
Investigation of the Acid/Basic Sites of Zeolite Trough Some Catalysed Nucleophilic Reactions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
Zeolites as Emerging Green Catalysts
2. Discussion
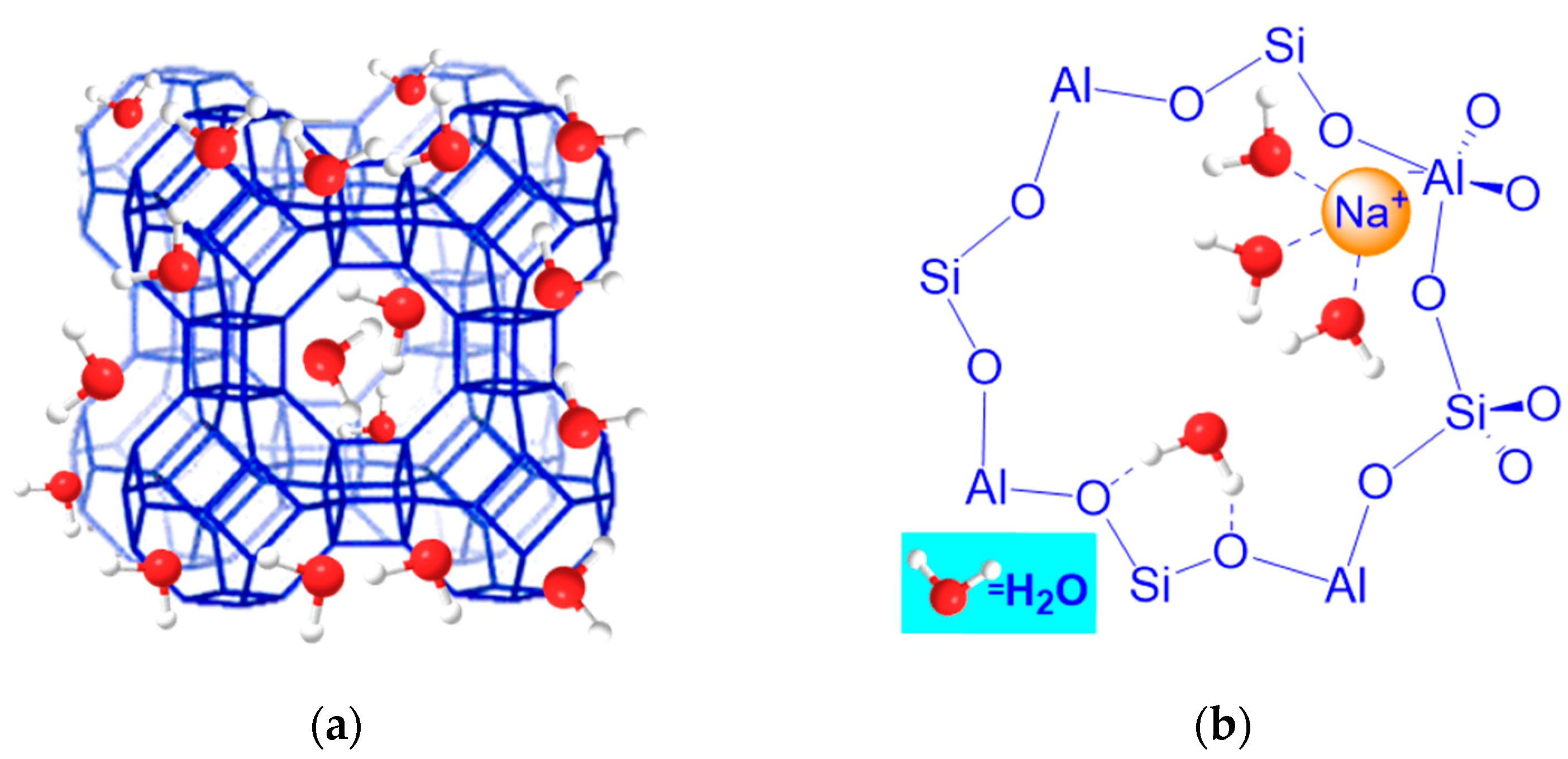
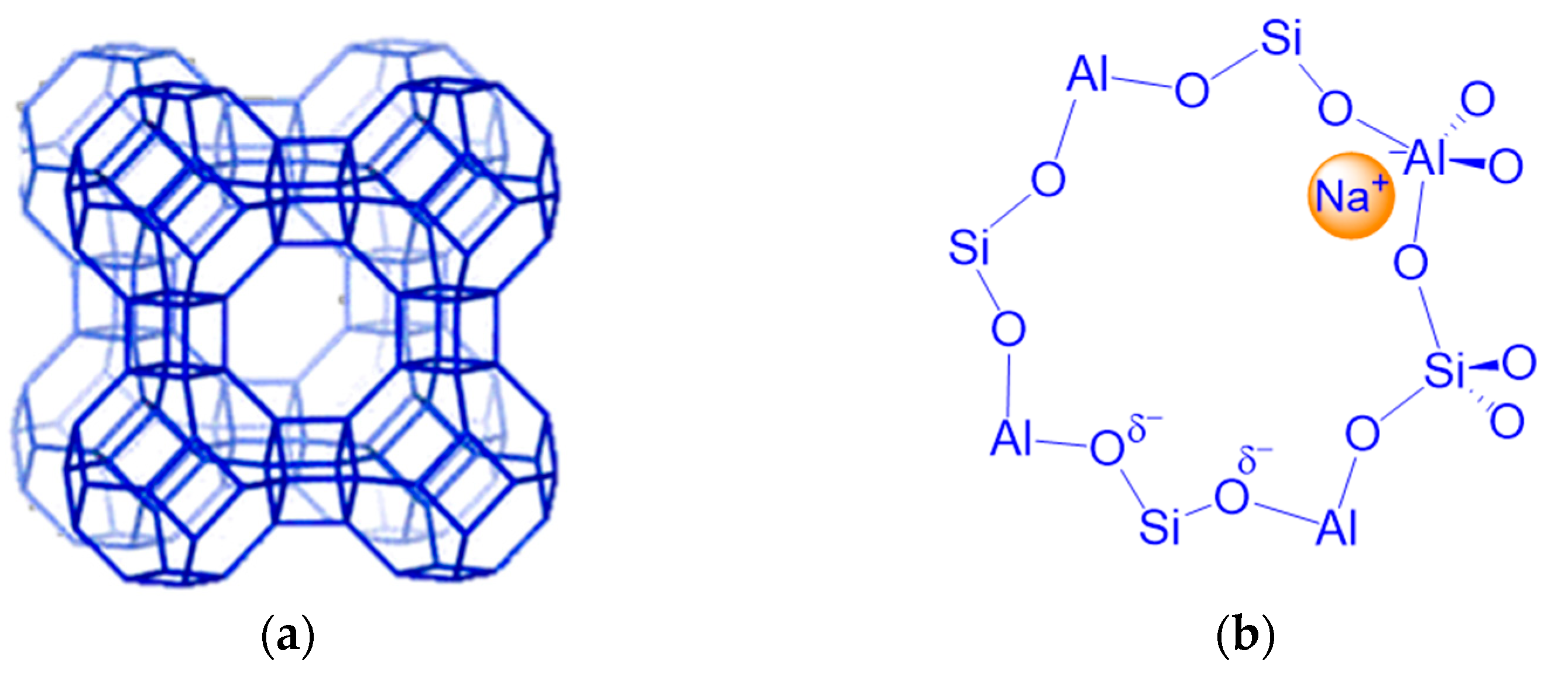
2.1. Heat Treatment of Zeolites and a Consequent Dehydration and Activation Process
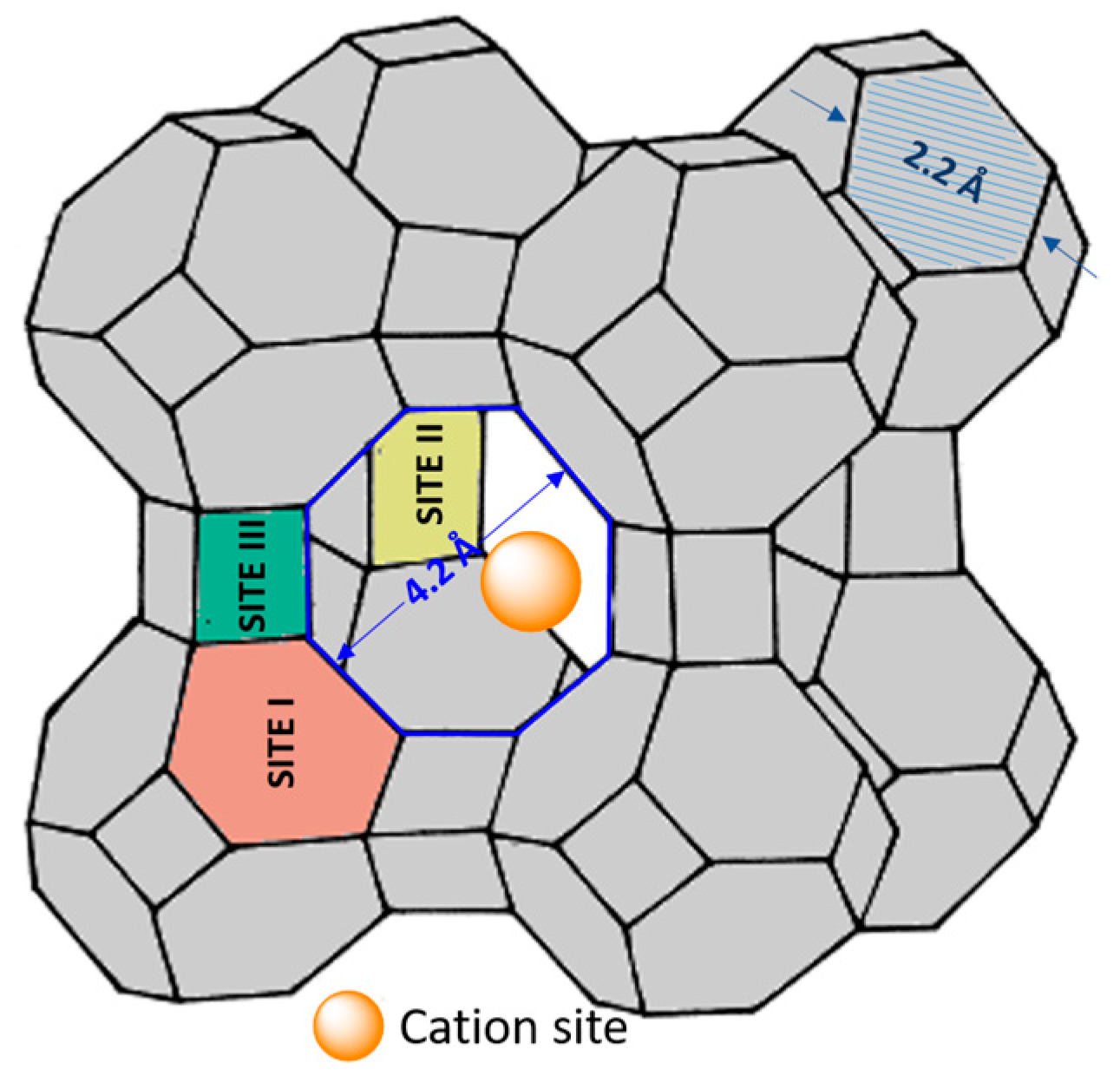
2.2. Basicity of the Lattice Oxygen Bridging Al and Si: Distribution of Aluminum in the Tetrahedra of Zeolite Crystal
2.3. Nucleophilic Substitution Reactions Catalyzed by Zeolite A: INSIGHT into the Basic Active Sites
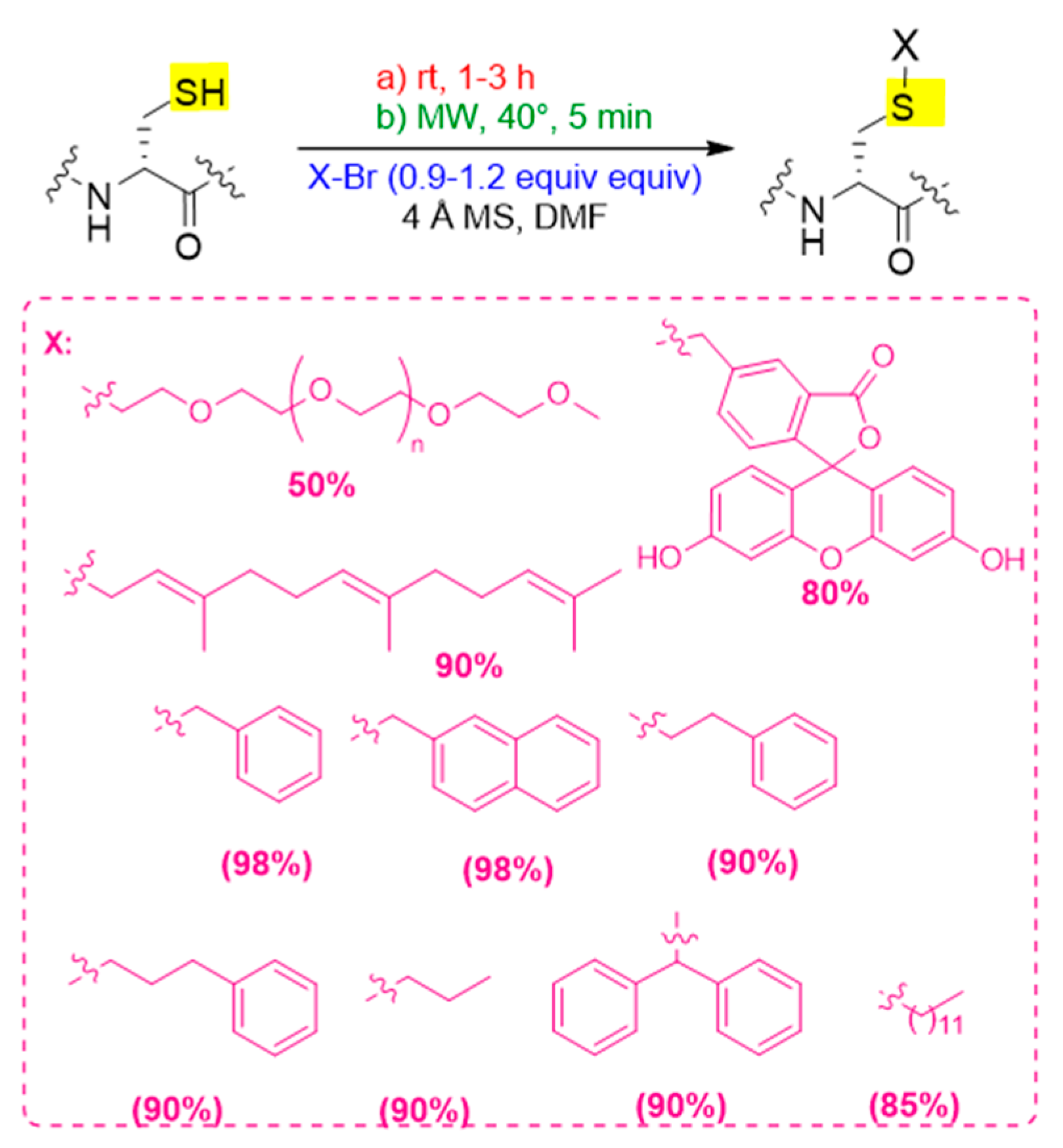
3. Nucleophilic Substitution Reactions Promoted by the Basic Catalysis of Zeolite-Na 4Å
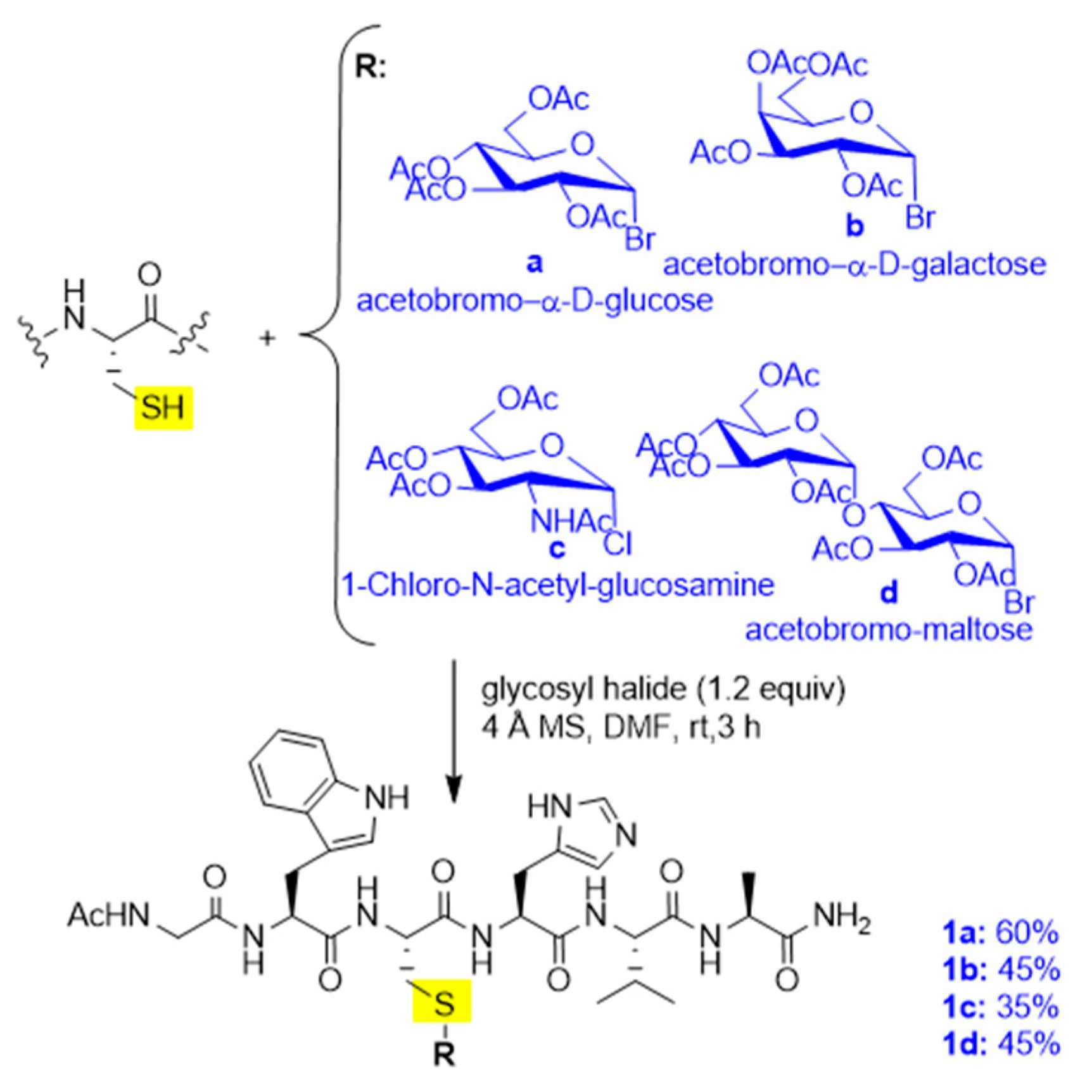
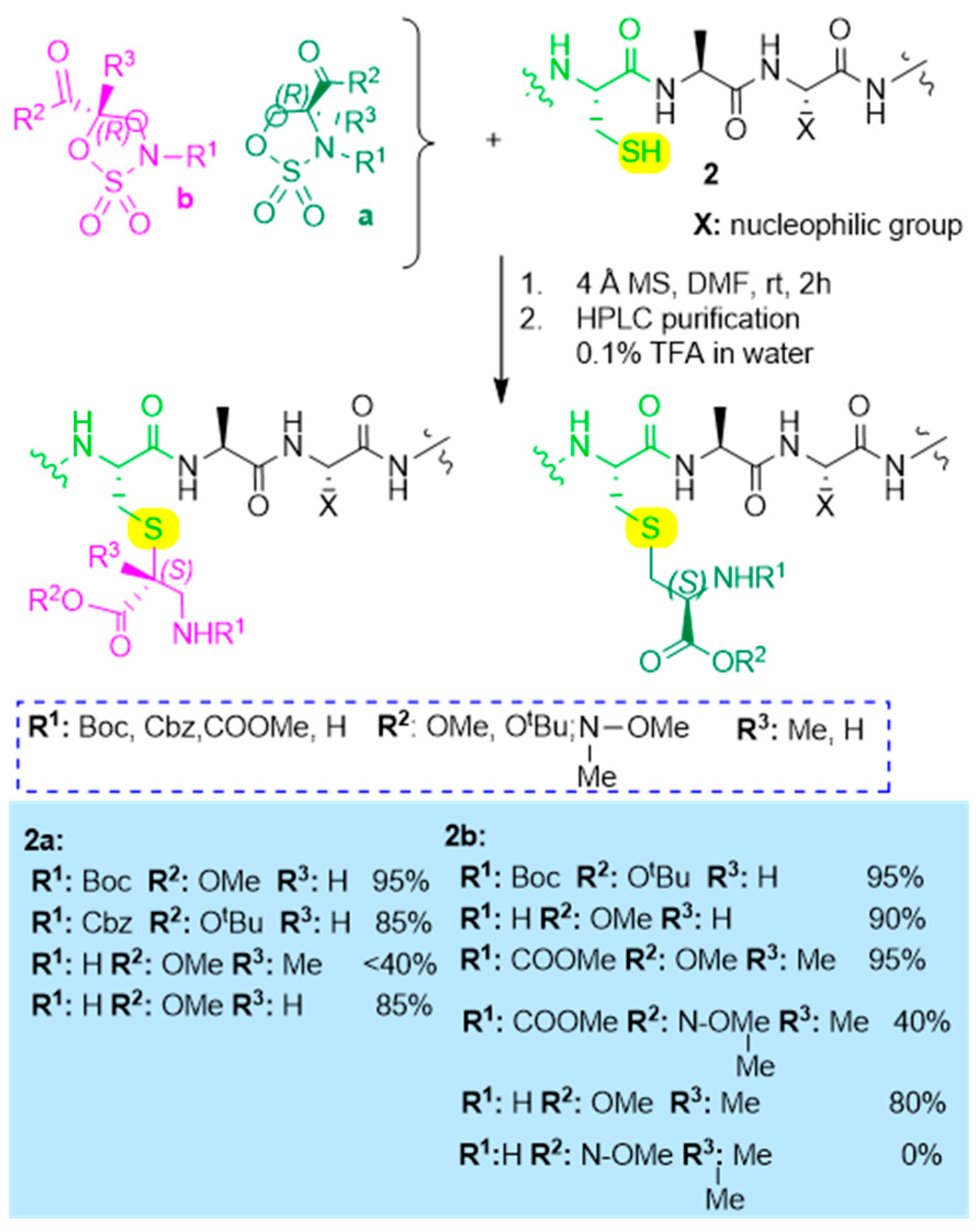
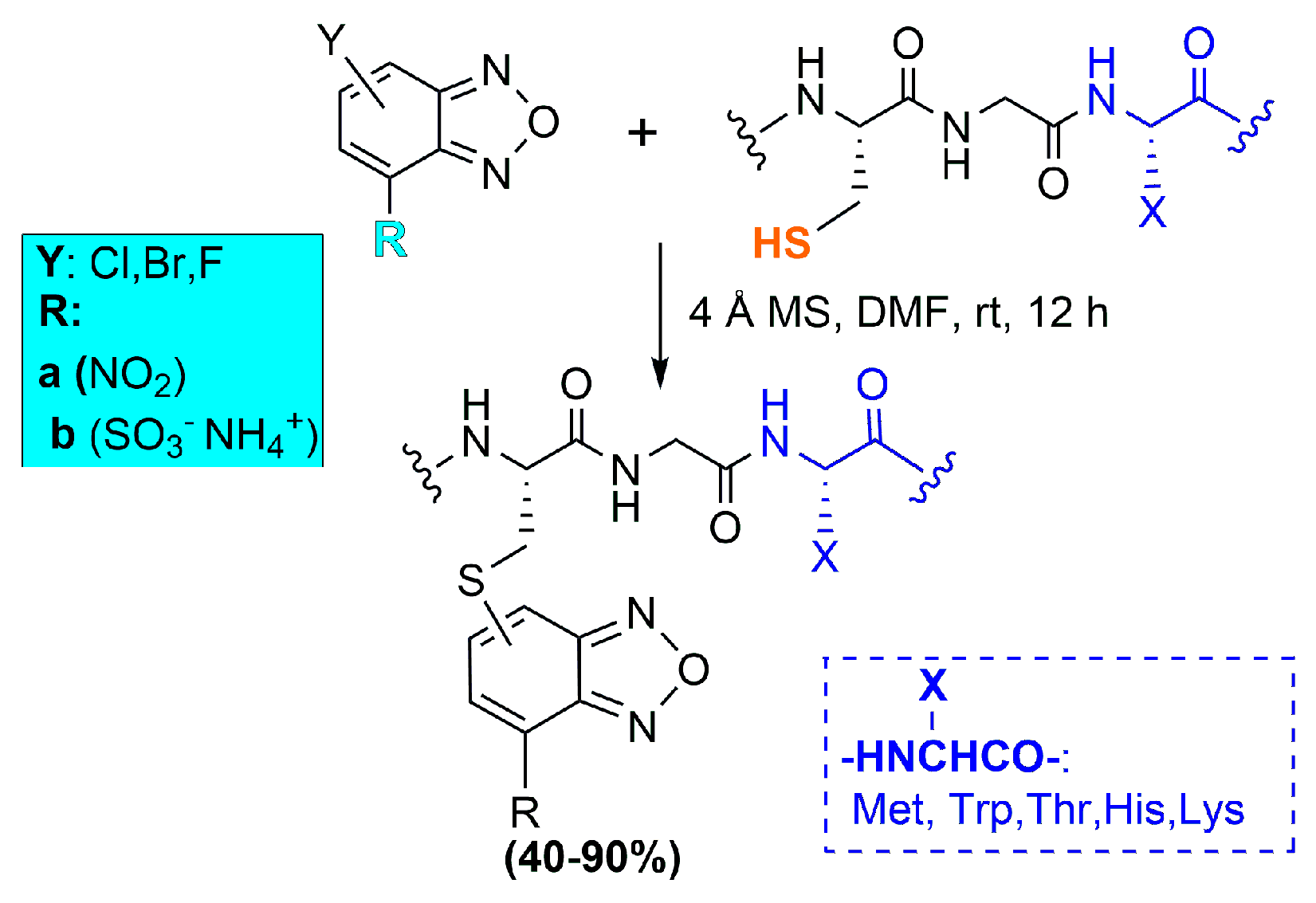
Solution and Solid-Phase Thio-Substitution of Peptide Molecules with Different Moieties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guisnet, M.; Gilson, J. Zeolites for Cleaner Technologies; Imperial College Press: London, UK, 2005; ISBN 1860943292. [Google Scholar] [CrossRef]
- Varna, R.S. Clay and Clay-Supported Reagents in Organic Synthesis. Tetrahedron 2002, 58, 1235–1255. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Yu, J. Applications of zeolites in sustainable chemistry. Chem 2017, 3, 928–949. [Google Scholar] [CrossRef] [Green Version]
- Corma, A. State of the art and future challenges of zeolites as catalysts. J. Catal. 2003, 216, 298–312. [Google Scholar] [CrossRef]
- Li, Y.; Yu, J. Emerging applications of zeolites in catalysis, separation and host–guest assembly. Nat. Rev. Mater. 2021, 6, 1156–1174. [Google Scholar] [CrossRef]
- Martínez, C.; Corma, A. Inorganic molecular sieves: Preparation, modification and industrial application in catalytic processes. Coord. Chem. Rev. 2011, 255, 1558–1580. [Google Scholar] [CrossRef] [Green Version]
- Corma, A. From microporous to mesoporous molecular sieve materials and their use in catalysis. Chem. Rev. 1997, 97, 2373–2419. [Google Scholar] [CrossRef]
- Verdoliva, V.; Saviano, M.; De Luca, S. Zeolites as Acid/Basic Solid Catalysts: Recent Synthetic Developments. Catalysts 2019, 9, 248. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Gao, M.; Yan, W.; Yu, J. Regulation of the Si/Al ratios and Al distributions of zeolites and its impact on properties. Chem. Sci. 2023; ahead of print. [Google Scholar] [CrossRef]
- Kaushik, V.K.; Vijayalakshmi, R.P.; Choudary, N.V.; Bhat, S.G.T. XPS studies on cation exchanged zeolite A. Micropor. Mesopor. Mater. 2002, 51, 139–144. [Google Scholar] [CrossRef]
- Julbe, A.; Drobek, M. Zeolite T-type. In Encyclopedia of Membranes; Drioli, E., Giorno, L., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; ISBN 9783662443231. [Google Scholar] [CrossRef]
- Ugal, J.R.; Hassan, K.H.; Ali, I.H. Preparation of type 4Å zeolite from Iraqi Kaolin: Characterization and properties measurements. J. Assoc. Arab Univ. Basic Appl. Sci. 2010, 9, 2–5. [Google Scholar] [CrossRef] [Green Version]
- McCusker, L.B.; Liebau, F.; Engelhardt, G. Nomenclature of structural and compositional characteristics of ordered microporous and mesoporous materials with inorganic hosts (IUPAC Recommendations 2001). Pure Appl. Chem. 2001, 73, 381–394. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yu, J. New stories of zeolite structures: Their descriptions, determinations, predictions, and evaluations. Chem. Rev. 2014, 114, 7268–7316. [Google Scholar] [CrossRef]
- Rabo, J.A. Unifying Principles in Zeolite Chemistry and Catalysis. Catal. Rev. Sci. Eng. 1981, 23, 293–313. [Google Scholar] [CrossRef]
- Fernández, A.-B.; Marinas, A.; Blasco, T.; Fornésa, V.; Corma, A. Insight into the active sites for the Beckmann rearrangement on porous solids by in situ infrared spectroscopy. J. Catal. 2006, 243, 270–277. [Google Scholar] [CrossRef]
- Muraoka, K.; Sada, Y.; Miyazaki, D.; Chaikittisilp, W.; Okubo, T. Linking synthesis and structure descriptors from a large collection of synthetic records of zeolite materials. Nat. Commun. 2019, 10, 4459. [Google Scholar] [CrossRef] [Green Version]
- Perez-Carbajo, J.; Balestra, S.R.G.; Calero, S.; Merkling, P.J. Effect of lattice shrinking on the migration of water within zeolite LTA. Micropor. Mesopor. Mater. 2020, 293, 109808. [Google Scholar] [CrossRef] [Green Version]
- Van Reeuwijk, L.P. The Thermal Dehydration of Natural Zeolites; Mededelingen Landbouwhogeschool Wageningen; van der Plas, L., Ed.; Communications Agricultural University: Wageningen, The Netherlands, 1974; pp. 74–79. [Google Scholar]
- Osuga, R.; Yokoi, T.; Kondo, J.N. Probing the basicity of lattice oxygen on H-form zeolites using CO2. J. Catal. 2019, 371, 291–297. [Google Scholar] [CrossRef]
- Zheng, A.; Li, S.; Liu, S.-B.; Deng, F. Acidic properties and structure–activity correlations of solid acid catalysts revealed by solid-state NMR spectroscopy. Acc. Chem. Res. 2016, 49, 655–663. [Google Scholar] [CrossRef]
- Loewenstein, W. The distribution of aluminum in the tetrahedra of silicates and aluminates. Am. Mineral. J. Earth Planet. Mater. 1954, 39, 92–96. [Google Scholar]
- Pauling, L. The principles determining the structure of complex ionic cristals. J. Am. Chem. Soc. 1929, 51, 1010–1026. [Google Scholar] [CrossRef]
- George, J.; Waroquiers, D.; Di Stefano, D.; Petretto, G.; Rignanese, G.-M.; Hautier, G. The Limited Predictive Power of the Pauling Rules. Chem. Angew. Int. Ed. 2020, 59, 7273–7618. [Google Scholar] [CrossRef] [Green Version]
- Verdoliva, V.; Saviano, M.; De Luca, S. Zeolites employed as basic catalyst for nucleophilic substitution reactions: An analysis of the adopted approach and hypothesized new perspectives. Inorg. Chim. Acta 2021, 528, 120630. [Google Scholar] [CrossRef]
- Calce, E.; Leone, M.; Monfregola, L.; De Luca, S. Chemical Modifications of Peptide Sequences via S-Alkylation Reaction. Org. Lett. 2013, 15, 5354–5357. [Google Scholar] [CrossRef] [PubMed]
- Calce, E.; De Luca, S. Microwave heating in peptide side chain modification via cysteine alkylation. Amino Acids 2016, 48, 2267–2271. [Google Scholar] [CrossRef] [PubMed]
- Calce, E.; Leone, M.; Monfregola, L.; De Luca, S. Lipidated peptides via post-synthetic thioalkylation promoted by molecular sieves. Amino Acids 2014, 46, 1899–1905. [Google Scholar] [CrossRef]
- Calce, E.; Leone, M.; Mercurio, F.A.; Monfregola, L.; De Luca, S. Solid-Phase S-Alkylaion Promoted by Molecular Sieves. Org. Lett. 2015, 17, 5646–5649. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Ocàriz, V.; Compañòn, I.; Aydillo, C.; Castro-Loṕez, J.; Jimènez-Barbero, J.; Hurtado-Guerrero, R.; Avenoza, A.; Zurbano, M.M.; Peregrina, J.M.; Busto, J.H.; et al. Design of α-S-neoglycopeptides derived from MUC1 with a flexible and solvent-exposed sugar moiety. J. Org. Chem. 2016, 81, 5929–5941. [Google Scholar] [CrossRef] [Green Version]
- Calce, E.; Digilio, G.; Menchise, V.; Saviano, M.; De Luca, S. Chemoselective Glycosylation of Peptides through S-Alkylation Reaction. Chem. Eur. J. 2018, 24, 6231–6238. [Google Scholar] [CrossRef]
- De Luca, S.; Digilio, G.; Verdoliva, V.; Saviano, M.; Menchise, V.; Tovillas, P.; Jiménez-Osés, G.; Peregrina, J.M. A Late-Stage synthetic approach to lanthionine-containing peptides via S-alkylation on cyclic sulfamidates promoted by molecular sieves. Org. Lett. 2018, 20, 7478–7482. [Google Scholar] [CrossRef]
- De Luca, S.; Digilio, G.; Verdoliva, V.; Tovillas, P.; Jiménez-Osés, G.; Peregrina, J.M. Lanthionine Peptides by S-Alkylation with Substituted Cyclic Sulfamidates Promoted by Activated Molecular Sieves: Effects of the Sulfamidate Structure on the Yield. J. Org. Chem. 2019, 84, 14957–14964. [Google Scholar] [CrossRef]
- Verdoliva, V.; Digilio, G.; Saviano, M.; De Luca, S. Thio-conjugation of substituted benzofurazans to peptides: Molecular sieves catalyze nucleophilic attack on unsaturated fused rings. Catal. Sci. Technol. 2021, 11, 1067. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verdoliva, V.; Saviano, M.; De Luca, S. Investigation of the Acid/Basic Sites of Zeolite Trough Some Catalysed Nucleophilic Reactions. Foundations 2023, 3, 72-81. https://doi.org/10.3390/foundations3010008
Verdoliva V, Saviano M, De Luca S. Investigation of the Acid/Basic Sites of Zeolite Trough Some Catalysed Nucleophilic Reactions. Foundations. 2023; 3(1):72-81. https://doi.org/10.3390/foundations3010008
Chicago/Turabian StyleVerdoliva, Valentina, Michele Saviano, and Stefania De Luca. 2023. "Investigation of the Acid/Basic Sites of Zeolite Trough Some Catalysed Nucleophilic Reactions" Foundations 3, no. 1: 72-81. https://doi.org/10.3390/foundations3010008