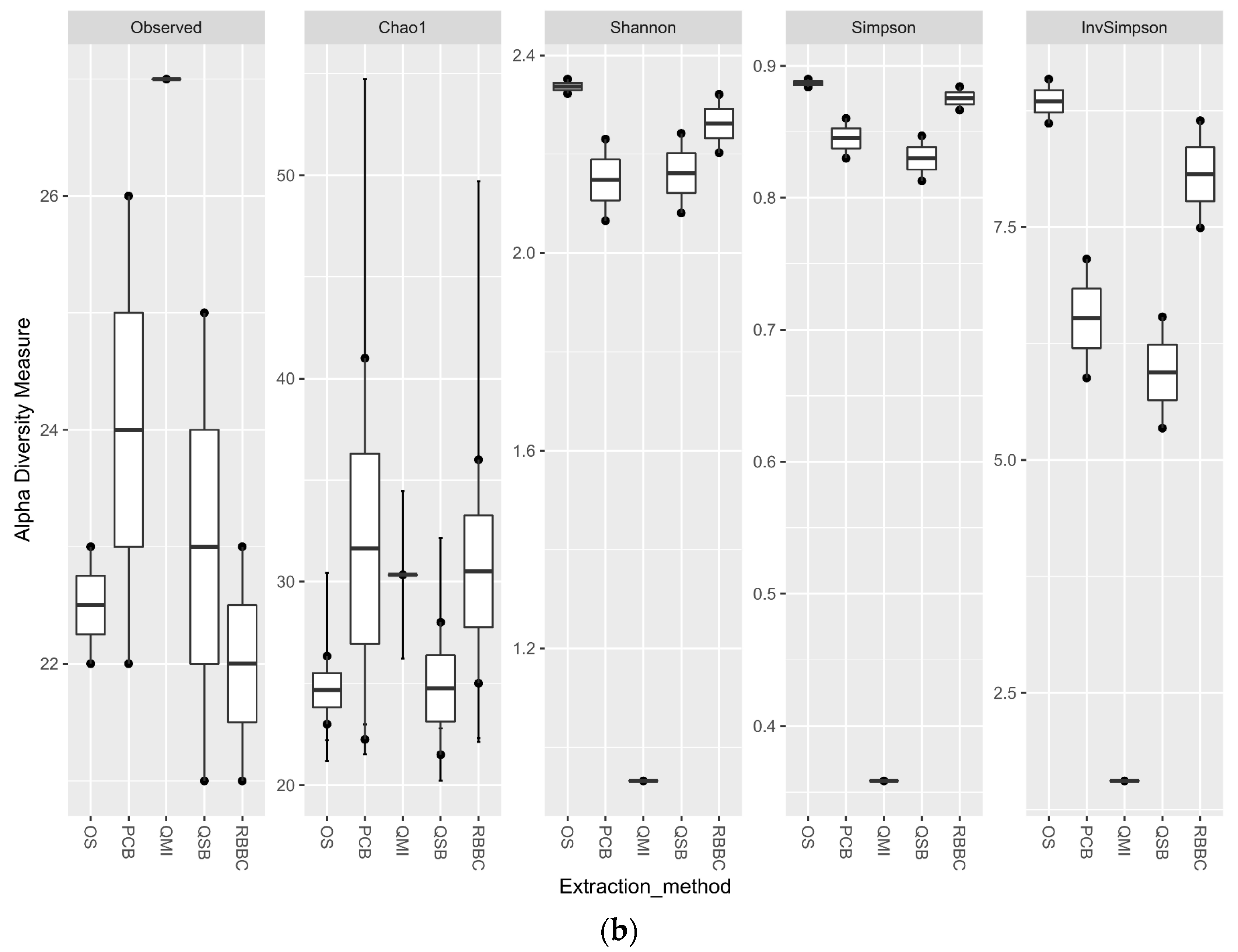
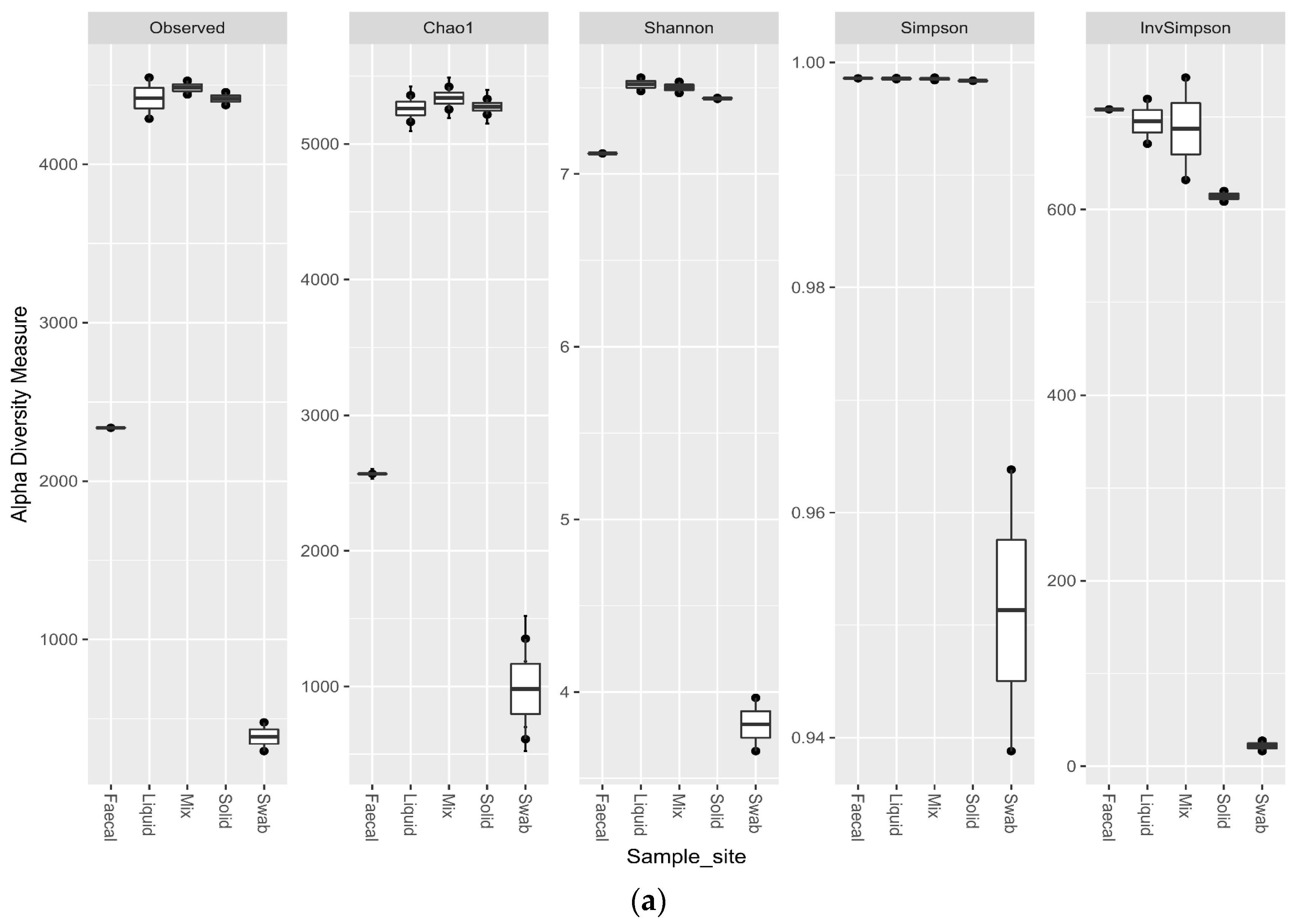
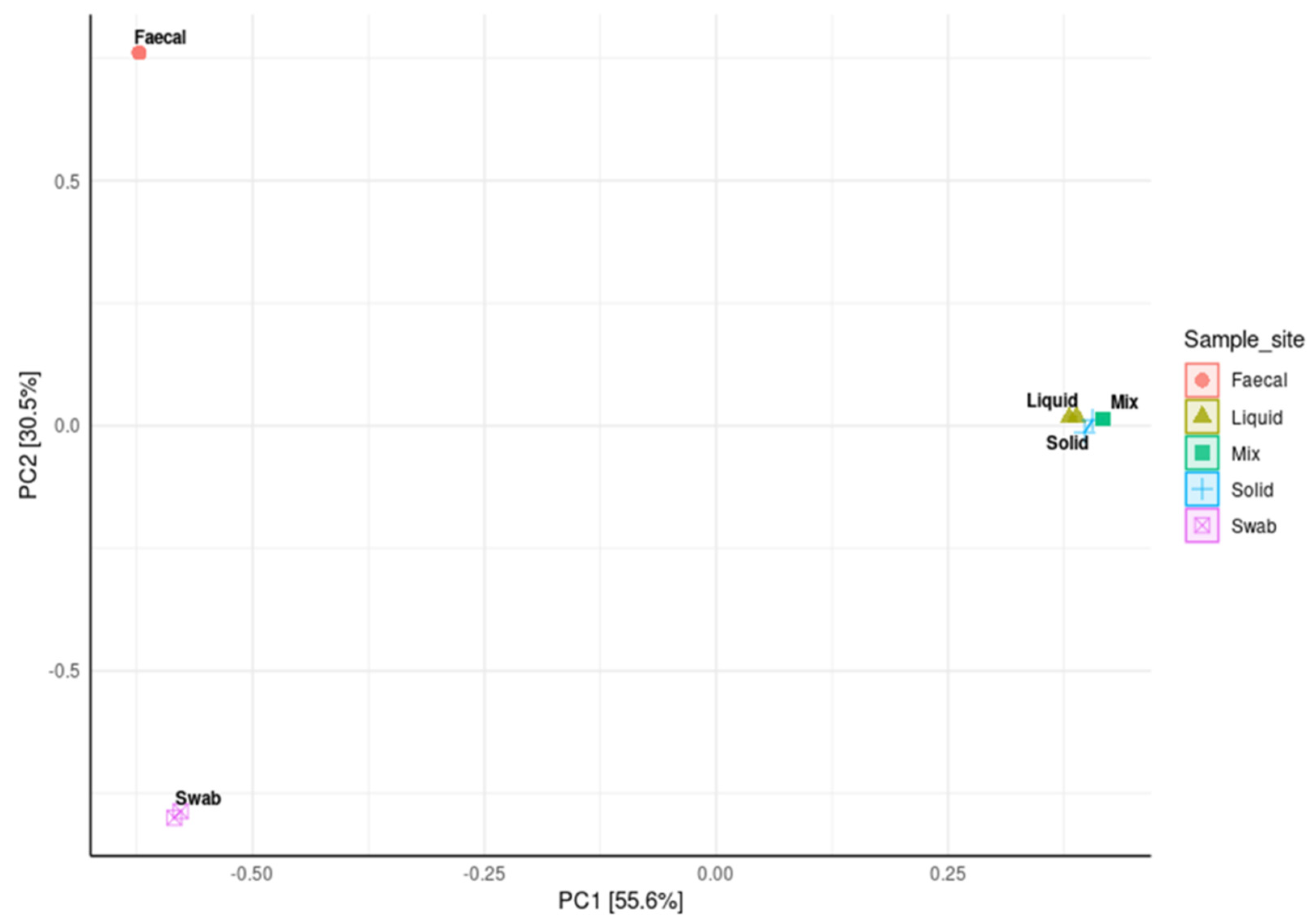
3.5. Impact of Sample Site Selection on Alpha Diversity and Similarity Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alexandratos, N.; Bruinsma, J. World Agriculture towards 2030/2050: The 2012 Revision; FAO: Rome, Italy, 2012. [Google Scholar]
- Karisa, B.; Moore, S.; Plastow, G. Analysis of biological networks and biological pathways associated with residual feed intake in beef cattle. Anim. Sci. J. 2014, 85, 374–387. [Google Scholar] [CrossRef]
- Thornton, P.K. Livestock production: Recent trends, future prospects. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 2853–2867. [Google Scholar] [CrossRef] [Green Version]
- Mizrahi, I. The Role of the Rumen Microbiota in Determining the Feed Efficiency of Dairy Cows. In Beneficial Microorganisms in Multicellular Life Forms; Rosenberg, E., Gophna, U., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 203–210. ISBN 978-3-642-21679-4. [Google Scholar]
- White, B.A.; Lamed, R.; Bayer, E.A.; Flint, H.J. Biomass utilization by gut microbiomes. Annu. Rev. Microbiol. 2014, 68, 279–296. [Google Scholar] [CrossRef]
- Gerber, P.J.; Steinfeld, H.; Henderson, B.; Mottet, A.; Opio, C.; Dijkman, J.; Falcucci, A.; Tempio, G. Tackling Climate Change through Livestock: A Global Assessment of Emissions and Mitigation Opportunities: Tackling Climate Change through Livestock: A Global Assessment of Emissions and Mitigation Opportunities; Food and Agriculture Organization of the United Nations (FAO): Rome, Italy, 2013; ISBN 92-5-107920-X. [Google Scholar]
- Johnson, K.A.; Johnson, D.E. Methane emissions from cattle. J. Anim. Sci. 1995, 73, 2483–2492. [Google Scholar] [CrossRef]
- Wallace, R.J.; Snelling, T.J.; McCartney, C.A.; Tapio, I.; Strozzi, F. Application of meta-omics techniques to understand greenhouse gas emissions originating from ruminal metabolism. Genet. Sel. Evol. 2017, 49, 9. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Peng, Y.-J.; Chen, Y.; Klinger, C.M.; Oba, M.; Liu, J.-X.; Le Guan, L. Assessment of microbiome changes after rumen transfaunation: Implications on improving feed efficiency in beef cattle. Microbiome 2018, 6, 62. [Google Scholar] [CrossRef] [Green Version]
- Creevey, C.J.; Kelly, W.J.; Henderson, G.; Leahy, S.C. Determining the culturability of the rumen bacterial microbiome. Microb. Biotechnol. 2014, 7, 467–479. [Google Scholar] [CrossRef] [Green Version]
- Lazarevic, V.; Gaïa, N.; Girard, M.; François, P.; Schrenzel, J. Comparison of DNA extraction methods in analysis of salivary bacterial communities. PLoS ONE 2013, 8, e67699. [Google Scholar] [CrossRef]
- Henderson, G.; Cox, F.; Kittelmann, S.; Miri, V.H.; Zethof, M.; Noel, S.J.; Waghorn, G.C.; Janssen, P.H. Effect of DNA extraction methods and sampling techniques on the apparent structure of cow and sheep rumen microbial communities. PLoS ONE 2013, 8, e74787. [Google Scholar] [CrossRef] [Green Version]
- Kittelmann, S.; Kirk, M.R.; Jonker, A.; McCulloch, A.; Janssen, P.H. Buccal swabbing as a noninvasive method to determine bacterial, archaeal, and eukaryotic microbial community structures in the rumen. Appl. Environ. Microbiol. 2015, 81, 7470–7483. [Google Scholar] [CrossRef] [Green Version]
- Vaidya, J.D.; van den Bogert, B.; Edwards, J.E.; Boekhorst, J.; van Gastelen, S.; Saccenti, E.; Plugge, C.M.; Smidt, H. The Effect of DNA Extraction Methods on Observed Microbial Communities from Fibrous and Liquid Rumen Fractions of Dairy Cows. Front. Microbiol. 2018, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Fliegerova, K.; Tapio, I.; Bonin, A.; Mrazek, J.; Callegari, M.L.; Bani, P.; Bayat, A.; Vilkki, J.; Kopečný, J.; Shingfield, K.J.; et al. Effect of DNA extraction and sample preservation method on rumen bacterial population. Anaerobe 2014, 29, 80–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapio, I.; Shingfield, K.J.; McKain, N.; Bonin, A.; Fischer, D.; Bayat, A.R.; Vilkki, J.; Taberlet, P.; Snelling, T.J.; Wallace, R.J. Oral Samples as Non-Invasive Proxies for Assessing the Composition of the Rumen Microbial Community. PLoS ONE 2016, 11, e0151220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herd, R.M.; Arthur, P.F. Physiological basis for residual feed intake. J. Anim. Sci. 2009, 87, E64–E71. [Google Scholar] [CrossRef] [PubMed]
- Nkrumah, J.D.; Okine, E.K.; Mathison, G.W.; Schmid, K.; Li, C.; Basarab, J.A.; Price, M.A.; Wang, Z.; Moore, S.S. Relationships of feedlot feed efficiency, performance, and feeding behavior with metabolic rate, methane production, and energy partitioning in beef cattle. J. Anim. Sci. 2006, 84, 145–153. [Google Scholar] [CrossRef]
- Shen, J.S.; Chai, Z.; Song, L.J.; Liu, J.X.; Wu, Y.M. Insertion depth of oral stomach tubes may affect the fermentation parameters of ruminal fluid collected in dairy cows. J. Dairy Sci. 2012, 95, 5978–5984. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Morales, E.; Arco-Pérez, A.; Martín-García, A.I.; Yáñez-Ruiz, D.R.; Frutos, P.; Hervás, G. Use of stomach tubing as an alternative to rumen cannulation to study ruminal fermentation and microbiota in sheep and goats. Anim. Feed. Sci. Technol. 2014, 198, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, O. Rumenocentesis: A useful technique for diagnosis and monitoring of rumen health in cattle. Livestock 2017, 22, 240–243. [Google Scholar] [CrossRef]
- Castillo, C.; Hernández, J. Ruminal Fistulation and Cannulation: A Necessary Procedure for the Advancement of Biotechnological Research in Ruminants. Animals 2021, 11, 1870. [Google Scholar] [CrossRef]
- Estellé, J. Benefits from the joint analysis of host genomes and metagenomes: Select the holobiont. J. Anim. Breed. Genet. 2019, 136, 75–76. [Google Scholar] [CrossRef] [Green Version]
- Zilber-Rosenberg, I.; Rosenberg, E. Role of microorganisms in the evolution of animals and plants: The hologenome theory of evolution. FEMS Microbiol. Rev. 2008, 32, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Schrader, C.; Schielke, A.; Ellerbroek, L.; Johne, R. PCR inhibitors-occurrence, properties and removal. J. Appl. Microbiol. 2012, 113, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Martin, J.C.; Scott, P.; Parkhill, J.; Flint, H.J.; Scott, K.P. 16S rRNA gene-based profiling of the human infant gut microbiota is strongly influenced by sample processing and PCR primer choice. Microbiome 2015, 3, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amore, R.; Ijaz, U.Z.; Schirmer, M.; Kenny, J.G.; Gregory, R.; Darby, A.C.; Shakya, M.; Podar, M.; Quince, C.; Hall, N. A comprehensive benchmarking study of protocols and sequencing platforms for 16S rRNA community profiling. BMC Genom. 2016, 17, 55. [Google Scholar] [CrossRef] [Green Version]
- Prodan, A.; Tremaroli, V.; Brolin, H.; Zwinderman, A.H.; Nieuwdorp, M.; Levin, E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 2020, 15, e0227434. [Google Scholar] [CrossRef] [Green Version]
- Amin, N.; Schwarzkopf, S.; Kinoshita, A.; Tröscher-Mußotter, J.; Dänicke, S.; Camarinha-Silva, A.; Huber, K.; Frahm, J.; Seifert, J. Evolution of rumen and oral microbiota in calves is influenced by age and time of weaning. Anim. Microbiome 2021, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Morrison, M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 2004, 36, 808–812. [Google Scholar] [CrossRef]
- Griffiths, R.I.; Whiteley, A.S.; O’Donnell, A.G.; Bailey, M.J. Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA- and rRNA-based microbial community composition. Appl. Environ. Microbiol. 2000, 66, 5488–5491. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Stoeck, T.; Bass, D.; Nebel, M.; Christen, R.; Jones, M.D.M.; Breiner, H.-W.; Richards, T.A. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 2010, 19 (Suppl. 1), 21–31. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Andersen, K.S.; Kirkegaard, R.H.; Karst, S.M.; Albertsen, M. Ampvis2: An R package to analyse and visualise 16S rRNA amplicon data. BioRxiv 2018, 299537. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Morton, J.T.; Marotz, C.; Washburne, A.; Silverman, J.; Zaramela, L.S.; Edlund, A.; Zengler, K.; Knight, R. Establishing microbial composition measurement standards with reference frames. Nat. Commun. 2019, 10, 2719. [Google Scholar] [CrossRef] [Green Version]
- Paulson, J.N.; Talukder, H.; Héctor, C.B. Longitudinal differential abundance analysis of microbial marker-gene surveys using smoothing splines. BioRxiv 2017, 099457. [Google Scholar] [CrossRef]
- McAllister, T.A.; Meale, S.J.; Valle, E.; Guan, L.L.; Zhou, M.; Kelly, W.J.; Henderson, G.; Attwood, G.T.; Janssen, P.H. RUMINANT NUTRITION SYMPOSIUM: Use of genomics and transcriptomics to identify strategies to lower ruminal methanogenesis. J. Anim. Sci. 2015, 93, 1431–1449. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.L.; Goopy, J.P.; Hegarty, R.S.; Oddy, V.H.; Thompson, A.N.; Toovey, A.F.; Macleay, C.A.; Briegal, J.R.; Woodgate, R.T.; Donaldson, A.J.; et al. Genetic and environmental variation in methane emissions of sheep at pasture. J. Anim. Sci. 2014, 92, 4349–4363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegarty, R.S.; Goopy, J.P.; Herd, R.M.; McCorkell, B. Cattle selected for lower residual feed intake have reduced daily methane production. J. Anim. Sci. 2007, 85, 1479–1486. [Google Scholar] [CrossRef]
- Ghanbari Maman, L.; Palizban, F.; Fallah Atanaki, F.; Elmi Ghiasi, N.; Ariaeenejad, S.; Ghaffari, M.R.; Hosseini Salekdeh, G.; Kavousi, K. Co-abundance analysis reveals hidden players associated with high methane yield phenotype in sheep rumen microbiome. Sci. Rep. 2020, 10, 4995. [Google Scholar] [CrossRef] [Green Version]
- Frey, J.C.; Pell, A.N.; Berthiaume, R.; Lapierre, H.; Lee, S.; Ha, J.K.; Mendell, J.E.; Angert, E.R. Comparative studies of microbial populations in the rumen, duodenum, ileum and faeces of lactating dairy cows. J. Appl. Microbiol. 2010, 108, 1982–1993. [Google Scholar] [CrossRef]
- Romero-Pérez, G.A.; Ominski, K.H.; McAllister, T.A.; Krause, D.O. Effect of environmental factors and influence of rumen and hindgut biogeography on bacterial communities in steers. Appl. Environ. Microbiol. 2011, 77, 258–268. [Google Scholar] [CrossRef] [Green Version]
- Flint, H.J.; Bayer, E.A.; Rincon, M.T.; Lamed, R.; White, B.A. Polysaccharide utilization by gut bacteria: Potential for new insights from genomic analysis. Nat. Rev. Microbiol. 2008, 6, 121–131. [Google Scholar] [CrossRef]
- Godoy-Vitorino, F.; Goldfarb, K.C.; Karaoz, U.; Leal, S.; Garcia-Amado, M.A.; Hugenholtz, P.; Tringe, S.G.; Brodie, E.L.; Dominguez-Bello, M.G. Comparative analyses of foregut and hindgut bacterial communities in hoatzins and cows. ISME J. 2012, 6, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Akonyani, Z.P.; Song, F.; Li, Y.; Qiqige, S.; Wu, J. Comparative Analysis of the Microbiota Between Rumen and Duodenum of Twin Lambs Based on Diets of Ceratoides or Alfalfa. Pol. J. Microbiol. 2021, 70, 175–187. [Google Scholar] [CrossRef]
- Ellis, J.E.; Williams, A.G.; Lloyd, D. Oxygen consumption by ruminal microorganisms: Protozoal and bacterial contributions. Appl. Environ. Microbiol. 1989, 55, 2583–2587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhao, S.; Zhang, Y.; Sun, P.; Bu, D.; Wang, J. New Primers Targeting Full-Length Ciliate 18S rRNA Genes and Evaluation of Dietary Effect on Rumen Ciliate Diversity in Dairy Cows. Curr. Microbiol. 2015, 71, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Fouts, D.E.; Szpakowski, S.; Purushe, J.; Torralba, M.; Waterman, R.C.; MacNeil, M.D.; Alexander, L.J.; Nelson, K.E. Next generation sequencing to define prokaryotic and fungal diversity in the bovine rumen. PLoS ONE 2012, 7, e48289. [Google Scholar] [CrossRef] [PubMed]
- Wallace, R.J.; Rooke, J.A.; Duthie, C.-A.; Hyslop, J.J.; Ross, D.W.; McKain, N.; de Souza, S.M.; Snelling, T.J.; Waterhouse, A.; Roehe, R. Archaeal abundance in post-mortem ruminal digesta may help predict methane emissions from beef cattle. Sci. Rep. 2014, 4, 5892. [Google Scholar] [CrossRef] [PubMed]
- Tajima, K.; Aminov, R.I.; Nagamine, T.; Matsui, H.; Nakamura, M.; Benno, Y. Diet-dependent shifts in the bacterial population of the rumen revealed with real-time PCR. Appl. Environ. Microbiol. 2001, 67, 2766–2774. [Google Scholar] [CrossRef] [Green Version]
- Olson, N.D.; Morrow, J.B. DNA extract characterization process for microbial detection methods development and validation. BMC Res. Notes 2012, 5, 668. [Google Scholar] [CrossRef] [Green Version]
- Min, B.R.; Gurung, N.; Shange, R.; Solaiman, S. Potential role of rumen microbiota in altering average daily gain and feed efficiency in meat goats fed simple and mixed pastures using bacterial tag-encoded FLX amplicon pyrosequencing. J. Anim. Sci. 2019, 97, 3523–3534. [Google Scholar] [CrossRef]
- Newbold, C.J.; de La Fuente, G.; Belanche, A.; Ramos-Morales, E.; McEwan, N.R. The Role of Ciliate Protozoa in the Rumen. Front. Microbiol. 2015, 6, 1313. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.G.; Coleman, G.S. The rumen protozoa. In The Rumen Microbial Ecosystem; Hobson, P.N., Stewart, C.S., Eds.; Springer: Dordrecht, The Netherlands, 1997; pp. 73–139. ISBN 978-94-010-7149-9. [Google Scholar]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extraction Method | Abbreviation | Comments | Reference |
|---|---|---|---|
| QIAamp Fast DNA Stool Mini Kit | QS | Followed manufacturer’s instructions | Qiagen®, Hilden, Germany |
| QIAamp Fast DNA Stool Mini Kit+ Bead Beating | QSB | As above, including bead beating step | Qiagen®, Hilden, Germany |
| QIAamp DNA Microbiome Kit | QM | Followed manufacturer’s instructions | Qiagen®, Hilden, Germany |
| QIAamp DNA Microbiome Kit + InhibitEX Buffer | QMI | As above, including use of InhibitEX buffer | Qiagen®, Hilden, Germany |
| Soil DNA Purification Kit | OS | Followed manufacturer’s instructions | Omni-International®, Kennesaw, GA, USA |
| Repeat Bead Beater and Column Method | RBBC | As stated in reference | [30] |
| Phenol Chloroform Extraction | PC | As stated in reference | [31] |
| Phenol Chloroform Extraction + Bead Beating | PCB | As above, including bead-beating step | [31] |
| Method | Swab | Liquid | Complete | Solid | Faecal |
|---|---|---|---|---|---|
| QS | |||||
| Concentration (ng/µL) | 1.05 ±1.05 | 22 ±1.5 | 57.75 ±12.05 | 87.75 ±26.1 | 86.6 ±3.4 |
| A260/280 | 2.63 | 2.01 | 1.97 | 1.94 | 1.97 |
| QSB | |||||
| Concentration (ng/µL) | 4.15 ±0.65 | 46.65 ±17.25 | 124.1 ±69.6 | 312.05 ±6.05 | 247.65 ±3.25 |
| A260/280 | 1.86 | 2.04 | 1.93 | 1.88 | 1.84 |
| QM | |||||
| Concentration (ng/µL) | 4.25 ±1.05 | 84.15 ±3.25 | 107.8 ±67.7 | 248.25 ±39.7 | 142.4 ±13.2 |
| A260/280 | 1.75 | 1.71 | 0.94 | 1.11 | 0.94 |
| QMI | |||||
| Concentration (ng/µL) | 4 ±0.6 | 61.05 ±6.65 | 61.25 ±3.95 | 36.35 ±13.25 | 47.5 ±13.5 |
| A260/280 | 1.81 | 1.88 | 1.54 | 1.32 | 1.37 |
| OS | |||||
| Concentration (ng/µL) | 3.15 ±2.1 | 102.9 ±16.5 | 228.9 ±19.8 | 299.85 ±44.25 | 152.8 ±4.5 |
| A260/280 | 1.83 | 1.8 | 1.79 | 1.79 | 1.35 |
| RBBC | |||||
| Concentration (ng/µL) | 7.44 ±1.6 | 147.3 ±2.6 | 315 ±113.5 | 513.9 ±37.4 | 335.4 ±23.7 |
| A260/280 | 1.83 | 1.8 | 1.57 | 1.58 | 1.35 |
| PC | |||||
| Concentration (ng/µL) | 4.72 ±1.48 | 11.75 ±0.45 | 20.85 ±2.75 | 30.85 ±4.55 | 13.5 ±1.1 |
| A260/280 | 1.9 | 1.86 | 1.93 | 1.92 | 1.84 |
| PCB | |||||
| Concentration (ng/µL) | 31.3 ±0.7 | 329.5 ±1.7 | 394.35 ±205.3 | 1050.55 ±193 | 834.4 ±51 |
| A260/280 | 2.27 | 1.89 | 1.92 | 1.96 | 1.87 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mott, A.C.; Schneider, D.; Hünerberg, M.; Hummel, J.; Tetens, J. Bovine Rumen Microbiome: Impact of DNA Extraction Methods and Comparison of Non-Invasive Sampling Sites. Ruminants 2022, 2, 112-132. https://doi.org/10.3390/ruminants2010007
Mott AC, Schneider D, Hünerberg M, Hummel J, Tetens J. Bovine Rumen Microbiome: Impact of DNA Extraction Methods and Comparison of Non-Invasive Sampling Sites. Ruminants. 2022; 2(1):112-132. https://doi.org/10.3390/ruminants2010007
Chicago/Turabian StyleMott, Alexander C., Dominik Schneider, Martin Hünerberg, Jürgen Hummel, and Jens Tetens. 2022. "Bovine Rumen Microbiome: Impact of DNA Extraction Methods and Comparison of Non-Invasive Sampling Sites" Ruminants 2, no. 1: 112-132. https://doi.org/10.3390/ruminants2010007