

A Concise Review of Prodigious Salinomycin and Its Derivatives Effective in Treatment of Breast Cancer: (2012–2022)
 ,
,  ,
,
Abstract
:1. Introduction
1.1. Triple Negative Breast Cancer (TNBC)
1.2. Epidemiology
1.3. Pathology and Molecular Features
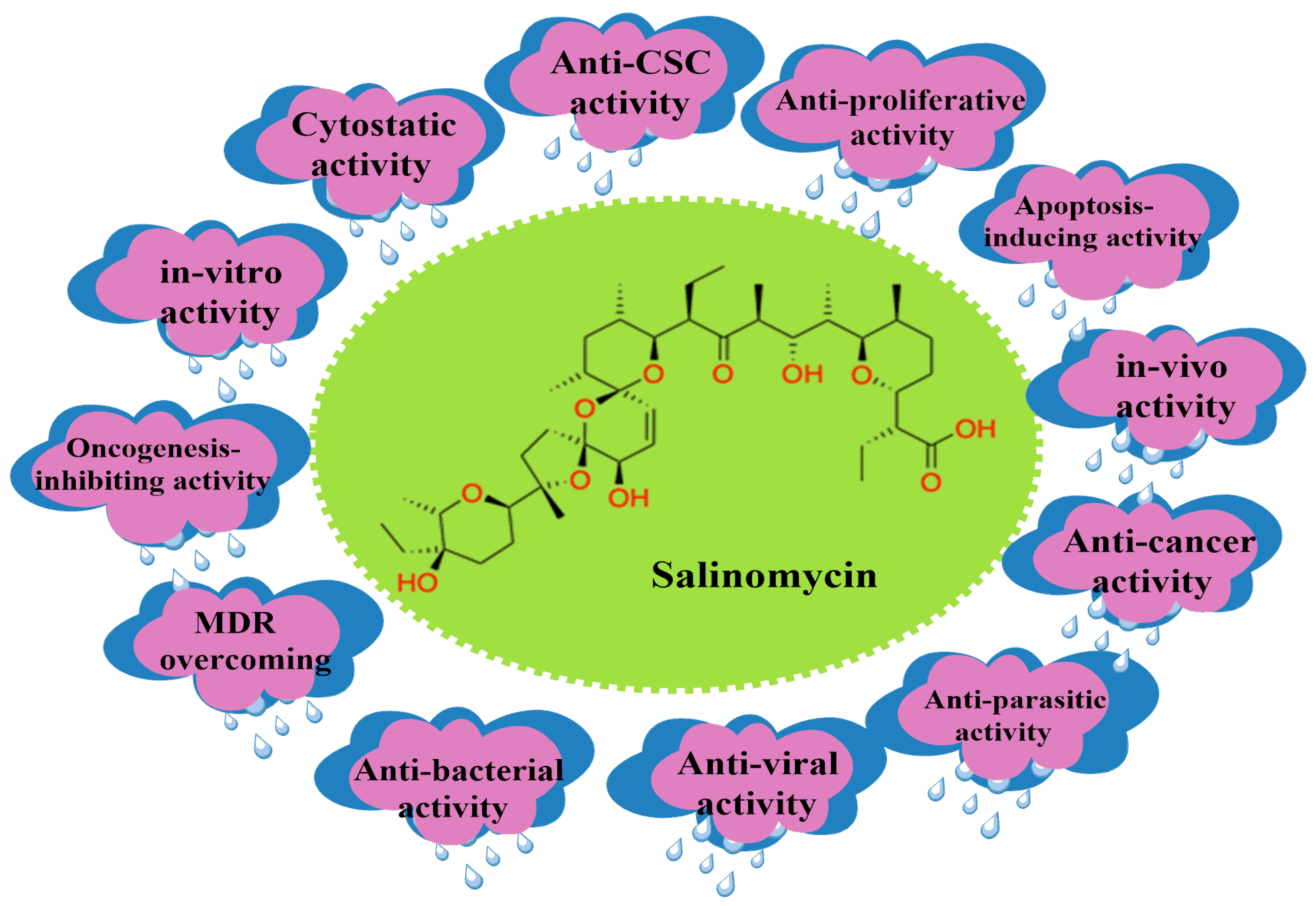
2. Salinomycin (SAL) and Its Bioactivity
2.1. SAL’s Mechanism of Action against CSCs
2.1.1. Apoptosis of CSCs
2.1.2. Interference of ATP-Binding Cassette (ABC) Transporters
2.1.3. Inhibition of Oxidative Phosphorylation and Glycolysis
2.1.4. Polyether Ionophore Effects on the Mitochondria
2.1.5. Induction of Autophagy, ROS, and DNA Damage
2.1.6. Endoplasmic Reticulum Stress
2.1.7. Inhibition of the Wnt Signaling Cascade
2.1.8. Sequestration of Iron in the Lysosome
2.1.9. Intracellular Binding Targets
2.1.10. Differentiation of CSCs and SAL Bioactivity
3. Clinical History
3.1. In Vitro Studies
3.2. In Vivo Studies
4. SAL’s Toxicological and Pharmacological Properties and Clinical Applications
4.1. In Human Cells
4.2. In Animals
4.3. Pharmacokinetics and Pharmacodynamics
4.4. Clinical Studies
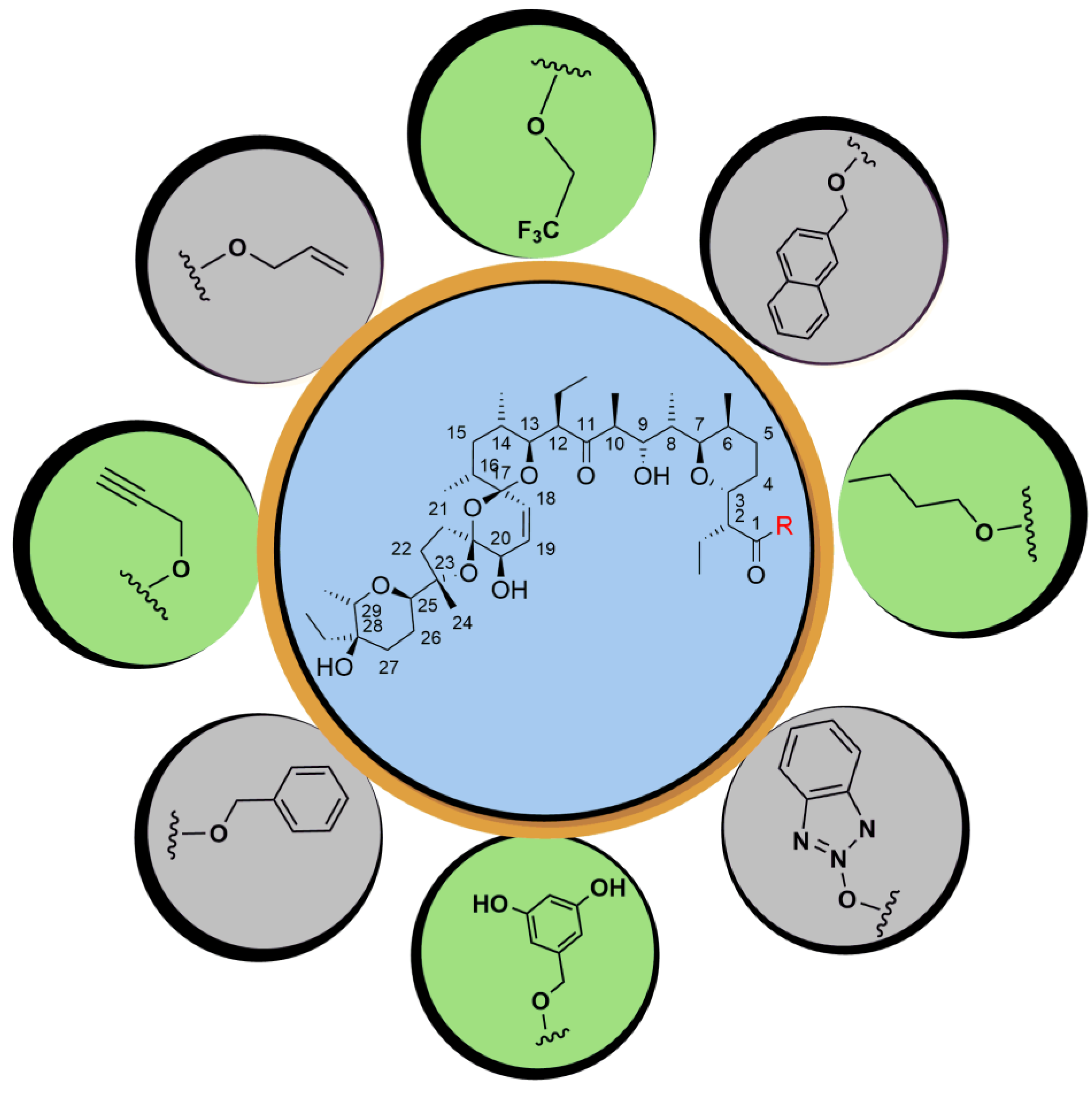
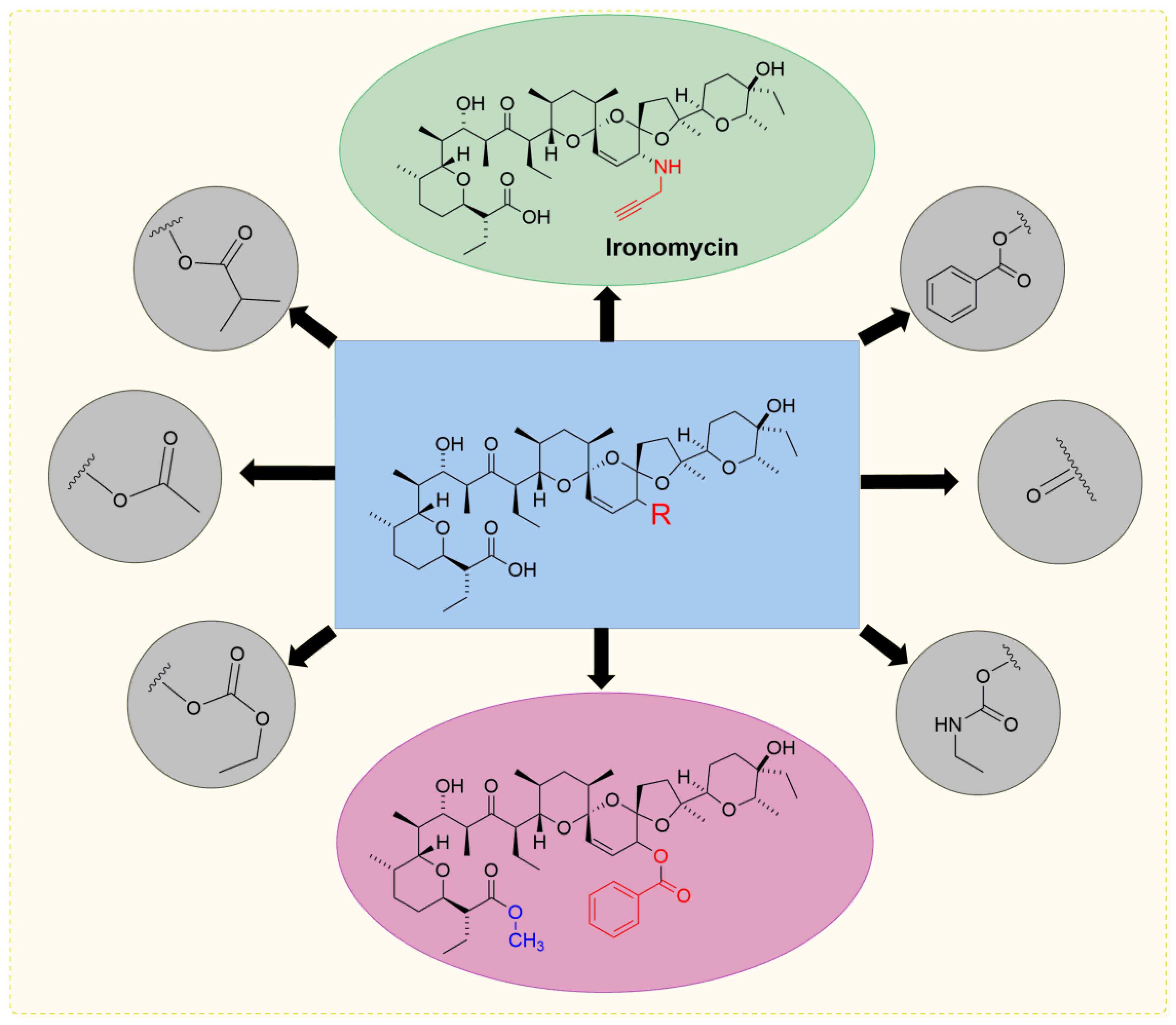
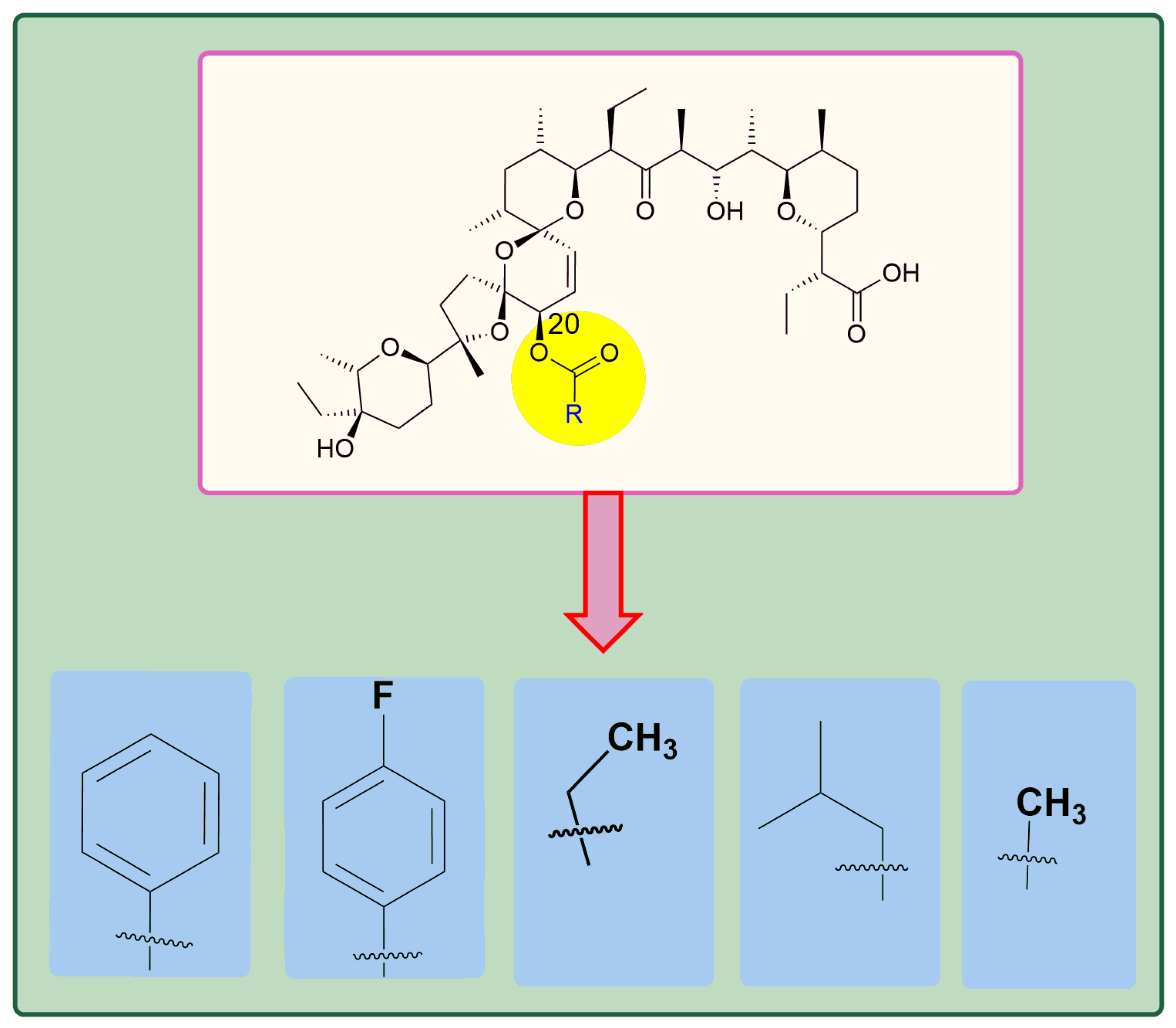
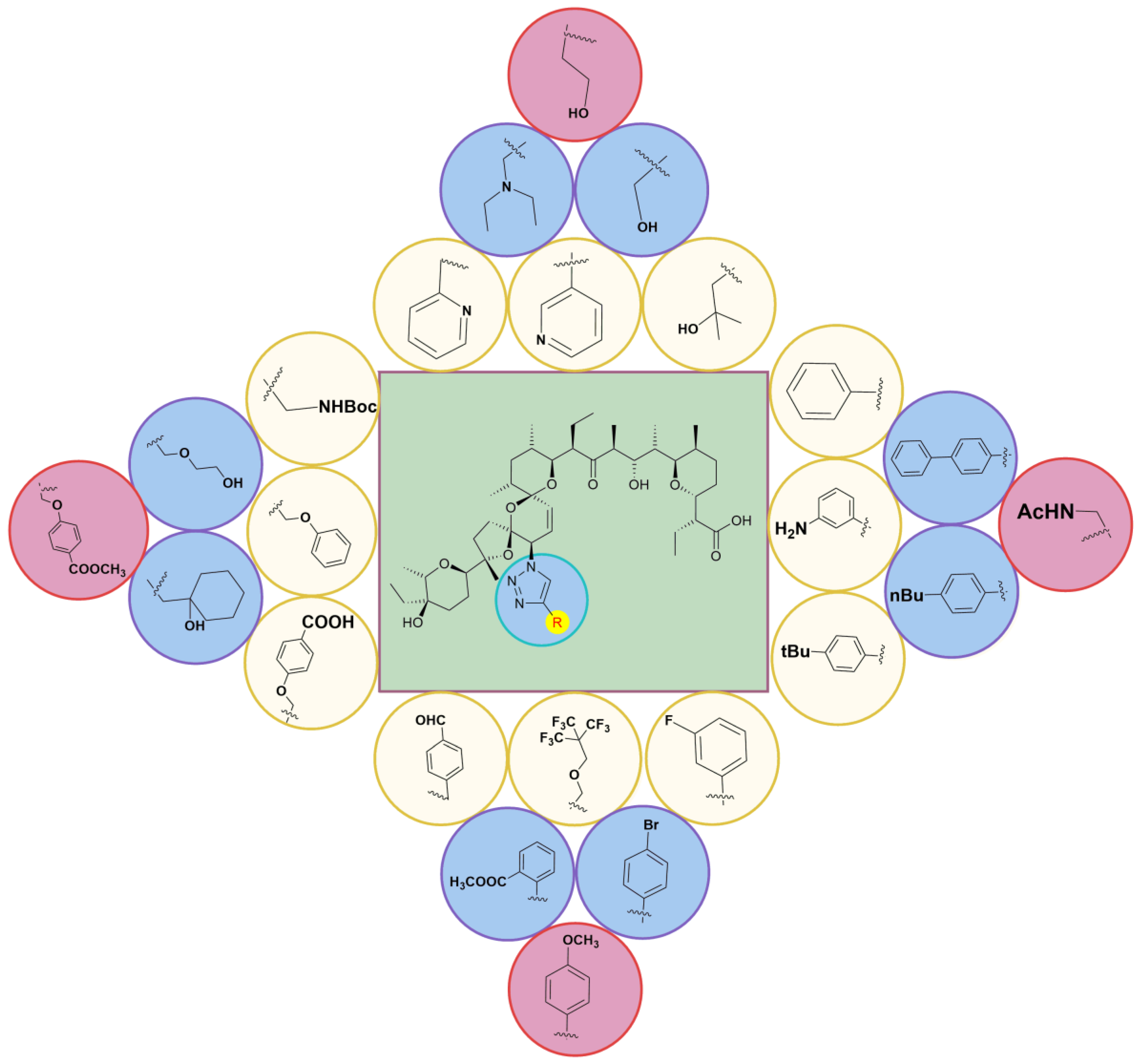
5. Physicochemical Analysis of SAL and Its Analogs
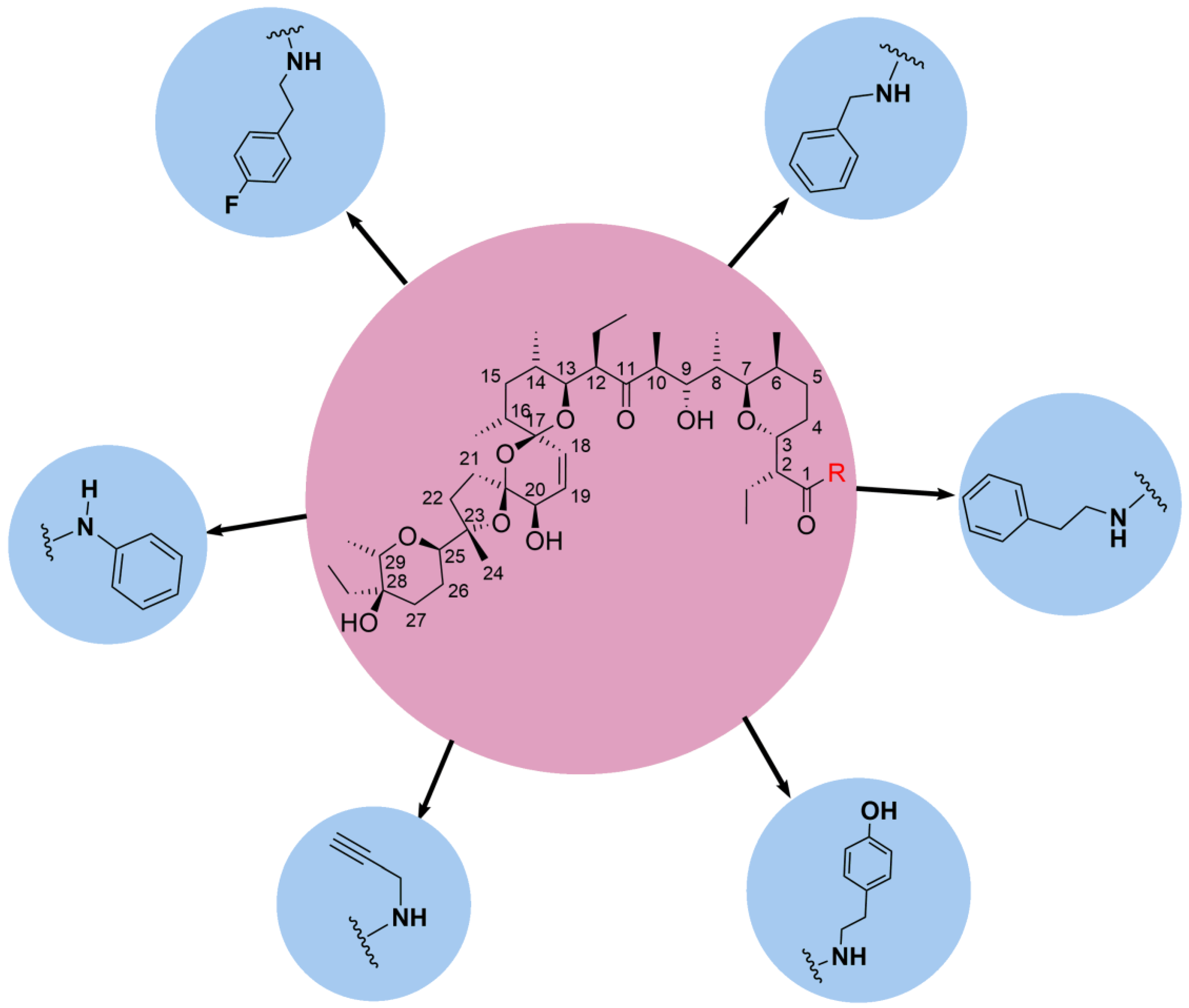
5.1. C1 Analogs
5.2. Chemistry of C20 Analogs
5.3. SAL Double-Modified Analogs
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qi, D.; Liu, Y.; Li, J.; Huang, J.H.; Hu, X.; Wu, E. Salinomycin as a potent anticancer stem cell agent: State of the art and future directions. Med. Res. Rev. 2022, 42, 1037–1063. [Google Scholar] [CrossRef]
- Von Suskil, M.; Sultana, S.N.; Elbezanti, W.O.; Al-Odat, O.S.; Chitren, R.C.; Tiwari, A.K.; Challagundla, K.B.; Srivastava, S.K.; Jonnalagadda, S.C.; Alpdogan-Budak, T.; et al. Bruton’s Tyrosine Kinase Targeting in Multiple Myeloma. Int. J. Mol. Sci. 2021, 22, 5707. [Google Scholar] [CrossRef]
- Hermann, P.C.; Sainz, B., Jr. Pancreatic cancer stem cells: A state or an entity. Semin. Cancer Bio. 2018, 53, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Nat. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [PubMed]
- Papaccio, F.; Paino, F.; Regad, T.; Papaccio, G.; Desiderio, V.; Tirino, V. Concise Review: Cancer Cells, Cancer Stem Cells and Mesenchymal Stem Cells: Influence in Cancer Development. Stem. Cells Transl. Med. 2017, 6, 2115–2125. [Google Scholar] [CrossRef]
- Nayak, A.; Warrier, N.M.; Kumar, P. Cancer Stem Cells and the Tumor Microenvironment: Targeting the Critical Crosstalk through Nanocarrier Systems. Stem Cell Rev. Rep. 2022, 18, 2209–2233. [Google Scholar] [CrossRef] [PubMed]
- Waghray, M.; Yalamanchili, M.; Dziubinski, M.; Zeinali, M.; Erkkinen, M.; Yang, H.; Schradle, K.A.; Urs, S.; Pasca Di Magliano, M.; Welling, T.H.; et al. GM-CSF Mediates Mesenchymal-Epithelial crosstalk in Pancreatic Cancer. Cancer Discov. 2016, 6, 886–899. [Google Scholar] [CrossRef]
- Raggi, C.; Mousa, H.S.; Correnti, M.; Sica, A.; Invernizz, P. Cancer stem cells and tumor-associated macrophages: A roadmap for multitargeted strategies. Oncogene 2016, 35, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.H.; Swathi, Y.; Tan, S.; Goh, J.; Seishima, R.; Murakami, K.; Oshima, M.; Tsuji, T.; Phuah, P.; Tan, L.T.; et al. AQP5 enriches for stem cells and cancer origins in the distal stomach. Nature 2020, 578, 437–443. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells cancer and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Lytle, N.K.; Barber, A.G.; Reya, T. Stem cell fate in Cancer growth, progression, and therapy resistance. Nat. Rev. Cancer 2018, 18, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem. Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Dosch, J.S.; Ziemke, E.K.; Shettigar, A.; Rehemtulla, A.; Sebolt-Leopold, J.S. Cancer Stem Marker Phenotypes Are Reversible and Functionally Homogeneous in a Preclinical Model of Pancreatic Cancer. Cancer Res. 2015, 75, 4582–4592. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qi, D.; Hsieh, T.C.; Huang, J.H.; Wu, J.M.; Wu, E. Trailblazing perspectives on targeting breast cancer stem cells. Pharmacol. Ther. 2021, 223, 107800. [Google Scholar] [CrossRef]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targetting signaling pathways and the immune microenvironment of cancer stem cells-a clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targetting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwsser, K.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef]
- Arfaoui, A.; Rioualen, C.; Azzoni, V.; Pinna, G.; Finetti, P.; Wicinski, J.; Josselin, E.; Macario, M.; Castellano, R.; Léonard-Stumpf, C.; et al. A genome-wide RNAi screen reveals essential therapeutic targets of breast cancer stem cells. EMBO Mol. Med. 2019, 11, 9930. [Google Scholar] [CrossRef]
- Elbezanti, W.O.; Al-Odat, O.S.; Chitren, R.; Singh, J.K.; Srivastava, S.K.; Gowda, K.; Amin, S.; Robertson, G.P.; Nemmara, V.V.; Jonnalagadda, S.C.; et al. Development of novel bruton’s tyrosine kinase inhibitor that exerts anti-cancer activities potentiates response of chemotherapeutic agents in multiple myeloma stem-like cells. Front. Pharmacol. 2022, 13, 1–18. [Google Scholar] [CrossRef]
- Singh, S.; Numan, A.; Maddiboyina, B.; Arora, S.; Riadi, Y.; Md, S. The emerging role of immune checkpoint inhibitors in the treatment of triple-negative breast cancer. Drug Discov. Today 2021, 26, 1721–1727. [Google Scholar] [CrossRef]
- Ataollahi, M.R.; Sharifi, J.; Paknahad, M.R.; Paknahad, A. Breast cancer and associated factors: A review. J. Med. Life 2015, 8, 6–11. [Google Scholar] [PubMed]
- Sharma, G.N.; Dave, R.; Sanadya, J.; Sharma, P.; Sharma, K.K. Various types and management of breast cancer: An overview. J. Adv. Pharm. Technol. Res. 2010, 1, 109–126. [Google Scholar]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 6, 394–424. [Google Scholar] [CrossRef]
- Jin, X.; Mu, P. Targeting breast cancer metastasis. Breast Cancer Basic Clin. Res. 2015, 9, 23–34. [Google Scholar] [CrossRef]
- Gupta, G.P.; Massagué, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef]
- Narod, S.A.; Iqbal, J.; Miller, A.B. Why have breast cancer mortality rates declined? J. Cancer Policy 2015, 5, 8–17. [Google Scholar] [CrossRef]
- Urbaniak, A.; Reed, M.R.; Fil, D.; Moorjani, A.; Heflin, S.; Antoszczak, M.; Sulik, M.; Huczynski, A.; Kupsik, M.R.; Eoff, L.; et al. Single and double modified salinomycin analogs target stem-like cells in 2D and 3D breast cancer models. J. Biomed. Pharmacother. 2021, 141, 111815. [Google Scholar] [CrossRef] [PubMed]
- Ismail-Khan, R.; Bui, M. A Review of Triple-Negative Breast Cancer. Cancer Control 2010, 17, 173–176. [Google Scholar] [CrossRef]
- Kaplan, H.G.; Malmgren, J.A.; Atwood, M.K. Impact of triple negative phenotype on breast cancer prognosis. In Proceedings of the 29th Annual San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 14–17 December 2006. [Google Scholar]
- Kuran, D.; Flis, S.; Antoszczak, M.; Piskorek, M.; Huczynski, A. Ester derivatives of Salinomycin efficiently eliminate breast cancer cells via ER-stress-induced apoptosis. Eur. J. Pharmacol. 2021, 893, 173824. [Google Scholar] [CrossRef]
- Rakha, E.A.; Reis-Filho, J.S.; Ellis, I.O. Basal-Like Breast Cancer: A Critical Review. J. Clin. Oncol. 2008, 26, 2568–2581. [Google Scholar] [CrossRef]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S.; et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. J. Am. Med. Assoc. 2006, 295, 2492–2502. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffery, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Mai, T.T.; Hamaï, A.; Hienzsch, A.; Cañeque, T.; Müller, S.; Wicinski, J.; Cabaud, O.; Leroy, C.; David, A.; Acevedo, V.; et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 2017, 9, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Antoszczak, M.; Urbaniak, A.; Delgado, M.; Maj, E.; Borgstrom, B.; Wietrzyk, J.; Huczynski, A.; Yua, Y.; Chambers, T.C. Biological activity of doubly modified salinomycin analogs-Evaluation in vitro and ex vivo. Eur. J. Med. Chem. 2018, 156, 510–523. [Google Scholar] [CrossRef]
- Haruyasu, K.; Otake, N.; Yonehara, H.; Sato, S.; Saito, Y. The structure of salinomycin, a new member of the polyether antibiotics. Tetrahedron Lett. 1973, 14, 4955–4958. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Shibuya, M.; Sugawara, H.; Kawaguchi, O.; Hirose, C. Salinomycin, a new polyether antibiotic. J. Antibio. 1974, 27, 814–821. [Google Scholar] [CrossRef]
- Piperno, A.; Marrazzo, A.; Scala, A.; Rescifina, A. Chemistry and Biology of Salinomycin and its Analogues. Ital. J. Chem. Sci. 2016, 19, 177–209. [Google Scholar] [CrossRef]
- Borgstrom, B.; Xiaoli, H.; Posta, M.; Hegardt, C.; Ordesson, S.; Strand, D. Synthetic modification of saliomycin: Selective O-acylation and biological evaluation. Chem. Commun. 2013, 49, 9944–9946. [Google Scholar] [CrossRef]
- Naujokat, C.; Steinhart, R. Salinomycin as a drug for targeting human cancer stem cells. J. Biomed. Biotech. 2012, 950658. [Google Scholar] [CrossRef]
- Jangamreddy, J.R.; Jain, M.V.; Hallbeck, A.L.; Roberg, K.; Lotfi, K.; Łos, M.J. Glucose starvation-mediated inhibition of salinomycin induced autophagy cancer cell specific cell death. Oncotarget 2015, 6, 10134–10145. [Google Scholar] [CrossRef] [PubMed]
- Mitani, M.; Yamanishi, T.; Miyazaki, Y.; Otake, N. Salinomycin effects on mitochondrial ion translocation and respiration. Antimicrob. Agents Chemother. 1976, 9, 655–660. [Google Scholar] [CrossRef]
- Zhao, S.J.; Wang, X.J.; Wu, Q.J.; Liu, C.; Li, D.A.; Fu, X.T.; Zhang, H.F.; Shao, L.R.; Sun, J.Y.; Sun, B.L.; et al. Induction of G1 Cell Cycle Arrest in Human Glioma Cells by Salinomycin Through Triggering ROS-Mediated DNA Damage In Vitro and In Vivo. Neurochem. Res. 2017, 42, 997–1005. [Google Scholar] [CrossRef]
- Huang, X.; Borgstro, B.; Kempengren, S.; Persson, L.; Hegardt, C.; Strand, D.; Oredsson, S. Breast cancer stem cell selectivity of synthetic nanomolar-active salinomycin analogs. BMC Cancer 2016, 16, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Borgström, B.; Stegmayr, J. The Molecular Basis of Inhibition of Stemlike Cancer Cells by salinomycin. ACS Cent. Sci. 2018, 4, 760–767. [Google Scholar] [CrossRef]
- Li, B.; Wu, J.; Tang, L.; Lian, X.; Li, Z.; Duan, W.; Qin, T.; Zhao, X.; Hu, Y.; Zhang, C.; et al. Synthesis and anti-tumor activity evaluation of salinomycin C20-O-alkyl/benzyl oxime derivatives. Org. Biomol. Chem. 2022, 20, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca2+ homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef]
- Lu, D.; Choi, M.Y.; Yu, J.; Castro, J.E.; Kipps, T.J.; Carson, D.A. Salinomycin inhibits Wnt signaling and selectively induces apoptosis in chronic lymphocytic leukemia cells. Proc. Natl. Acad. Sci. USA 2011, 108, 13253–13257. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, L.; Xiong, Y. Salinomycin exerts anti-colorectal cancer activity by targeting Beta-catenin/T-Cell factor complex. Br. J. Pharmacol. 2019, 176, 3390–3406. [Google Scholar] [CrossRef]
- Hamaï, A.; Cañeque, T.; Müller, S. An iron hand over cancer stem cells. Autophagy 2017, 13, 1465–1466. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhou, S.; Qi, D.; Xiang, S.H.; Wong, E.T.; Wang, X.; Fonkem, E.; Hsieh, T.C.; Yang, J.; Kirmani, B.; et al. Nucleolin is a Functional Binding Protein for Salinomycin in Neuroblastoama Stem Cells. J. Am. Chem. Soc. 2019, 141, 3613–3622. [Google Scholar] [CrossRef]
- Versini, A.; Colombeau, L.; Hinzsch, A.; Gaillet, C.; Retailleau, P.; Debieu, S.; Muller, S.; Caneque, T.; Rodriguez, R. Salinomycin Derivatives Kill Breast Cancer Stem Cells by Lysosomal Iron Targeting. Chem-A. Eur. J. 2020, 26, 7416–7424. [Google Scholar] [CrossRef] [PubMed]
- Fähling, M.; Steege, A.; Perlewitz, A.; Nafz, B.; Mrowka, R.; Persson, P.B.; Thiele, B.J. Role of nucleolin in postranscriptional control of MMP-9 expression. Biochim. Biophys. Acta 2005, 1731, 32–40. [Google Scholar] [CrossRef]
- Bhatia, S.; Reister, S.; Mahotka, C.; Meisel, R.; Borkhardt, A.; Grinstein, E. Control of AC133/CD133 and impact on human hematopoietic progenitor cells through nucleolin. Leukemia 2015, 29, 2208–2220. [Google Scholar] [CrossRef] [PubMed]
- Grinstein, E.; Du, Y.; Santourlidis, S.; Christ, J.; Uhrberg, M.; Wernet, P. Nucleolin Regulates Gene Expression in CD34-positive Hematopoietic Cells. J. Biol. Chem. 2007, 282, 12439–12449. [Google Scholar] [CrossRef]
- Pipeline—Hillstream Biopharma, Inc. Available online: https://hillstreambio.com/pipeline/ (accessed on 12 April 2022).
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1–33. [Google Scholar] [CrossRef]
- Paredes, F.; Williams, H.C.; San Martin, A. Metabolic adaptation in hypoxia and cancer. Cancer Lett. 2021, 502, 133–142. [Google Scholar] [CrossRef]
- An, H.; Kim, J.Y.; Lee, N.; Cho, Y.; Oh, E.; Seo, J.H. Salinomycin possesses anti-tumor activity and inhibits breast cancer stem-like cells via an apoptosis-independent pathway. Biochem. Biophys. Res. Commun. 2015, 466, 696–703. [Google Scholar] [CrossRef]
- Tefas, L.R.; Barbalata, C.; Tefas, C.; Tomuta, I. Salinomycin-Based drug delivery system: Overcoming the hurdles in cancer Therapy. Pharmaceutics 2021, 13, 1120. [Google Scholar] [CrossRef]
- Magrath, J.W.; Raney, W.R.; Kim, Y. In vitro demonstration of salinomycin as a novel chemotherapeutic agent for the treatment of Sox2-positive glioblastoma cancer stem cells. Oncol. Rep. 2020, 44, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Yin, X.; Zhou, X.; Niu, X.; Wang, Y.; Su, M. Salinomycin-Loaded High-Density Lipoprotein Exerts Promising Anti-Ovarian Cancer Effects by Inhibiting Epithelial-Mesenchymal Transition. Int. J. Nanomed. 2022, 17, 4059–4071. [Google Scholar] [CrossRef] [PubMed]
- Dewangan, J.; Srivastava, S.; Srikanta, K.R. Salinomycin Inhibits Breast Cancer Progression via Targeting HIF-1α/VEGF Mediated Tumor Angiogenesis in Vitro and in Vivo. Biochem. Pharmacol. 2019, 164, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; He, L.; Dai, W.Q.; Xu, Y.P.; Wu, D.; Lin, C.L.; Wu, S.M.; Cheng, P.; Zhang, Y.; Shen, M.; et al. Salinomycin inhibits proliferation and induces apoptosis of hepatocellular carcinoma in vitro and in vivo. E-Bio Med. (PLoS ONE) 2012, 7, 506–538. [Google Scholar] [CrossRef]
- Wang, F.; Zheng, Z.; Guan, J.; Qi, D.; Zhou, S.; Shen, X.; Wang, F.; Wenkert, D.; Kirmani, B.; Solouki, T.; et al. Identification of a panel of genes as a prognostic biomarker for glioblastoma. E-Bio Med. (PLoS ONE) 2018, 37, 68–77. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, F.; Zhang, Y.; Austin, J. Salinomycin Suppresses PDGFRβ, MYC, and Notch Signaling in Human Medulloblastoma. Pharmacol. Ther. 2014, 3, 1020. [Google Scholar]
- Boehmerle, W.; Endres, M. salinomycin induces calpain and cytochrome-c mediated neuronal cell death. Cell Death Dis. 2011, 2, e168. [Google Scholar] [CrossRef]
- Fuchs, D.; Heinold, A.; Opelz, G.; Daniel, V.; Naujokat, C. Salinomycin induces apoptosis and overcomes apoptosis resistance in human cancer cells. J. Biochem. Biophys. Res. Commun. 2009, 390, 743–749. [Google Scholar] [CrossRef]
- Scherzed, A.; Hackenberg, S.; Froelich, K.; Rak, K.; Technau, A.; Radeloff, A.; Nöth, U.; Koehler, C.; Hagen, R.; Kleinsasser, N. Effects of salinomycin on human bone marrow-derived mesenchymal stem cells in vitro. Toxicol. Lett. 2013, 218, 207–214. [Google Scholar] [CrossRef]
- Shen, H.; Sun, C.C.; Kang, L.; Tan, X.; Shi, P.; Wang, L.; Liu, E.; Gong, J. Low dose salinomycin inhibits breast cancer metastasis by repolarizing tumor hijacked macrophages toward the M1 phenotype. Eur. J. Pharm. Sci. 2021, 157, 105629. [Google Scholar] [CrossRef]
- Jiang, J.; Li, H.; Qaed, E. Salinomycin, as an autophagy modulator—A new avenue to anticancer: A review. J. Exp. Clin. Cancer Res. 2018, 37, 26. [Google Scholar] [CrossRef] [PubMed]
- Scherzad, A.; Hackenberg, S.; Schramm, C.; Froelich, K.; Ginzkey, C.; Rudolf, H.; Kleinsasser, N. Geno-and cytotoxicity of salinomycin in human nasal mucosa and peripheral blood lymphocytes. Toxicol. Vitro 2015, 29, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.E.; Polverini, P.J.; Kunkel, S.L.; Harlow, L.A.; DiPietro, L.A.; Elner, V.M.; Elner, S.G.; Strieter, R.M. Interleukin-8 as a macrophage-derived mediator of angiogenesis. J. Sci. 1992, 258, 1798–1801. [Google Scholar] [CrossRef]
- Arenberg, D.A.; Kunkel, S.L.; Polverini, P.J.; Glass, M.; Burdick, M.D.; Strieter, R.M. Inhibition of interleukin-8 reduces tu- morigenesis of human non-small cell lung cancer in SCID mice. J. Clin. Investig. 1996, 97, 2792–2802. [Google Scholar] [CrossRef] [PubMed]
- Szkudlarek-Mikho, M.; Saunders, R.A.; Yap, S.F.; Ngeow, Y.F.; Chin, K.V. Salinomycin, a polyether ionophoric antibiotic, inhibits adipogenesis. Biochem. Biophys. Res. Commun. 2012, 428, 487–493. [Google Scholar] [CrossRef]
- Scherzad, A.; Hackenberg, S.; Froelich, K.; Rak, K.; Hagen, R.; Taeger, J.; Bregenzer, M.; Kleinsasser, N. Chronic exposure of low dose salinomycin inhibits MSC migration capability in vitro. Biomed. Rep. 2016, 4, 325–330. [Google Scholar] [CrossRef]
- Potter, L.M.; Blake, J.P.; Blair, M.E.; Bliss, B.A.; Denbow, D.M. Salinomycin toxicity in turkeys. Poult. Sci. 1986, 65, 1955–1959. [Google Scholar] [CrossRef]
- Chaudhari, P.J.; Bari, S.B.; Surana, S.J.; Nagar, A. Discovery and Anticancer Activity of Novel 1,3,4-Thiadiazole- and Aziridine-Based Indolin-2-ones via in silico Design Followed by Supramolecular Green Synthesis. ACS Omega 2022, 7, 17270–177294. [Google Scholar] [CrossRef]
- Salinomycin toxicity causes deaths of calves on two Scottish dairy farms. Vet. Rec. 2012, 170, 118–121. [CrossRef]
- Holliman, A.; Howie, F.; Payne, J.; Scholes, S. Salinomycin toxicity in dairy calves. Vet. Rec. 2011, 169, 561. [Google Scholar] [CrossRef]
- Plumlee, K.H.; Johnson, B.; Galey, F.D. Acute salinomycin toxicosis of pigs. J. Clin. Investig. 1995, 7, 419–420. [Google Scholar] [CrossRef] [PubMed]
- Ashrafihelan, J.J.; Eisapour, H.; Erfani, A.M.; Kalantary, A.A.; Amoli, J.S.; Mozafari, M. High mortality due to accidental salinomycin intoxication in sheep. Interdiscip. Toxicol. 2014, 7, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Rudrapal, M.; Issahaku, A.; Agoni, C.; Nagar, A.; Nedale, A.B.; Lokwani, D.K. In silico screening of phytopolyphenolics for the identification of bioactive compounds as novel protease inhibitors effective against SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 40, 10437–10453. [Google Scholar] [CrossRef]
- Van der Linde-Sipman, J.S.; Van den Ingh, T.S.G.A.M.; Van Nes, J.J.; Verhagen, H.; Kersten, J.G.T.M.; Beynen, A.C.; Plekkringa, R. Salinomycin-induced polyneuropathy in cats: Morphologic and epidemiologic data. Vet. Pathol. 1999, 36, 152–156. [Google Scholar] [CrossRef]
- Shalaby, M.A.; el-Sanousi, A.A.; Yehia, M.M.; Naser, A.; Reda, I.M. The effect of salinomycin on the immune response of chicks. Dtsch. Tierarztl. Wochenschr. 1993, 5, 182–185. [Google Scholar]
- Shuang, Z.; Wang, F.; Wong, E.T.; Fonkem, E.; Hsieh, T.-C.; Wu, J.M.; Wu, E. Salinomycin: A novel anti-cancer agent with known anti-coccidial activities. Curr. Med. Chem. 2013, 20, 4095–4101. [Google Scholar] [CrossRef]
- Ojo, O.O.; Bhadauria, S.; Rath, S.K. Dose-dependent adverse effects of salinomycin on male reproductive organs and fertility in mice. E-Bio Med. (PLoS ONE) 2013, 8, e69086, Erratum in 2019, 14, e0226872. [Google Scholar] [CrossRef] [PubMed]
- Resham, K.; Patel, P.N.; Thummuri, D.; Guntuku, L.; Shah, V.; Bambal, R.B.; Naidu, V.G.M. Preclinical drug metabolism and pharmacokinetics of salinomycin, a potential candidate for targeting human cancer stem cells. Chem. Biol. Interact. 2015, 240, 146–152. [Google Scholar] [CrossRef]
- Paulus, E.F.; Kurz, M.; Matter, H.; Vertesy, L. Solid State Solution Structure of the Salinomycin-Sodium Complex: Stabilization of Different Conformers for an Ionophore in Different Environments. J. Am. Chem. Soc. 1998, 120, 8209. [Google Scholar] [CrossRef]
- Urbaniaka, A.; Delgado, M.; Antoszczak, M.; Huczyński, A.; Chambers, T.C. Salinomycin derivatives exhibit activity against primary acute lymphoblastic leukemia (ALL) cells in vitro. Biomed. Pharmacother. 2018, 99, 384–390. [Google Scholar] [CrossRef]
- Li, B.; Wu, J.; Zhang, W.; Zhongwen, L.; Chen, G.; Zhou, Q.; Wu, S. Synthesis and biological activity of salinomycin-hydroxamic acid conjugates. Bioorg. Med. Chem. Lett. 2017, 27, 1624–1626. [Google Scholar] [CrossRef]
- Borgstrom, B.; Huang, X.; Hegardt, C.; Oredsson, S.; Strand, D. Structure-activity relationships in salinomycin: Cytotoxicity and phenotype selectivity of semi-synthetic derivatives. Chem.–A Eur. J. 2017, 23, 2077–2083. [Google Scholar] [CrossRef] [PubMed]
- Czerwonka, D.; Urbaniak, A.; Sobczak, S.; Pina-Ovidedo, S.; Chambers, T.C.; Antoszczak, M.; Huczynski, A. Synthesis and anticancer activity of tertiary amides of salinomycin and their C20-oxo analogues. Chem. Med. Chem. 2020, 15, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wu, J.; Li, B.; Wu, H.; Wang, L.; Hao, J.; Wu, S.; Zhou, Q. Structure-activity & structure-toxicity relationship study of salinomycin diastereoisomers and their benzoylated derivatives. Org. Biomol. Chem. 2016, 14, 2840–2845. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wu, J.; Li, B.; Xia, J.; Wu, H.; Wang, L.; Hao, J.; Zhou, Q.; Wu, S. Synthesis and biological activity evaluation of 20-epi-salinomycin and its 20-O-acyl derivatives. RSC. Adv. 2016, 6, 41885. [Google Scholar] [CrossRef]
- Shi, Q.; Li, Y.; Bo, S.; Li, X.; Zhao, P.; Liu, Q.; Yang, Z.; Cong, H.; Deng, H.; Chen, M.; et al. Discovery of a 19F MRI sensitive salinomycin derivative with high cytotoxicity towards cancer cells. Chem. Commun. 2016, 52, 5136. [Google Scholar] [CrossRef]
- Zhao, P.; Dong, S.; Bhattacharyya, J.; Chen, M. iTEP Nanoparticle-Delivered Salinomycin Displays an Enhanced Toxicity of Cancer Stem Cells in Orthotopic Breast Tumors. Mol. Pharm. 2014, 11, 2703. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C.W. Cu-Catalyzed Azide-Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Li, Y.; Shi, Q.; Shao, J.; Yuan, Y.; Yang, Z.; Chen, S.; Zhou, X.; Wen, S.; Jiang, Z.X. Synthesis and biological evaluation of 20-epi-amino-20-deoxysalinomycin derivatives. Eur. J. Med. Chem. 2018, 148, 279–290. [Google Scholar] [CrossRef]
- Antoszczak, M. A medicinal chemistry perspective on salinomycin as a potent anticancer and anti-CSCs agent. Eur. J. Med. Chem. 2019, 164, 366–377. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
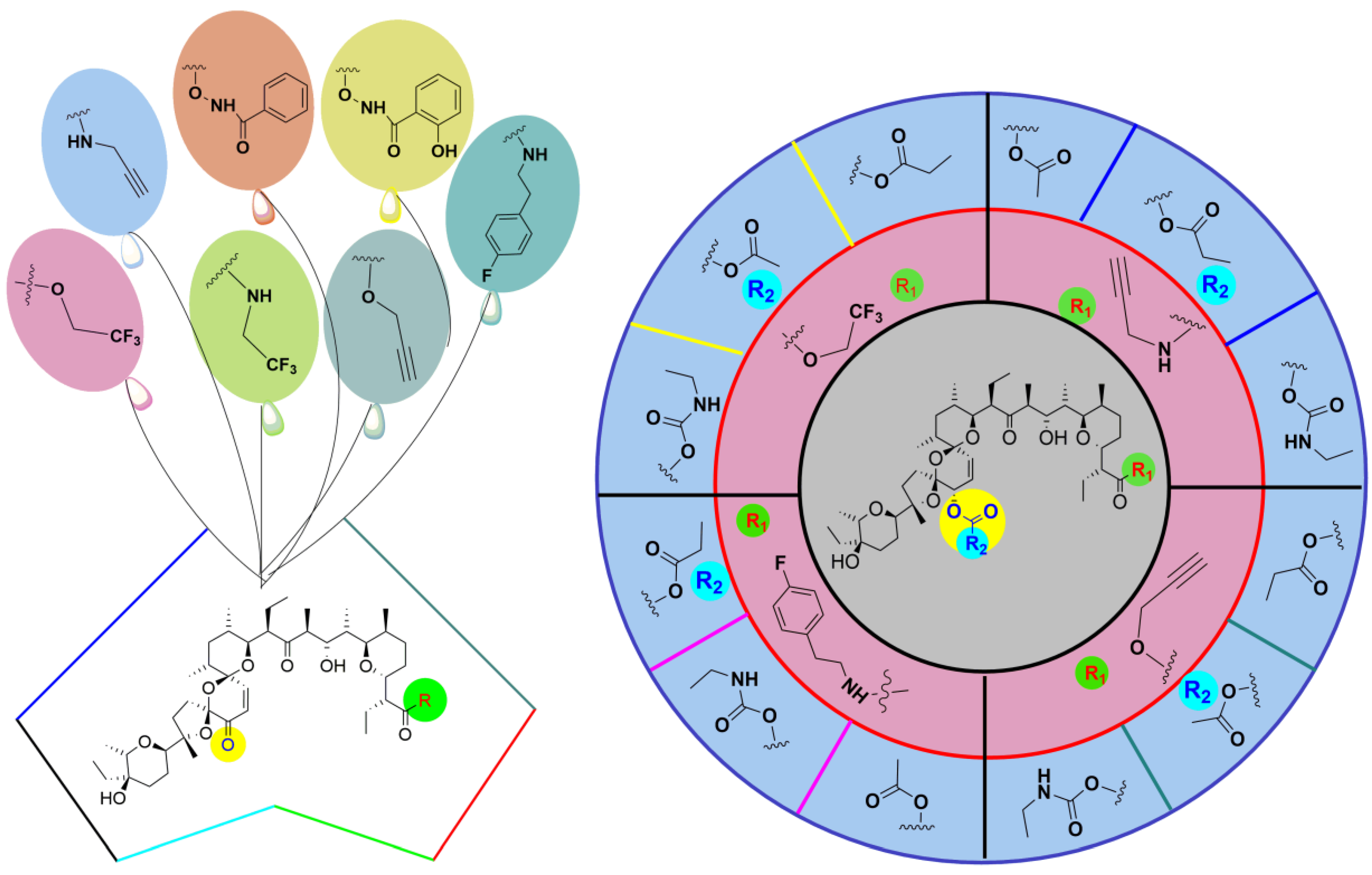{kind=link}
| Tumor Cell Line | In Vitro (IC50/EC50) | References |
|---|---|---|
| Chronic Lymphocytic Leukemia (CCL) | 100-fold | [49] |
| CD-24 low Breast Cancer | 0.4 µm | [1,43,51] |
| CSCs-high NB | 1.2 µm | [1] |
| CSCs-high GBM | 1.25 µm | [60] |
| Medulloblastoma | 0.1–2 µm | [61] |
| CSCs-high Pancreatic | 0.5–2 µm | [1] |
| Cell Type | In Vitro (IC50/EC50) | Reference |
|---|---|---|
| Human Bone Marrow Mesenchymal Stem Cells (hBMSC) | 30 µm | [71] |
| Human Bone Marrow Mesenchymal Stem Cells (hBMSC) Chronic Exposure | 0.1 μM | [78] |
| Human Nasal Mucosa | 10–20 µm | [74] |
| Animal | In Vivo (LD50) | Reference |
|---|---|---|
| Rats | 0.4 µm | [21] |
| Hens | 60 mg/kg | [82] |
| Broiler Chickens and Laying Hens | 108 mg/kg and 104 mg/kg | [83] |
| Horses | 0.6 µg/kg | [83] |
| Mice | 18 mg/kg (Intraperitoneally) 50 mg/kg (Orally) | [39] |
| Tumor Cell Line | C1-Modification | In Vitro Activity Compared to SAL | Reference |
|---|---|---|---|
| Doxorubicin-resistant Lovo Colon Cancer | Esterification | 8-fold | [87] |
| Vincristine-resistant Human Promyelocytic Leukemia (HL-60) | Esterification | 8-fold | [87] |
| Acute Lymphoblastic Leukemia Cells | 1,1,1-trifluoro-2-methoxyethane | 2-fold | [88] |
| Triple Negative Breast Cancer (MDA-MB-231) | 2,2,2-trifluoroethyl Ester | 3-fold | [32] |
| Triple Negative Breast Cancer (MDA-MB-231) | Benzotriazole Ester | 5-fold | [32] |
| Triple Negative Breast Cancer (MDA-MB-231) | p-brominated Hydroxamic acid | 7-fold | [89] |
| Human Gastric Carcinoma (HCG-27) | p-brominated Hydroxamic acid | 7-fold | [89] |
| Colon, Gastric and Triple Negative Breast Cancers | Phenyl, Phenol and Octanoyl hydroxamic acid | 2-3-fold | [102] |
| Cancer Type | C 17, C 20 or C 21- Modification | In Vitro Activity Compared to SAL | References |
|---|---|---|---|
| CSCs-high Breast Cancer | C 20-Amine (IRO) | 10-fold | [43,51] |
| Colon and Breast Cancers | 17, 21-di-epi-20-O-Bz-SAL Sodium salt | 2-fold | [102] |
| Triple Negative Breast Cancer (MDA-MB-231) | 20-epi-O-acylated | 2-10-fold | [95] |
| Gastric Cancer (HGC-27) | |||
| Colorectal Cancer (HT-29) | |||
| Triple Negative Breast Cancer (MDA-MB-231) | Benzoyl | 10-fold | [93] |
| Gastric Cancer (HGC-27) | |||
| Colorectal Cancer (HT-29) | |||
| Murine Breast Cancer (4T1) | Perfluoro-tert-butyl Ether Triazole | 2-fold | [96] |
| Human Glioblastoma (U87) | Tert-butyl Benzene | ||
| 3-pyridine | |||
| Perfluoro-tert-butyl Ether | |||
| Epithelial Colorectal Adenocarcinoma (MCF-7) | Diphenyl Triazole | 2.9-fold | |
| Perfluoro-tert-butyl Amide | 2-fold | [54,97] | |
| Colon Carcinoma (CaCo2) | Perfluoro-tert-butyl Ether Triazole | 29.5-fold | [96] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soni, V.; Nagar, A.; Bardiya, R.; Mara, J.; Von Suskil, L.; Rose, S.; Sonawane, C. A Concise Review of Prodigious Salinomycin and Its Derivatives Effective in Treatment of Breast Cancer: (2012–2022). Int. J. Transl. Med. 2023, 3, 217-245. https://doi.org/10.3390/ijtm3020016
Soni V, Nagar A, Bardiya R, Mara J, Von Suskil L, Rose S, Sonawane C. A Concise Review of Prodigious Salinomycin and Its Derivatives Effective in Treatment of Breast Cancer: (2012–2022). International Journal of Translational Medicine. 2023; 3(2):217-245. https://doi.org/10.3390/ijtm3020016
Chicago/Turabian StyleSoni, Viren, Akhil Nagar, Ruchita Bardiya, Jacob Mara, Lukas Von Suskil, Sabrina Rose, and Chetankumar Sonawane. 2023. "A Concise Review of Prodigious Salinomycin and Its Derivatives Effective in Treatment of Breast Cancer: (2012–2022)" International Journal of Translational Medicine 3, no. 2: 217-245. https://doi.org/10.3390/ijtm3020016