
Chromatin Organization during C. elegans Early Development

Department of Molecular, Cellular and Developmental Biology, University of Michigan, Ann Arbor, MI 48109, USA
*
Author to whom correspondence should be addressed.
DNA 2024, 4(1), 64-83; https://doi.org/10.3390/dna4010004
Submission received: 28 December 2023
/
Revised: 5 February 2024
/
Accepted: 19 February 2024
/
Published: 22 February 2024
(This article belongs to the Special Issue DNA Organization in Model Organisms)
Abstract
:Embryogenesis is characterized by dynamic chromatin remodeling and broad changes in chromosome architecture. These changes in chromatin organization are accompanied by transcriptional changes, which are crucial for the proper development of the embryo. Several independent mechanisms regulate this process of chromatin reorganization, including the segregation of chromatin into heterochromatin and euchromatin, deposition of active and repressive histone modifications, and the formation of 3D chromatin domains such as TADs and LADs. These changes in chromatin structure are directly linked to developmental milestones such as the loss of developmental plasticity and acquisition of terminally differentiated cell identities. In this review, we summarize these processes that underlie this chromatin reorganization and their impact on embryogenesis in the nematode C. elegans.
1. Introduction
Chromatin organization is a highly dynamic and precisely regulated process in the developing Caenorhabditis elegans embryo. After pronuclear fusion, the embryo exists in a totipotent state. As embryonic cells divide and differentiate, they establish cell-specific gene expression programs. This is accompanied by major remodeling of the chromatin structure. Several mechanisms are involved in this process. One of these is the formation of heterochromatin and euchromatin [1]. As embryogenesis progresses, the chromatin segregates into two distinct states: the actively transcribed euchromatin and the transcriptionally silent heterochromatin [1]. This occurs at specific points in the timeline of embryo development, and disruption of these events in embryogenesis results in functional consequences for the development of the embryo and the health of the adult worms. Formation of these chromatin states is also accompanied by the deposition of active and repressive histone marks that reinforce the appropriate transcriptional state for genomic loci [2,3]. Active histone modifications are generally associated with chromatin that have a higher rate of transcription, and conversely repressive histone modifications are generally associated with a silent chromatin state.
Spatial organization of chromatin within the nucleus is influenced by the establishment of topologically associating domains (TADs) and lamina-associating domains (LADs) on the chromatin. TADs are sub-megabase-scale chromatin structures that separate the genome into self-interacting domains [4,5]. As defined by interaction frequencies observed through Hi-C and multiplexed fluorescence in situ hybridization (FISH), DNA elements within a self-interacting domain share more interactions inside their domain compared to interactions with DNA elements outside their domains. LADs are heterochromatic genomic regions that are physically anchored to the nuclear lamina and they spatially sequester silent regions of the chromatin to the nuclear periphery [6]. Both these genomic structures regulate the organization of chromatin and are responsible, to some extent, for creating permissive and repressive environments for the transcription of appropriate genes in the appropriate context within embryogenesis.
For hermaphrodite embryos, there is the additional complication of dosage compensation. Dosage compensation in C. elegans is necessary to equalize the transcriptional output of the two hermaphrodite X chromosomes to that of the single male X chromosome [7]. It is a process that significantly alters the chromosome architecture on the X chromosome, its histone modification landscape and transcriptional output during embryo development. This review looks at the most recent evidence that explores the timeline of formation of key structural features on chromatin and the mechanisms that regulate the various aspects of chromatin architecture and organization throughout C. elegans early development.
2. Histone H3K9 Methylation-Mediated Heterochromatin Formation
There are broadly two types of chromatin inside an interphase nucleus, euchromatin and heterochromatin. Euchromatin refers to transcriptionally active “open” chromatin, and heterochromatin refers to transcriptionally inactive “closed” chromatin. As zygotic transcription in C. elegans is activated in 4-cell embryos [8], chromatin starts segregating into these two distinct forms from its previously uncondensed state. Heterochromatin is formed de novo as cells start differentiating and acquiring their specific cell identities [1], in a process that is precisely timed. Under transmission electron microscopy (TEM), heterochromatin appears as electron-dense regions (EDRs) inside the nucleus [9]. EDRs first appear in embryos during initiation of gastrulation at the 28-cell stage as numerous electron-dense puncta appear dispersed throughout the nucleus. As gastrulation progresses, these dense puncta begin coalescing into fewer but larger electron dense compartments throughout the nucleus. In late-stage embryos (>200 cells), large EDRs are clearly visible and are localized to the nuclear periphery [9].
In C. elegans, the most well-studied heterochromatin-associated repressive histone marks are H3K9me3 and H3K27me3. H3K9me3 is associated with, and required for the formation of repressive heterochromatin in many organisms [10,11,12]. H3K9me2/me3 histone marks are both dynamically regulated during early embryogenesis in C. elegans and their deposition on the chromosomes coincides with the timeline of EDR formation [9]. Histone H3K9me modifications promote the formation of heterochromatin through multiple mechanisms: they are responsible for the formation of active and inactive genomic compartments [13,14], compaction of individual chromosome regions [14] and recruitment of pathways that lead to the perinuclear anchoring of H3K9me2/me3-enriched genomic regions [13,14,15,16,17,18].
MET-2 is an H3K9 histone methyltransferase that is responsible for the deposition of H3K9me1/me2 marks [16]. In met-2 mutant embryos, EDRs corresponding to heterochromatin start forming much later in embryogenesis [9]. EDR puncta only start appearing at the late gastrulation stage, and do not appear as dense as WT EDRs at this stage of development. In late-stage met-2 mutant embryos, the EDRs coalesce to form larger territories, but these territories occupy a significantly smaller proportion of the nuclear volume than WT EDRs [9]. MET-2 is localized in the cytosol in early embryos and is transported inside the nucleus at the onset of gastrulation in 20–50 cell embryos by its cofactor LIN-65 [9,18]. Inducing premature nuclear accumulation of MET-2 using a nuclear localization signal (NLS) leads to a premature increase in H3K9me2 on the chromatin [9]. This suggests that heterochromatin could also form earlier in embryogenesis by inducing precocious nuclear accumulation of MET-2, but heterochromatin formation through visualizing EDRs has not been directly tested under these conditions. Similar to its C. elegans homolog, the mammalian homolog of MET-2, SETDB1, is known to promote heterochromatin formation though H3K9me3 [11].
MET-2 and its binding partners, LIN-65 and ARLE-14, form nuclear hubs during gastrulation of the embryo [9,18]. These hubs co-localize with H3K9me2 but exclude active marks such as H3K4me3 [9]. Loss of LIN-65 delays the deposition of repressive H3K9me1/me2/me3 marks on the chromatin during development [9]. Loss of LIN-65 also destabilizes MET-2 and prevents the formation of MET-2 nuclear foci [9,18]. Loss of ARLE-14 delays only the deposition of H3K9me1/me2 without affecting MET-2 stability or nuclear hub formation. Interestingly, ARLE-14 does stabilize catalytically deficient MET-2 and strengthens its association with chromatin [19]. In lin-65 mutants, repetitive DNA elements are significantly de-repressed [9,18]. In arle-14 mutants, there is evidence for modest de-repression of repetitive elements in one study [9] but repetitive elements tested in another study did not show de-repression [18]. In sum, these indicate that heterochromatin formation may not be robust in lin-65 mutants, and may also be affected in arle-14 mutants. Furthermore, the LIN-65-mediated nuclear accumulation of MET-2 was found to be the rate-limiting step in the deposition of H3K9me2 on chromatin. Heterozygous lin-65/+ embryos with only one copy of lin-65 had only half the levels of H3K9me2 in stage-matched gastrula embryos [9]. The nuclear accumulation of MET-2 at the gastrulation stage was also found to be controlled by the length of the S phase [20]. Increasing and decreasing the length of the S phase in early embryos by growing worms at different temperatures affected the rate of timing of MET-2 nuclear accumulation. Lower temperatures led to precocious nuclear accumulation of all three binding partners and growing worms at higher temperatures led to delayed accumulation [20].
SET-25 is another H3K9 histone methyltransferase that is responsible for the deposition of H3K9me3. In set-25 mutants, there is a complete loss of heterochromatin, as visualized by EDRs [20]. The nucleus throughout gastrulation appears as uniform and devoid of EDRs as it does post-fertilization. Even in >200 cell embryos, there is no detectable formation of heterochromatin [20]. Despite this, set-25 mutant embryos are viable and the embryos are able to terminate developmental plasticity, suggesting that heterochromatin formation is not required for initiating cellular differentiation [20]. met-2 mutants lack virtually all embryonic H3K9me1/me2 but still have deposition of H3K9me3, albeit at significantly reduced levels [9]. This is because SET-25 can be recruited to genomic loci by two independent mechanisms, a MET-2-dependent pathway which targets repetitive elements and satellite repeats and a NRDE-3-dependent pathway which targets transposons and insertions [21,22]. In the absence of met-2, H3K9me3 can still be formed de novo through the NRDE-3-dependent pathway [21] and, thus, the reduced heterochromatin that is present in met-2 mutants can be attributed to this pathway.
3. Other Mechanisms of Heterochromatin Formation
Unlike in Drosophila or humans, where H3K9me3 and H3K27me3 are enriched on different chromosomal regions, H3K9me3, H3K27me3 and H3K23me3 tend to co-occur on stable heterochromatin in C. elegans [23,24,25,26]. The role of H3K27me3 is relatively well-characterized in embryos. In contrast to H3K9me2/me3, which are dynamically regulated throughout embryonic development, H3K27me3 is inherited in the embryos from modifications already present on both the oocyte and the sperm [27,28]. In germ cells, the H3K27me3 mark is deposited by the polycomb repressive complex 2 (PRC2), composed of MES-2, MES-3 and MES-6 [29]. PRC2 enables the de novo formation of H3K27me3 during larval development of germ cells [27].
In the embryonic stages, H3K27me3 inherited from parental germ cells is maintained on chromosomes through the first few rounds of cell divisions. During embryogenesis, this mark is enriched over genomic loci that were silent in their parental germlines [28]. Existing H3K27me3 is then propagated during successive embryonic cell divisions by the PRC2 complex [28,30]. The MES-2-mediated propagation of H3K27me3 on chromatin in embryos undergoing gastrulation is important for chromatin compaction at this stage in development. Extrachromosomal arrays and endogenous chromatin loci were both found to be physically de-compacted in 100-cell embryos with a mes-2 mutation compared to WT, as measured by immunofluorescence experiments [30]. This phenotype was accompanied by a decrease in H3K27me3 deposition [30].
Some demethylases and interactors of the PRC2 complex also regulate the deposition of H3K27me3 on the chromatin. UTX-1 is an H3K27me3 demethylase that is broadly expressed in embryos and is responsible for regulating the levels of H3K27me2/me3 [31]. Three KDM6 demethylase family members, JMJD-3.1, JMJD-3.2 and JMJD-3.3, present in C. elegans embryos function redundantly with each other to demethylate H3K27me3 as well [31]. The PRC2 complex can be targeted to genomic loci during embryogenesis by LET-418 [32], the C. elegans Mi2 homolog that is a component of the nucleosome remodeling and deacetylase (NuRD) complex critically responsible for embryonic development in mammals [33,34,35]. In the absence of LET-418, H3K27me3 is specifically reduced at LET-418 target genes [32]. JMJD-1.2 is another demethylase that regulates three heterochromatin marks, H3K9me2, H3K23me2 and H3K27me2, present in embryos and was found to protect DNA in early embryos from replication stress [36], though the mechanism for this has not been elucidated. Some other less studied repressive histone marks present on heterochromatin during embryogenesis include H3K23me2 [24] and H3K56me3 [37]. In addition to H3K23 methylation, other H3K23 modifications are also widely present in embryos, as measured by mass spectrometry [24], though their roles in development, if any, are less known.
Some histone variants are also known to play important roles in facilitating the process of heterochromatin formation by enabling the deposition of repressive histone modifications. In metazoans, both H3 and H3.3 are expressed at various stages of development and the presence of these different histone variants potentiates the deposition of specific histone modifications. In the case of H3.3, active histone marks such as H3K4me3 and H3K36me2 are catalyzed preferentially [38,39,40,41,42]. In contrast, the canonical H3 favors the deposition of repressive histone marks associated with heterochromatin such as H3K9me2/me3 and H3K27me2/me3 [39]. In early embryogenesis from 2-cell to 50-cell stages, cells inside the embryos are enriched in H3.3 and have very low levels of H3 and therefore H3K9me. This is a holdover from germ cells where H3.3 is maintained at a high level in both oocytes and sperm [42]. Class I and Class II H3 genes are specific gene clusters that produce H3 and have distinct patterns of expression and regulation during embryogenesis [42]. Class I and Class II H3 begin accumulating on the chromatin at the 2-cell stage and at the onset of gastrulation, respectively, and H3 slowly starts replacing H3.3 and allows the gradual accumulation of H3K9me2/3 [42]. At the late embryo stages, H3 is highly enriched on the entire embryo, except for the P lineage germ cells that retain H3.3 enrichment [42]. This process is conserved in mammals, where H3.3 inherited from germ cells is replaced with other histone H3 variants [43].
Another important player in regulating heterochromatin is linker histone H1, transcribed from his-24 [44], which promotes the deposition of repressive histone marks on the chromatin during embryogenesis [45]. HIS-24 is cytoplasmic in both the maternal and paternal germ cells, and immediately after fertilization is rapidly translocated to the nucleus where it associates with the chromatin at the pronucleus stage [45]. HIS-24 continues to be associated with the chromatin through embryogenesis in all cells, with the exception of the Z2 and Z3 primordial germ cells (PGCs) [45]. This mechanism of germ cell cytoplasmic retention and rapid nuclear translocation is lost in mutants for mes-2, mes-3, mes-4, mes-6 and sir-2.1, suggesting the involvement of these proteins in this pathway [45]. his-24 mutant worms were unable to silence extrachromosomal transgenes due to a loss of H3K9me2 repressive mark and aberrant gain of H3K4me3 active mark [44,45]. A his-24 mutation also exacerbated the defect in H3K27me3 deposition on the chromosomes of germ cells in mes-3 mutants [45]. Taken together, these results suggest that linker histone H1 may promote the accumulation of repressive histone marks in early embryogenesis, though the mechanism and the potential involvement of the PRC2 complex components remain unclear. In addition to his-24, there are seven other linker histone variants in C. elegans, hil-1 through hil-7. These additional variants do not have any known roles in embryogenesis [46].
4. Developmental Regulation of Active Chromatin
After fertilization and pronuclear fusion, the chromosomes exist in a decondensed state until the onset of the gastrula stage where they begin partitioning into active and inactive compartments. Prior to the onset of gastrulation, many active epigenetic marks inherited from parental germ cells are present on the chromosomes. These include methylation marks such as H3K4me2 [47] and H3K79me2 [48], and acetylation marks such as H2BK12ac, H3K23ac, H3K27ac, H4K16ac, H4K5ac, H4K8ac, and H4K12ac [48]. After the first few rounds of DNA replication, the previous parent-of-origin specific distribution of histone marks on the chromatin is completely altered. Acetylation marks are largely equalized over all the chromosomes [48]. The well-studied H3K4me2/me3 marks, in contrast, show more dynamic regulation in early embryos.
H3K4me3 is enriched at transcription start sites (TSS) of active genes, whereas H3K4me2 tends to be uniformly distributed over the gene body [49]. At the resolution of microscopy, H3K4me2/me3 are present uniformly and at relatively high levels in early 2–4-cell embryos, except for the X chromosome where it is depleted in embryos [50,51,52]. At the 8-cell stage, H3K4me3 begins being enriched on some blastomeres in a lineage-specific manner. H3K4me3 is completely lost in the germline blastomere, and some of the somatic blastomeres [50]. In contrast, it is highly enriched on the AB descendants at eight cells and remains enriched in 80-cell embryos in all the cells of the AB lineage [50]. The AB blastomere is formed from the first cell division in the C. elegans embryo. The AB lineage gives rise to the nervous system, the hypodermis and about half of the pharyngeal tissues [53].
The C. elegans mixed-lineage leukemia (MLL) complex, which has methyltransferase activity targeting H3K4 and demethylase activity targeting H3K27, is responsible for this lineage-specific regulation [50,54,55]. Different components of the MLL complex are differentially required for H3K4 methylation at different developmental stages. The two important components of the complex required in embryos are SET-2 and ASH-2. SET-2 is the C. elegans homolog of mammalian SET-1, which is a core member of the mammalian MLL complex. ASH-2, which was initially suggested to be a core member of the C. elegans MLL complex [54], was later found to be dispensable for MLL activity at specific stages of development [56], indicating it is an ancillary binding partner of the MLL complex rather than a core component. During embryonic development, both ASH-2 and SET-2 are required for H3K4me3 deposition, but ASH-2 also functions in a SET-2-independent pathway for H3K4me2 deposition [56]. SET-2 is specifically required for accumulation of H3K4me2 in PGCs at later stages of embryogenesis [56]. Another member of the MLL complex, SET-16, also has H3K4 methyltransferase activity in vitro [54,56]. However, depletion of SET-16 causes embryonic lethality [57], making it difficult to elucidate its potential role in regulating H3K4me during development. UTX-1, an H3K27me2/me3 demethylase, forms a complex with SET-16 and its expression is required for SET-16 expression and vice versa [31], suggesting cooperation between pathways regulating active and repressive histone marks during embryogenesis.
HTZ-1 is a histone H2A variant that is required for proper embryo development [58]. HTZ-1 is maternally loaded into embryos and incorporated into chromatin, starting at the 4-cell stage. Maternally loaded HTZ-1 mRNA is actively converted into protein, and the levels of HTZ-1 increase with the progress of embryogenesis [58]. It is enriched on the chromatin upstream of a subset of developmentally important actively transcribed genes at their TSS, where it regulates transcription by RNA PolII [58]. Depletion of HTZ-1 in embryos results in developmental defects, such as embryonic lethality and early larval arrest, likely due to mis-expression of these developmental regulators [58]. The various relevant histone variants and their roles in embryogenesis have been summarized in Table 1.
5. Antagonism between Repressive and Active Histone Marks
Active and repressive histone marks tend to, with some exceptions, be enriched on mutually exclusive genomic regions in C. elegans. The histone methyltransferases that are responsible for regulating the deposition of these marks frequently interact and antagonize each other to establish the epigenetic landscape of the C. elegans early embryo. The MLL complex, with well-characterized interactions with both active and repressive histone post-translational modifications (PTMs), is one of the prime examples of this balancing act. The MLL complex-mediated H3K4me2/me3 deposition sustains transcription at its target loci in embryos [2]. In addition, the MLL complex member SET-16 also associates with H3K27me2/me3 demethylase UTX-1 [31]. Despite this interaction, the two marks can be toggled independently in embryos [54]. utx-1 mutants exhibit significant embryonic lethality, which may suggest that the demethylase activity is important for embryogenesis. However, mutant embryos with catalytically deficient UTX-1 which cannot remove H3K27me2/me3, are viable and develop into healthy adults. In fact, rather than its demethylase activity, it is the interaction between UTX-1 and SET-16 that seems to be crucial for embryo development [31].
A more direct example of antagonism is the exclusion of active marks on heterochromatin through the formation of MET-2 nuclear foci on chromatin. In addition to the established role of MET-2 in H3K9me-mediated heterochromatin formation, catalytically deficient met-2 mutants (met-2 CD) that are unable to catalyze the methylation of H3K9 are still able to maintain some of their function in regulating heterochromatin, suggesting that MET-2 has an H3K9me-independent role during embryogenesis [19]. Catalytically inactive met-2 only has a partial effect on the de-repression of heterochromatin genes, where 28% of genes de-repressed in met-2 null mutants remained repressed in met-2 CD mutants [19]. Evidence from ChIP-seq suggests that the formation of MET-2 nuclear foci themselves, without catalyzing H3K9me1/me2 formation, are important for excluding the active histone marks H3K9ac and H3K27ac at met-2 target genomic loci [19]. met-2 CD mutants do not gain active marks on met-2 target genomic loci, which prevents most of these loci from being de-repressed even in the absence of the repressive H3K9me histone marks [19]. MET-2 cofactors LIN-65 and ARLE-14 are important for forming and stabilizing these MET-2 foci and strengthening their association with the chromatin [19]. While the mechanism behind foci formation is not known, MET-2 and LIN-65 both contain disordered domains [9,18,19], which suggests that liquid–liquid phase separation may be a contributor [59,60].
Antagonism between MES-4-mediated H3K36me3 and PRC2-mediated H3K27me3 is important for maintaining appropriate patterns of gene expression in germ cells [28]. Early embryos inherit both the active H3K36me3 and repressive H3K27me3 from germ cells, where these marks occupy mutually exclusive genomic loci [28]. In these early embryos, germline-expressed genes carried H3K36me3 whereas soma-specific genes carried H3K27me3 [28]. The loss of MES-4 and its associated H3K36me3 mark led to the acquisition of repressive H3K27me3 on germline-specific genes in embryos. Other genomic loci carrying MES-4-independent H3K36me3 did not acquire any repressive marks. This evidence shows that MES-4 activity on germline-specific genes repels the activity of the repressive MES-2/3/6 complex [28]. Histone modifications covered in this review as well as their associated functions have been summarized in Table 2.
6. Spatial Organization of Chromatin inside the Nucleus
As the embryos differentiate and heterochromatin is formed, the chromatin is dramatically reorganized spatially inside the embryonic nuclei. This reorganization segregates active chromatin from silent chromatin, and this process is primarily driven by pathways that anchor heterochromatic regions to the nuclear periphery where they are relatively inaccessible to transcriptional machinery. The specific regions of chromatin that are sequestered to the nuclear lamina are also called lamina-associated domains (LADs) [6]. In C. elegans, LADs occur on all the distal regions of autosomes, called chromosome “arms”, and only the left distal arm on the X chromosome [65]. LADs are enriched in repetitive sequences and transcriptionally inactive genes, and they tend to have deposition of H3K27me3 and H3K9me2/me3 repressive marks [16,25,62,65].
LADs are tethered to the inner nuclear lamina and were first defined in C. elegans by their association with LEM-2, an inner membrane transmembrane protein [65]. LEM-2 ChIP-seq experiments show that the regions of heterochromatin that directly associate with the lamina, called LEM-2 subdomains, are punctuated by gaps. These gaps in LEM-2 subdomains are more frequent and bigger in size as they move further away from the chromosome arms and are enriched in transcriptionally active genes. Inactive genes in embryonic LADs, and specifically ones that occur on LEM-2 subdomains that interact directly with the nuclear lamina, tend to remain transcriptionally silent through development [65].
Histone H3K9 methylation is required for the peripheral anchoring of heterochromatin. MET-2 and SET-25 function redundantly to position heterochromatic arrays at the periphery in early embryos in an H3K9me2/me3-dependent mechanism [16,18]. Heterochromatin anchoring is completely lost in met-2; set-25 double mutants [16,18]. However, there are differing reports of the effect of H3K9 HMT single mutants in regulating anchoring of heterochromatic arrays compared to endogenous heterochromatin. In the study by Towbin et al. (2012), met-2 mutants did not show any defect in heterochromatic array anchoring but set-25 mutants showed a partial defect [16]. LEM-2 ChIP experiments in the study by Delaney et al. (2019) found complete ablation of anchoring of endogenous heterochromatin in met-2 mutants, but no effect in set-25 [18]. MET-2 and SET-25 can independently methylate certain targets [21], which can be used to explain some of these differences. SET-25 was shown to be able to methylate heterochromatic arrays independently of MET-2 [21], which can explain the absence of the de-tethering phenotype in met-2 mutants in Towbin et al. (2012). However, on endogenous targets set-25 requires met-2 for methylation, which is corroborated by experiments from Delaney et al. (2019). Delaney et al. (2019) also found LIN-65, a cofactor of MET-2, to be required to maintain perinuclear anchoring in early embryos [18]. CEC-4 is a chromodomain protein that binds to H3K9me deposited by MET-2 and SET-25 and anchors the H3K9 methylated chromatin to the nuclear lamina [63,64]. CEC-4 is required for peripheral anchoring of heterochromatin in early embryos, but not at later stages of development [64]. Mechanisms of tethering differ between species [71], but the requirement of H3K9me2 seems to be a conserved feature seen in several model organisms [72,73].
MRG-1 regulates perinuclear anchoring in larval cells and is also active during embryogenesis [17]. In mrg-1 mutant embryos, heterochromatic arrays are de-compacted but maintain their association with the nuclear lamina due to the presence of CEC-4-mediated anchoring [17]. Perinuclear anchoring is lost in both embryonic and larval stages in cec-4; mrg-1 double mutants [17]. The formation of LADs in embryos also requires balancing active and repressive chromatin marks. In this process, MRG-1 plays a central role where it prevents CBP-1-mediated active H3K27ac mark from spreading to silent regions of the genome [17]. MRG-1 associates with euchromatic regions of the genome, where it is thought to bind to the active H3K36me2/me3 marks. In C. elegans and other metazoans, it also forms a complex with HAT and HDAC complexes [74,75,76]. CBP-1 is a cofactor for several transcription factors and promotes several pathways of somatic cell differentiation during embryogenesis [77]. In Cabianca et al. (2019), the authors quantified the localization and de-compaction of heterochromatic foci formed by integrated arrays to show that the loss of MRG-1-mediated sequestering of CBP-1 to euchromatic loci led to de-tethering from the nuclear lamina and de-compaction of condensed chromatin. ChIP-qPCR data also showed the gain of H3K27ac active mark on heterochromatin. MET-1 and MES-4-mediated deposition of H3K36me2/me3 marks on euchromatin is also required for proper tethering of heterochromatin [17]. Cabianca et al. (2019) suggests that this may be through the MRG-1 pathway as MRG-1 is thought to be able to bind these histone marks, though the exact mechanism remains unclear [17]. Exogenous heterochromatic arrays tested for peripheral anchoring in early embryos showed de-repression in H3K9me mutants, but also in mutants for several other histone modification pathways that do not affect peripheral localization of these arrays [16]. Additionally, cec-4 mutants did not show de-repression of heterochromatic genes [64]. Together, this suggests that independent pathways of gene repression through histone modifications and CEC-4-mediated peripheral anchoring are active in early embryogenesis.
Telomeres present at the ends of chromosomes are also peripherally localized inside the nucleus. The peripheral enrichment of telomeric regions is evident from 20–50 cell embryos, and increases with the progression of embryogenesis [78]. The pathways regulating telomeric positioning in embryos are independent and distinct from those regulating sub-telomeric heterochromatin. Telomere localization in embryos is dependent on SUN-1, GEI-17 and the shelterin protein POT-1. In the larval stages of development, there is evidence of additional redundant pathways as telomeric regions localize to the periphery in L1 larvae, even in the absence of embryonic tethering pathways [78]. POT-1 associates directly with telomeric DNA and directs their association to the periphery in a SUN-1-dependent manner. GEI-17, a SUMO E3 ligase, was also found to be required for this function [78]. However, while the sumoylation of proteins in this pathway is well characterized in yeast [79], its role in C. elegans remains to be elucidated. Both pathways regulating the spatial organization of DNA during embryo development have been summarized in Figure 1.
7. Emergence of Topologically Associating Domains (TADs)
Topologically associating domains (TADs) are sub-megabase-scale 3D chromatin structures that are a conserved feature of chromatin organization [4,5]. TADs are composed of chromatin domains that have higher frequencies of interaction within the DNA sequences inside the domain and have relatively low interaction frequencies with DNA sequences in other domains. These interactions that define TADs are highly stochastic and occur at low absolute frequencies [76,80,81,82], but these small interactions are higher than expected by probability and specifically regulated by transcriptional states and epigenetic landscapes. Most TADs are demarcated by boundaries that isolate neighboring domains [83,84,85]. In other organisms, CTCF proteins mark these TAD boundaries and act as transcriptional insulators. However, C. elegans does not have a CTCF ortholog, and no other protein that may perform a similar function has been identified [86]. Eigenvector deconvolution of TAD domains is commonly used to categorize TADs into two compartments: the transcriptionally active “A” compartment and the silent “B” compartment [83].
Recent advancements in chromosome conformation capture technologies such as Hi-C have enabled us to study the formation of these local 3D structures and global TAD distribution through development. Using Hi-C in mixed-stage C. elegans embryos, strong TADs were found to be present on the X chromosome [87]. TADs on the autosomes are also present at this stage, although the boundaries are significantly weaker. TADs are more regularly spaced on the X chromosome compared to the autosomes and resemble mammalian TADs. TAD formation on the X is dependent on the dosage compensation complex (DCC). In DCC mutants, previously isolated TAD domains have increased contact frequencies with each other, and clearly demarcated TAD boundaries are lost [68,87]. The role of TAD formation in hermaphrodite dosage compensation is explored in more detail later in this review.
Methylation of histone H3K9 by MET-2 and SET-25 also affects TAD formation in embryos [14]. TADs are significantly weaker and less insulated in met-2; set-25 mutants that have lost H3K9me2/me3 repressive marks. cec-4 mutants also exhibited weaker TADs, though the effect was much less substantial than in met-2; set-25 [14], suggesting that both H3K9me deposition and perinuclear anchoring are independently contributing to TAD formation during embryogenesis. This weakening of TADs and compartments leads to some genes being mis-segregated into the wrong compartment, i.e., genes previously in an active A compartment being mis-segregated into an inactive B compartment; however, this does not seem to lead to any significant changes in gene expression [14].
Hi-C analysis in late-stage embryos showed that the autosomes assume a conformation where the two arms of an autosome form into inactive B compartments, and the middle transcriptionally active regions forms the active A compartment [87]. The two B compartments have high interaction frequency within their domains but also with each other. Another study used chromosome tracing with a multiplexed FISH approach to study the onset of TAD and compartment formation on C. elegans autosomes during early embryonic development [15]. This approach relies on FISH probes spanning 100 kb genomic windows within known TADs to capture the 3D conformation of individual chromosomes in single cells. The results showed that the conventional conformation of the autosomes is seen to first arise at the gastrulation stage during embryogenesis [15]. These conventional compartments are not present in autosomes in early embryos (2–25 cells), where the two B compartments are isolated from each other and separated spatially by the middle A compartment in an unconventional “barbell” conformation. Though the B compartments are separated in early embryos, local folding and increased contact frequency within the individual B compartments was evident even in these early embryos [15]. This indicates that formation of compartments is a very early step in embryogenesis.
This study also showed that the CEC-4-mediated anchoring of heterochromatin induces stretching of the chromosomes in early embryos. CEC-4 is involved in the perinuclear anchoring of heterochromatin domains through H3K9me [64] and this function of CEC-4 was found to be an important mediator of compartment size and overall chromosome compaction [15]. Using the multiplexed FISH approach, cec-4 mutant early embryos seem to have more compacted chromosomes due to the loss of stretching. This is in contrast to older embryos and adults, where heterochromatic arrays and the endogenous X chromosome are de-compacted due to the loss of CEC-4 [17,63,64].
8. Epigenetic Modifications and the Loss of Developmental Plasticity
Most somatic cells progressively lose developmental plasticity after the gastrula stage and acquire their terminal identities at the end of gastrulation, which occurs around 6 h after fertilization [88,89,90,91,92,93]. The reorganization of chromatin from its uncompartmentalized state into euchromatin and heterochromatin at the onset of gastrulation coincides with this loss of developmental plasticity in C. elegans [20,30,88].
In C. elegans, MET-2 and SET-25 both regulate the timing of developmental plasticity loss and differentiation. In mammals, loss of the MET-2 homolog SETDB1 leads to the loss of cellular differentiation and aids in somatic cellular reprogramming [94]. In the context of cellular reprogramming, H3K9me3 was found to block TFs from accessing broad chromatin domains whose expression was necessary to induce pluripotency [95]. In C. elegans, the loss of developmental plasticity was tested directly in met-2 and set-25 single mutant embryos in a cell fate challenge assay, where they ectopically express a transcription factor, hlh-1, that promotes the differentiation of embryonic cells to body wall muscle cells [91]. Ectopic expression of hlh-1 before the loss of developmental plasticity promotes the differentiation of all embryonic cells into body wall muscle cells [91]. In this assay, MET-2 was found to promote the loss of developmental plasticity, as significantly more met-2 mutant embryos retained their developmental plasticity at the 100-cell stage compared to WT [20]. In contrast, SET-25 was found to inhibit loss of plasticity as set-25 mutant embryos were less plastic than WT at 100 cells [20]. These contrasting effects of met-2 and set-25 in embryos suggests that lower levels of H3K9me3, a phenotype present in both mutants, is not driving the loss of plasticity. Instead, the regulation of plasticity is likely through H3K9me2 as this mark is completely lost in met-2-mutant embryos but is modestly increased in set-25 mutants [20]. This differentiation assay provides evidence that met-2 and set-25 both regulate developmental plasticity, likely through their modulation of H3K9me2 and its subsequent effects on the timing of heterochromatin formation, where the mis-regulation of the histone mark alters the timing of the onset of heterochromatin formation [20].
The mammalian homologs of MET-2 and SET-25 are SETDB1 and EHMT2, respectively. Loss of these methyltransferases during mammalian embryogenesis results in embryonic lethality [96,97]. However, unlike the mammalian homologs, mutant C. elegans embryos that are null for met-2 and set-25 have developmental delays and lower viability than WT but are still able to develop into adult worms [19,20]. Both these mutants are still able to eventually lose developmental plasticity and begin differentiating, indicating that there may be other regulators of plasticity at later stages of development. set-25 mutants are particularly striking in their complete inability to form heterochromatin even at the late-embryonic 200-cell stage [20]. Even with a complete loss of heterochromatin at these stages, the mutant worms are in fact less plastic than WT [20]. Additionally, RNA-seq experiments have shown that disruption of H3K9me causes gene-specific changes by modulating transcription factor activity [61]. These gene expression changes were independent of chromatin compaction, and were enriched in tissue-specific transcriptional programs [61]. These results suggest that in C. elegans, heterochromatin formation and the loss of plasticity can be decoupled.
While cec-4-mutant worms are healthy and appear WT, they respond uniquely to cell fate challenge assays. The authors used the same cell fate challenge assay described earlier, which uses the TF hs:hlh-1 to promote differentiation of mid-stage embryos into muscle cells. A significant fraction of cec-4 mutant embryos, which lack the peripheral anchoring of heterochromatin, continued to differentiate into normal embryos and hatched into L1s despite widespread expression of the transcription factor [64]. The authors reported that these hatched L1s appeared abnormal and had a low survival rate [64]. The authors suggest that this could be due to the failure of cec-4 mutants to stabilize the ectopic differentiation program initiated by hlh-1 [64]. However, several H3K9me pathway mutants tested in this assay also exhibit changes in developmental plasticity during embryogenesis [20], suggesting an alternative hypothesis that cec-4 mutant embryos could be less developmentally plastic.
Similar to H3K9me2, PRC2-mediated deposition of repressive H3K27me3 mark by EZH2 was also found to be required for the reprogramming and establishment of pluripotency [98]. In mammals, both of these marks are required for normal embryonic development, and mutant embryos lacking the methyltransferases for these repressive marks die early in gestation [94,96,98]. In C. elegans, PRC2 complex member MES-2, which is required for H3K27me2/me3 [29], promotes the transition from a developmentally plastic state to differentiated embryos [30]. Using the cell fate challenge assay, loss of mes-2 was found to increase the number of plastic cells in early 100-cell embryos. There were also significant changes in the transcriptome for developmental regulators of cell fate and differentiation for both mes-2 and mes-3 mutants, which suggested that the PRC2 complex is required for promoting a differentiated cell fate [30].
Another important mechanism involved in the loss of developmental plasticity at the gastrula stage is the loss of histone variant H3.3 inherited from the oocyte, and the enrichment of canonical H3 in the developing embryo, which facilitates the deposition of repressive histone marks on the chromatin that are linked to the process of heterochromatin formation [42]. C. elegans mutants that lacked the ability to enrich H3 in somatic cells had a defect in timely loss of developmental plasticity. This was tested in a cell fate challenge assay using ectopic expression of the TF che-1, which promotes differentiation into neuronal cell fate. Without the incorporation of H3 and its associated H3K9me2/me3 and H3K27me2/me3 repressive marks, embryos maintained developmental plasticity much later into embryogenesis [42].
9. Establishment of Dosage Compensation on the X Chromosome
The process of dosage compensation is unique for the C. elegans sex chromosomes compared to other organisms. Expression of transcripts from the two X chromosomes of hermaphrodite worms are repressed by half to match that of the single male X chromosome [7]. Establishment of dosage compensation and repression of the X chromosomes involves chromatin reorganization during early embryogenesis and its onset coincides with gastrulation and the loss of pluripotency [70,99]. This process is initiated by X signal elements such as sex-1, sex-2, ceh-39 and fox-1, and autosomal signal elements such as sea-1 and sea-2, that are transcribed from the sex chromosome and autosomes respectively [100,101,102,103,104,105,106]. The ratio of X:A signal elements determines whether XOL-1 is activated or repressed during early embryogenesis [107]. XOL-1 is the master regulator of sex determination and dosage compensation in C. elegans [107]. In hermaphrodites with two X chromosomes, the X-signal elements are able to repress XOL-1 which then promotes hermaphrodite sex development pathways as well as the initiation of dosage compensation on the X chromosomes [104].
The dosage compensation complex (DCC) consisting of a condensin IDC and several accessory proteins is responsible for carrying out the process of dosage compensation [108,109]. Condensin IDC is a C. elegans-specific ortholog of the conserved metazoan condensin complexes [109]. DPY-27, a core component of the condensin IDC, is visibly enriched on the X chromosome shortly after the onset of gastrulation in 30–50 cell embryos [70,110]. The DCC loads onto binding sites on the X chromosomes and spreads along the entire chromosome [111,112,113]. Loading of condensin IDC on the X chromosome alone is sufficient to initiate the process of chromosome compaction through chromatin looping [87,114], which leads to global gene repression on the X [115] and is thought to lead to TAD formation [87,114].
The loading of the DCC onto the X initiates the process of dosage compensation, which also involves the formation of TADs on the X [87,116]. In dosage compensation (DC)-deficient embryos, TAD boundaries and compartment formation are weaker [68,87]. DCC recruitment sites called rex sites were found to mark strong TAD boundaries on the X [87,114]. Removing rex sites abolished these TAD boundaries and conversely the introduction of a new rex site on the X created a novel TAD boundary [114,116]. However, abolishing almost all of the TAD structure on the X chromosomes in embryos did not significantly affect gene expression due to dosage compensation, suggesting that TAD formation is not the major driver of DCC-mediated repression [116].
Dosage compensation, as measured by RNA-seq on staged embryo populations, was not seen to be completely established during embryogenesis [66]. This was tested by measuring the transcriptional output from X chromosomes of hermaphrodites compared to samples of mixed hermaphrodites and males. In all the embryo stages sampled in the study, the hermaphrodite X chromosomes were upregulated when normalized against X chromosomes from mixed-sex embryos [66]. Even though condensin IDC loading on the X occurs shortly after the onset of gastrulation, establishment of dosage compensation cannot be accurately measured by RNA-seq due to the presence of mRNAs already transcribed in the embryo before this stage. However, in early embryos, there is evidence for the partial dosage compensation of zygotic genes that are newly transcribed in embryos after zygotic gene activation at the 4-cell stage. Depletion of dpy-27 by RNAi in early- and mixed-stage embryos resulted in the significant de-repression of X chromosomes over WT at those stages, suggesting that the DCC is actively establishing dosage compensation throughout embryogenesis [66].
In addition to chromatin looping by the condensin IDC, several members of the DCC are either directly involved in or recruit other proteins that carry out ancillary mechanisms that reinforce X chromosome compaction, gene repression and tethering of the X chromosomes to the nuclear lamina [63,69,117]. One of these mechanisms is mediated by H4K20me1, a repressive histone mark that becomes enriched on the X chromosomes in hermaphrodites during embryogenesis [66,67]. DPY-21, one of the members of the DCC, is a H4K20 demethylase that is responsible for this X-specific enrichment of H4K20me1 [68]. Deposition of H4K20me1 and its enrichment on the X chromosomes is initiated in bean stage embryos, though enrichment only peaks later in late 3-fold embryos [66,67,70]. RNA-seq experiments on dpy-21 mutants suggests that DPY-21 is required for the establishment of dosage compensation, and that loss of dpy-21 has a more pronounced effect on X expression during mid to late embryogenesis [66].
Additionally, a DCC-independent mechanism also contributes to shaping the chromatin of the X chromosomes of early embryos. The active histone mark H4K16ac is depleted on the X chromosome in all stages of embryogenesis, starting at 4-cell embryos [69,70]. The H4K16ac mark is inherited in the embryo from germ cells, where it is depleted from the X chromosome. The depletion of H4K16ac in embryos precedes the loading of the DCC, and at this stage it is dependent on MES proteins MES-2, MES-3 and MES-4, which are components of the PRC2 complex [70]. After DCC loading during early gastrulation, the depletion becomes DCC-dependent [69,70]. In mes-2 mutants, DPY-27 loading onto the X chromosomes is delayed compared to WT. In mes-2 and mes-4 mutants, enrichment of H4K20me1 on the X chromosome is also delayed [70]. The delay in onset of dosage compensation in mes-2/mes-4 mutants is likely related to their function in regulating the expression of early zygotic genes such as sdc-2 that are important for the initiation of dosage compensation. The timeline of dosage compensation and other major mechanisms of embryonic chromatin reorganization are summarized in Figure 2.
10. Conclusions and Future Directions
The chromatin landscape is dynamically regulated in C. elegans early embryogenesis. Separation of the chromatin into heterochromatin and euchromatin is one of the early steps of embryogenesis. Evidence suggests that de novo heterochromatin formation, which is mediated by the deposition of repressive histone marks, may be important for regulating the precise timing of termination of developmental plasticity and onset of cell differentiation programs in early embryos. However, while heterochromatin formation does regulate the timing of these processes, it is dispensable for differentiation and the acquisition of terminal cell fate. The precise mechanisms of how deposition of repressive histone marks leads to heterochromatin formation and consequently how heterochromatin formation may influence the loss of developmental plasticity remain to be elucidated.
The pathways that are responsible for the tethering of heterochromatin to the nuclear lamina play important roles in maintaining appropriate transcriptional silencing of those genomic regions at specific stages of embryonic development. While tethering is dependent on H3K9me deposition, evidence shows that deposition of repressive histone modifications and anchoring by tethering pathways contribute independently to transcriptional silencing of heterochromatin. Loss of proteins that mediate this peripheral anchoring of heterochromatin during embryogenesis seem to also affect the robustness of cell fate and differentiation pathways. The mechanisms behind how these pathways are being influenced by lamina tethering of silent domains, and whether these mechanisms overlap with the influence of heterochromatin formation on plasticity, is an interesting question for further research.
Initiation of dosage compensation on the hermaphrodite X chromosomes is an important process that contributes to chromatin organization during embryogenesis. This process involves many mechanisms that are also known to modulate autosome architecture during embryo development. Loading of the DCC onto the X chromosome at the onset of gastrulation is followed by chromosome compaction and a reduction in transcription through pathways of chromatin condensation by condensin IDC, as well as the deposition of repressive histone modifications, depletion of active histone modifications and peripheral tethering to the nuclear lamina. While the onset of termination of plasticity coincides with the first detectable loading of the DCC components onto the X chromosome and both processes involve PRC2-mediated histone modifications, whether these two pathways are directly related has not yet been tested.
Recent advances in chromosome conformation capture technologies have allowed us to observe the highly dynamic process of TAD formation during embryogenesis. It has also allowed the study of some of the mechanisms that regulate these processes in embryos. However, the functional significance of TAD formation at these stages is still an open question. From studies in other model organisms and in C. elegans adult tissues, it is evident that the disruption of the TAD structures does not necessarily lead to altered transcription. Rather, the relationship between the transcriptional state of the chromatin and TAD structures is complex and the transcriptional state may be governed by several redundant pathways. With this background, the importance and functional relevance of TAD structure and TAD formation during embryogenesis requires further examination.
Author Contributions
Conceptualization, E.J. and G.C.; writing—original draft preparation, E.J.; writing—review and editing, E.J. and G.C.; supervision, G.C.; project administration, G.C.; funding acquisition, G.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Institutes of Health grant number R01 GM13385801 and National Science Foundation grant number MCB1923206.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Politz, J.C.R.; Scalzo, D.; Groudine, M. Something Silent This Way Forms: The Functional Organization of the Repressive Nuclear Compartment. Annu. Rev. Cell Dev. Biol. 2013, 29, 241–270. [Google Scholar] [CrossRef]
- Ruthenburg, A.J.; Allis, C.D.; Wysocka, J. Methylation of Lysine 4 on Histone H3: Intricacy of Writing and Reading a Single Epigenetic Mark. Mol. Cell 2007, 25, 15–30. [Google Scholar] [CrossRef]
- Ahringer, J.; Gasser, S.M. Repressive Chromatin in Caenorhabditis elegans: Establishment, Composition, and Function. Genetics 2018, 208, 491–511. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological Domains in Mammalian Genomes Identified by Analysis of Chromatin Interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-Dimensional Folding and Functional Organization Principles of the Drosophila Genome. Cell 2012, 148, 458–472. [Google Scholar] [CrossRef]
- Gonzalez-Sandoval, A.; Gasser, S.M. On TADs and LADs: Spatial Control Over Gene Expression. Trends Genet. 2016, 32, 485–495. [Google Scholar] [CrossRef]
- Meyer, B.J.; Casson, L.P. Caenorhabditis elegans Compensates for the Difference in X Chromosome Dosage between the Sexes by Regulating Transcript Levels. Cell 1986, 47, 871–881. [Google Scholar] [CrossRef]
- Tadros, W.; Lipshitz, H.D. The Maternal-to-Zygotic Transition: A Play in Two Acts. Dev. Camb. Engl. 2009, 136, 3033–3042. [Google Scholar] [CrossRef]
- Mutlu, B.; Chen, H.-M.; Moresco, J.J.; Orelo, B.D.; Yang, B.; Gaspar, J.M.; Keppler-Ross, S.; Yates, J.R.; Hall, D.H.; Maine, E.M.; et al. Regulated Nuclear Accumulation of a Histone Methyltransferase Times the Onset of Heterochromatin Formation in C. Elegans Embryos. Sci. Adv. 2018, 4, eaat6224. [Google Scholar] [CrossRef]
- Becker, J.S.; Nicetto, D.; Zaret, K.S. H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Genet. 2016, 32, 29–41. [Google Scholar] [CrossRef]
- Padeken, J.; Methot, S.P.; Gasser, S.M. Establishment of H3K9-Methylated Heterochromatin and Its Functions in Tissue Differentiation and Maintenance. Nat. Rev. Mol. Cell Biol. 2022, 23, 623–640. [Google Scholar] [CrossRef]
- Montavon, T.; Shukeir, N.; Erikson, G.; Engist, B.; Onishi-Seebacher, M.; Ryan, D.; Musa, Y.; Mittler, G.; Meyer, A.G.; Genoud, C.; et al. Complete Loss of H3K9 Methylation Dissolves Mouse Heterochromatin Organization. Nat. Commun. 2021, 12, 4359. [Google Scholar] [CrossRef]
- van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef]
- Bian, Q.; Anderson, E.C.; Yang, Q.; Meyer, B.J. Histone H3K9 Methylation Promotes Formation of Genome Compartments in Caenorhabditis elegans via Chromosome Compaction and Perinuclear Anchoring. Proc. Natl. Acad. Sci. USA 2020, 117, 11459–11470. [Google Scholar] [CrossRef]
- Sawh, A.N.; Shafer, M.E.R.; Su, J.-H.; Zhuang, X.; Wang, S.; Mango, S.E. Lamina-Dependent Stretching and Unconventional Chromosome Compartments in Early C. Elegans Embryos. Mol. Cell 2020, 78, 96–111.e6. [Google Scholar] [CrossRef]
- Towbin, B.D.; González-Aguilera, C.; Sack, R.; Gaidatzis, D.; Kalck, V.; Meister, P.; Askjaer, P.; Gasser, S.M. Step-Wise Methylation of Histone H3K9 Positions Heterochromatin at the Nuclear Periphery. Cell 2012, 150, 934–947. [Google Scholar] [CrossRef]
- Cabianca, D.S.; Muñoz-Jiménez, C.; Kalck, V.; Gaidatzis, D.; Padeken, J.; Seeber, A.; Askjaer, P.; Gasser, S.M. Active Chromatin Marks Drive Spatial Sequestration of Heterochromatin in C. Elegans Nuclei. Nature 2019, 569, 734–739. [Google Scholar] [CrossRef]
- Delaney, C.E.; Methot, S.P.; Guidi, M.; Katic, I.; Gasser, S.M.; Padeken, J. Heterochromatic Foci and Transcriptional Repression by an Unstructured MET-2/SETDB1 Co-Factor LIN-65. J. Cell Biol. 2019, 218, 820–838. [Google Scholar] [CrossRef]
- Delaney, C.E.; Methot, S.P.; Kalck, V.; Seebacher, J.; Hess, D.; Gasser, S.M.; Padeken, J. SETDB1-like MET-2 Promotes Transcriptional Silencing and Development Independently of Its H3K9me-Associated Catalytic Activity. Nat. Struct. Mol. Biol. 2022, 29, 85–96. [Google Scholar] [CrossRef]
- Mutlu, B.; Chen, H.-M.; Gutnik, S.; Hall, D.H.; Keppler-Ross, S.; Mango, S.E. Distinct Functions and Temporal Regulation of Methylated Histone H3 during Early Embryogenesis. Development 2019, 146, dev174516. [Google Scholar] [CrossRef]
- Padeken, J.; Methot, S.; Zeller, P.; Delaney, C.E.; Kalck, V.; Gasser, S.M. Argonaute NRDE-3 and MBT Domain Protein LIN-61 Redundantly Recruit an H3K9me3 HMT to Prevent Embryonic Lethality and Transposon Expression. Genes Dev. 2021, 35, 82–101. [Google Scholar] [CrossRef]
- Zeller, P.; Padeken, J.; van Schendel, R.; Kalck, V.; Tijsterman, M.; Gasser, S.M. Histone H3K9 Methylation Is Dispensable for Caenorhabditis elegans Development but Suppresses RNA:DNA Hybrid-Associated Repeat Instability. Nat. Genet. 2016, 48, 1385–1395. [Google Scholar] [CrossRef]
- Ho, J.W.K.; Jung, Y.L.; Liu, T.; Alver, B.H.; Lee, S.; Ikegami, K.; Sohn, K.-A.; Minoda, A.; Tolstorukov, M.Y.; Appert, A.; et al. Comparative Analysis of Metazoan Chromatin Organization. Nature 2014, 512, 449–452. [Google Scholar] [CrossRef]
- Vandamme, J.; Sidoli, S.; Mariani, L.; Friis, C.; Christensen, J.; Helin, K.; Jensen, O.N.; Salcini, A.E. H3K23me2 Is a New Heterochromatic Mark in Caenorhabditis elegans. Nucleic Acids Res. 2015, 43, 9694–9710. [Google Scholar] [CrossRef]
- Liu, T.; Rechtsteiner, A.; Egelhofer, T.A.; Vielle, A.; Latorre, I.; Cheung, M.-S.; Ercan, S.; Ikegami, K.; Jensen, M.; Kolasinska-Zwierz, P.; et al. Broad Chromosomal Domains of Histone Modification Patterns in C. elegans. Genome Res. 2011, 21, 227–236. [Google Scholar] [CrossRef]
- Garrigues, J.M.; Sidoli, S.; Garcia, B.A.; Strome, S. Defining Heterochromatin in C. Elegans through Genome-Wide Analysis of the Heterochromatin Protein 1 Homolog HPL-2. Genome Res. 2015, 25, 76–88. [Google Scholar] [CrossRef]
- Gaydos, L.J.; Wang, W.; Strome, S. H3K27me and PRC2 Transmit a Memory of Repression across Generations and during Development. Science 2014, 345, 1515–1518. [Google Scholar] [CrossRef]
- Gaydos, L.J.; Rechtsteiner, A.; Egelhofer, T.A.; Carroll, C.R.; Strome, S. Antagonism between MES-4 and Polycomb Repressive Complex 2 Promotes Appropriate Gene Expression in C. Elegans Germ Cells. Cell Rep. 2012, 2, 1169–1177. [Google Scholar] [CrossRef]
- Bender, L.B.; Cao, R.; Zhang, Y.; Strome, S. The MES-2/MES-3/MES-6 Complex and Regulation of Histone H3 Methylation in C. elegans. Curr. Biol. CB 2004, 14, 1639–1643. [Google Scholar] [CrossRef]
- Yuzyuk, T.; Fakhouri, T.H.I.; Kiefer, J.; Mango, S.E. The Polycomb Complex Protein Mes-2/E(z) Promotes the Transition from Developmental Plasticity to Differentiation in C. Elegans Embryos. Dev. Cell 2009, 16, 699–710. [Google Scholar] [CrossRef]
- Vandamme, J.; Lettier, G.; Sidoli, S.; Schiavi, E.D.; Jensen, O.N.; Salcini, A.E. The C. Elegans H3K27 Demethylase UTX-1 Is Essential for Normal Development, Independent of Its Enzymatic Activity. PLoS Genet. 2012, 8, e1002647. [Google Scholar] [CrossRef]
- Käser-Pébernard, S.; Pfefferli, C.; Aschinger, C.; Wicky, C. Fine-Tuning of Chromatin Composition and Polycomb Recruitment by Two Mi2 Homologues during C. Elegans Early Embryonic Development. Epigenetics Chromatin 2016, 9, 1–17. [Google Scholar] [CrossRef]
- Reynolds, N.; Latos, P.; Hynes-Allen, A.; Loos, R.; Leaford, D.; O’Shaughnessy, A.; Mosaku, O.; Signolet, J.; Brennecke, P.; Kalkan, T.; et al. NuRD Suppresses Pluripotency Gene Expression to Promote Transcriptional Heterogeneity and Lineage Commitment. Cell Stem Cell 2012, 10, 583–594. [Google Scholar] [CrossRef]
- O’Shaughnessy-Kirwan, A.; Signolet, J.; Costello, I.; Gharbi, S.; Hendrich, B. Constraint of Gene Expression by the Chromatin Remodelling Protein CHD4 Facilitates Lineage Specification. Development 2015, 142, 2586–2597. [Google Scholar] [CrossRef]
- Hendrich, B.; Guy, J.; Ramsahoye, B.; Wilson, V.A.; Bird, A. Closely Related Proteins MBD2 and MBD3 Play Distinctive but Interacting Roles in Mouse Development. Genes Dev. 2001, 15, 710–723. [Google Scholar] [CrossRef]
- Myers, T.R.; Amendola, P.G.; Lussi, Y.C.; Salcini, A.E. JMJD-1.2 Controls Multiple Histone Post-Translational Modifications in Germ Cells and Protects the Genome from Replication Stress. Sci. Rep. 2018, 8, 3765. [Google Scholar] [CrossRef]
- Jack, A.P.M.; Bussemer, S.; Hahn, M.; Pünzeler, S.; Snyder, M.; Wells, M.; Csankovszki, G.; Solovei, I.; Schotta, G.; Hake, S.B. H3K56me3 Is a Novel, Conserved Heterochromatic Mark That Largely but Not Completely Overlaps with H3K9me3 in Both Regulation and Localization. PLoS ONE 2013, 8, e51765. [Google Scholar] [CrossRef]
- McKittrick, E.; Gafken, P.R.; Ahmad, K.; Henikoff, S. Histone H3.3 Is Enriched in Covalent Modifications Associated with Active Chromatin. Proc. Natl. Acad. Sci. USA 2004, 101, 1525–1530. [Google Scholar] [CrossRef]
- Loyola, A.; Bonaldi, T.; Roche, D.; Imhof, A.; Almouzni, G. PTMs on H3 Variants before Chromatin Assembly Potentiate Their Final Epigenetic State. Mol. Cell 2006, 24, 309–316. [Google Scholar] [CrossRef]
- Hake, S.B.; Garcia, B.A.; Duncan, E.M.; Kauer, M.; Dellaire, G.; Shabanowitz, J.; Bazett-Jones, D.P.; Allis, C.D.; Hunt, D.F. Expression Patterns and Post-Translational Modifications Associated with Mammalian Histone H3 Variants. J. Biol. Chem. 2006, 281, 559–568. [Google Scholar] [CrossRef]
- Kreher, J.; Takasaki, T.; Cockrum, C.; Sidoli, S.; Garcia, B.A.; Jensen, O.N.; Strome, S. Distinct Roles of Two Histone Methyltransferases in Transmitting H3K36me3-Based Epigenetic Memory Across Generations in Caenorhabditis elegans. Genetics 2018, 210, 969–982. [Google Scholar] [CrossRef]
- Gleason, R.J.; Guo, Y.; Semancik, C.S.; Ow, C.; Lakshminarayanan, G.; Chen, X. Developmentally Programmed Histone H3 Expression Regulates Cellular Plasticity at the Parental-to-Early Embryo Transition. Sci. Adv. 2023, 9, eadh0411. [Google Scholar] [CrossRef]
- Ishiuchi, T.; Abe, S.; Inoue, K.; Yeung, W.K.A.; Miki, Y.; Ogura, A.; Sasaki, H. Reprogramming of the Histone H3.3 Landscape in the Early Mouse Embryo. Nat. Struct. Mol. Biol. 2021, 28, 38–49. [Google Scholar] [CrossRef]
- Jedrusik, M.A.; Schulze, E. A Single Histone H1 Isoform (H1.1) Is Essential for Chromatin Silencing and Germline Development in Caenorhabditis elegans. Development 2001, 128, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Jedrusik, M.A.; Schulze, E. Linker Histone HIS-24 (H1.1) Cytoplasmic Retention Promotes Germ Line Development and Influences Histone H3 Methylation in Caenorhabditis elegans. Mol. Cell. Biol. 2007, 27, 2229–2239. [Google Scholar] [CrossRef]
- Jedrusik-Bode, M. Histone H1 and Heterochromatin Protein 1 (HP1) Regulate Specific Gene Expression and Not Global Transcription. Worm 2013, 2, e23703. [Google Scholar] [CrossRef]
- Arico, J.K.; Katz, D.J.; van der Vlag, J.; Kelly, W.G. Epigenetic Patterns Maintained in Early Caenorhabditis elegans Embryos Can Be Established by Gene Activity in the Parental Germ Cells. PLoS Genet. 2011, 7, e1001391. [Google Scholar] [CrossRef]
- Samson, M.; Jow, M.M.; Wong, C.C.L.; Fitzpatrick, C.; Aslanian, A.; Saucedo, I.; Estrada, R.; Ito, T.; Park, S.R.; Iii, J.R.Y.; et al. The Specification and Global Reprogramming of Histone Epigenetic Marks during Gamete Formation and Early Embryo Development in C. elegans. PLoS Genet. 2014, 10, e1004588. [Google Scholar] [CrossRef]
- Rechtsteiner, A.; Ercan, S.; Takasaki, T.; Phippen, T.M.; Egelhofer, T.A.; Wang, W.; Kimura, H.; Lieb, J.D.; Strome, S. The Histone H3K36 Methyltransferase MES-4 Acts Epigenetically to Transmit the Memory of Germline Gene Expression to Progeny. PLoS Genet. 2010, 6, e1001091. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Fisher, K.; Poulin, G.B. Lineage Specific Trimethylation of H3 on Lysine 4 during C. Elegans Early Embryogenesis. Dev. Biol. 2011, 355, 227–238. [Google Scholar] [CrossRef]
- Reuben, M.; Lin, R. Germline X Chromosomes Exhibit Contrasting Patterns of Histone H3 Methylation in Caenorhabditis elegans. Dev. Biol. 2002, 245, 71–82. [Google Scholar] [CrossRef]
- Kelly, W.G.; Schaner, C.E.; Dernburg, A.F.; Lee, M.-H.; Kim, S.K.; Villeneuve, A.M.; Reinke, V. X-Chromosome Silencing in the Germline of C. elegans. Development 2002, 129, 479–492. [Google Scholar] [CrossRef]
- Riddle, D.L.; Blumenthal, T.; Meyer, B.J.; Priess, J.R. Specification of Cell Fates in the AB Lineage. In C. elegans II, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Fisher, K.; Southall, S.M.; Wilson, J.R.; Poulin, G.B. Methylation and Demethylation Activities of a C. Elegans MLL-like Complex Attenuate RAS Signalling. Dev. Biol. 2010, 341, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Agger, K.; Cloos, P.A.C.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 Are Histone H3K27 Demethylases Involved in HOX Gene Regulation and Development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Bedet, C.; Robert, V.J.P.; Simonet, T.; Dunkelbarger, S.; Rakotomalala, C.; Soete, G.; Korswagen, H.C.; Strome, S.; Palladino, F. Caenorhabditis elegans Chromatin-Associated Proteins SET-2 and ASH-2 Are Differentially Required for Histone H3 Lys 4 Methylation in Embryos and Adult Germ Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8305–8310. [Google Scholar] [CrossRef] [PubMed]
- Andersen, E.C.; Horvitz, H.R. Two C. Elegans Histone Methyltransferases Repress Lin-3EGF Transcription to Inhibit Vulval Development. Development 2007, 134, 2991–2999. [Google Scholar] [CrossRef] [PubMed]
- Whittle, C.M.; McClinic, K.N.; Ercan, S.; Zhang, X.; Green, R.D.; Kelly, W.G.; Lieb, J.D. The Genomic Distribution and Function of Histone Variant HTZ-1 during C. Elegans Embryogenesis. PLoS Genet. 2008, 4, e1000187. [Google Scholar] [CrossRef] [PubMed]
- Borcherds, W.; Bremer, A.; Borgia, M.B.; Mittag, T. How Do Intrinsically Disordered Protein Regions Encode a Driving Force for Liquid–Liquid Phase Separation? Curr. Opin. Struct. Biol. 2021, 67, 41–50. [Google Scholar] [CrossRef]
- Saito, M.; Hess, D.; Eglinger, J.; Fritsch, A.W.; Kreysing, M.; Weinert, B.T.; Choudhary, C.; Matthias, P. Acetylation of Intrinsically Disordered Regions Regulates Phase Separation. Nat. Chem. Biol. 2019, 15, 51–61. [Google Scholar] [CrossRef]
- Methot, S.P.; Padeken, J.; Brancati, G.; Zeller, P.; Delaney, C.E.; Gaidatzis, D.; Kohler, H.; van Oudenaarden, A.; Großhans, H.; Gasser, S.M. H3K9me Selectively Blocks Transcription Factor Activity and Ensures Differentiated Tissue Integrity. Nat. Cell Biol. 2021, 23, 1163–1175. [Google Scholar] [CrossRef]
- Towbin, B.D.; Meister, P.; Pike, B.L.; Gasser, S.M. Repetitive Transgenes in C. Elegans Accumulate Heterochromatic Marks and Are Sequestered at the Nuclear Envelope in a Copy-Number- and Lamin-Dependent Manner. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 555–565. [Google Scholar] [CrossRef]
- Snyder, M.J.; Lau, A.C.; Brouhard, E.A.; Davis, M.B.; Jiang, J.; Sifuentes, M.H.; Csankovszki, G. Anchoring of Heterochromatin to the Nuclear Lamina Reinforces Dosage Compensation-Mediated Gene Repression. PLoS Genet. 2016, 12, e1006341. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sandoval, A.; Towbin, B.D.; Kalck, V.; Cabianca, D.S.; Gaidatzis, D.; Hauer, M.H.; Geng, L.; Wang, L.; Yang, T.; Wang, X.; et al. Perinuclear Anchoring of H3K9-Methylated Chromatin Stabilizes Induced Cell Fate in C. Elegans Embryos. Cell 2015, 163, 1333–1347. [Google Scholar] [CrossRef]
- Ikegami, K.; Egelhofer, T.A.; Strome, S.; Lieb, J.D. Caenorhabditis elegans Chromosome Arms Are Anchored to the Nuclear Membrane via Discontinuous Association with LEM-2. Genome Biol. 2010, 11, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.; Kranz, A.-L.; Su, A.; Winterkorn, L.H.; Albritton, S.E.; Ercan, S. Developmental Dynamics of X-Chromosome Dosage Compensation by the DCC and H4K20me1 in C. elegans. PLoS Genet. 2015, 11, e1005698. [Google Scholar] [CrossRef]
- Vielle, A.; Lang, J.; Dong, Y.; Ercan, S.; Kotwaliwale, C.; Rechtsteiner, A.; Appert, A.; Chen, Q.B.; Dose, A.; Egelhofer, T.; et al. H4K20me1 Contributes to Downregulation of X-Linked Genes for C. Elegans Dosage Compensation. PLoS Genet. 2012, 8, e1002933. [Google Scholar] [CrossRef]
- Brejc, K.; Bian, Q.; Uzawa, S.; Wheeler, B.S.; Anderson, E.C.; King, D.S.; Kranzusch, P.J.; Preston, C.G.; Meyer, B.J. Dynamic Control of X Chromosome Conformation and Repression by a Histone H4K20 Demethylase. Cell 2017, 171, 85–102.e23. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.B.; Snyder, M.J.; Custer, L.M.; Csankovszki, G. Caenorhabditis elegans Dosage Compensation Regulates Histone H4 Chromatin State on X Chromosomes. Mol. Cell. Biol. 2012, 32, 1710–1719. [Google Scholar] [CrossRef]
- Custer, L.M.; Snyder, M.J.; Flegel, K.; Csankovszki, G. The Onset of C. Elegans Dosage Compensation Is Linked to the Loss of Developmental Plasticity. Dev. Biol. 2014, 385, 279–290. [Google Scholar] [CrossRef]
- Manzo, S.G.; Dauban, L.; van Steensel, B. Lamina-Associated Domains: Tethers and Looseners. Curr. Opin. Cell Biol. 2022, 74, 80–87. [Google Scholar] [CrossRef]
- Poleshko, A.; Smith, C.L.; Nguyen, S.C.; Sivaramakrishnan, P.; Wong, K.G.; Murray, J.I.; Lakadamyali, M.; Joyce, E.F.; Jain, R.; Epstein, J.A. H3K9me2 Orchestrates Inheritance of Spatial Positioning of Peripheral Heterochromatin through Mitosis. eLife 2019, 8, e49278. [Google Scholar] [CrossRef] [PubMed]
- See, K.; Kiseleva, A.A.; Smith, C.L.; Liu, F.; Li, J.; Poleshko, A.; Epstein, J.A. Histone Methyltransferase Activity Programs Nuclear Peripheral Genome Positioning. Dev. Biol. 2020, 466, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Takano-Maruyama, M.; Pereira-Smith, O.M.; Gaufo, G.O.; Tominaga, K. MRG15, a Component of HAT and HDAC Complexes, Is Essential for Proliferation and Differentiation of Neural Precursor Cells. J. Neurosci. Res. 2009, 87, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Pardo, P.S.; Leung, J.K.; Lucchesi, J.C.; Pereira-Smith, O.M. MRG15, a Novel Chromodomain Protein, Is Present in Two Distinct Multiprotein Complexes Involved in Transcriptional Activation*. J. Biol. Chem. 2002, 277, 50860–50866. [Google Scholar] [CrossRef]
- Hajduskova, M.; Baytek, G.; Kolundzic, E.; Gosdschan, A.; Kazmierczak, M.; Ofenbauer, A.; Beato del Rosal, M.L.; Herzog, S.; ul Fatima, N.; Mertins, P.; et al. MRG-1/MRG15 Is a Barrier for Germ Cell to Neuron Reprogramming in Caenorhabditis elegans. Genetics 2019, 211, 121–139. [Google Scholar] [CrossRef]
- Shi, Y.; Mello, C. A CBP/P300 Homolog Specifies Multiple Differentiation Pathways in Caenorhabditis elegans. Genes Dev. 1998, 12, 943–955. [Google Scholar] [CrossRef]
- Ferreira, H.C.; Towbin, B.D.; Jegou, T.; Gasser, S.M. The Shelterin Protein POT-1 Anchors Caenorhabditis elegans Telomeres through SUN-1 at the Nuclear Periphery. J. Cell Biol. 2013, 203, 727–735. [Google Scholar] [CrossRef]
- Ferreira, H.C.; Luke, B.; Schober, H.; Kalck, V.; Lingner, J.; Gasser, S.M. The PIAS Homologue Siz2 Regulates Perinuclear Telomere Position and Telomerase Activity in Budding Yeast. Nat. Cell Biol. 2011, 13, 867–874. [Google Scholar] [CrossRef]
- Flyamer, I.M.; Gassler, J.; Imakaev, M.; Brandão, H.B.; Ulianov, S.V.; Abdennur, N.; Razin, S.V.; Mirny, L.A.; Tachibana-Konwalski, K. Single-Nucleus Hi-C Reveals Unique Chromatin Reorganization at Oocyte-to-Zygote Transition. Nature 2017, 544, 110–114. [Google Scholar] [CrossRef]
- Cardozo Gizzi, A.M.; Cattoni, D.I.; Nollmann, M. TADs or No TADS: Lessons from Single-Cell Imaging of Chromosome Architecture. J. Mol. Biol. 2020, 432, 682–693. [Google Scholar] [CrossRef]
- Stevens, T.J.; Lando, D.; Basu, S.; Atkinson, L.P.; Cao, Y.; Lee, S.F.; Leeb, M.; Wohlfahrt, K.J.; Boucher, W.; O’Shaughnessy-Kirwan, A.; et al. 3D Structures of Individual Mammalian Genomes Studied by Single-Cell Hi-C. Nature 2017, 544, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, W.; Abdennur, N.; Goloborodko, A.; Pekowska, A.; Fudenberg, G.; Loe-Mie, Y.; Fonseca, N.A.; Huber, W.; Haering, C.H.; Mirny, L.; et al. Two Independent Modes of Chromatin Organization Revealed by Cohesin Removal. Nature 2017, 551, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Nora, E.P.; Goloborodko, A.; Valton, A.-L.; Gibcus, J.H.; Uebersohn, A.; Abdennur, N.; Dekker, J.; Mirny, L.A.; Bruneau, B.G. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 2017, 169, 930–944.e22. [Google Scholar] [CrossRef] [PubMed]
- Heger, P.; Marin, B.; Schierenberg, E. Loss of the Insulator Protein CTCF during Nematode Evolution. BMC Mol. Biol. 2009, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Crane, E.; Bian, Q.; McCord, R.P.; Lajoie, B.R.; Wheeler, B.S.; Ralston, E.J.; Uzawa, S.; Dekker, J.; Meyer, B.J. Condensin-Driven Remodelling of X Chromosome Topology during Dosage Compensation. Nature 2015, 523, 240–244. [Google Scholar] [CrossRef]
- Lambert, J.; Lloret-Fernández, C.; Laplane, L.; Poole, R.J.; Jarriault, S. Chapter Five—On the Origins and Conceptual Frameworks of Natural Plasticity—Lessons from Single-Cell Models in C. elegans. In Current Topics in Developmental Biology; Jarriault, S., Podbilewicz, B., Eds.; Nematode Models of Development and Disease; Academic Press: Cambridge, MA, USA, 2021; Volume 144, pp. 111–159. [Google Scholar]
- Wood, W.B. Evidence from Reversal of Handedness in C. Elegans Embryos for Early Cell Interactions Determining Cell Fates. Nature 1991, 349, 536–538. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Fukushige, T.; McGhee, J.D.; Rothman, J.H. Reprogramming of Early Embryonic Blastomeres into Endodermal Progenitors by a Caenorhabditis elegans GATA Factor. Genes Dev. 1998, 12, 3809–3814. [Google Scholar] [CrossRef]
- Fukushige, T.; Krause, M. The Myogenic Potency of HLH-1 Reveals Wide-Spread Developmental Plasticity in Early C. Elegans Embryos. Development 2005, 132, 1795–1805. [Google Scholar] [CrossRef]
- Djabrayan, N.J.-V.; Dudley, N.R.; Sommermann, E.M.; Rothman, J.H. Essential Role for Notch Signaling in Restricting Developmental Plasticity. Genes Dev. 2012, 26, 2386–2391. [Google Scholar] [CrossRef]
- Bucher, E.A.; Seydoux, G. Gastrulation in the Nematode Caenorhabditis elegans. Semin. Dev. Biol. 1994, 5, 121–130. [Google Scholar] [CrossRef]
- Chen, J.; Liu, H.; Liu, J.; Qi, J.; Wei, B.; Yang, J.; Liang, H.; Chen, Y.; Chen, J.; Wu, Y.; et al. H3K9 Methylation Is a Barrier during Somatic Cell Reprogramming into iPSCs. Nat. Genet. 2013, 45, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Soufi, A.; Donahue, G.; Zaret, K.S. Facilitators and Impediments of the Pluripotency Reprogramming Factors’ Initial Engagement with the Genome. Cell 2012, 151, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Dodge, J.E.; Kang, Y.-K.; Beppu, H.; Lei, H.; Li, E. Histone H3-K9 Methyltransferase ESET Is Essential for Early Development. Mol. Cell. Biol. 2004, 24, 2478–2486. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Sugimoto, K.; Nozaki, M.; Ueda, J.; Ohta, T.; Ohki, M.; Fukuda, M.; Takeda, N.; Niida, H.; Kato, H.; et al. G9a Histone Methyltransferase Plays a Dominant Role in Euchromatic Histone H3 Lysine 9 Methylation and Is Essential for Early Embryogenesis. Genes Dev. 2002, 16, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.F.; Piccolo, F.M.; Tsubouchi, T.; Sauer, S.; Ryan, N.K.; Bruno, L.; Landeira, D.; Santos, J.; Banito, A.; Gil, J.; et al. ESCs Require PRC2 to Direct the Successful Reprogramming of Differentiated Cells toward Pluripotency. Cell Stem Cell 2010, 6, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Hsu, D.R.; Chuang, P.-T.; Meyer, B.J. DPY-30, a Nuclear Protein Essential Early in Embryogenesis for Caenorhabditis elegans Dosage Compensation. Development 1995, 121, 3323–3334. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.R.; Jow, M.M.; Meyer, B.J. The T-Box Transcription Factor SEA-1 Is an Autosomal Element of the X:A Signal That Determines C. Elegans Sex. Dev. Cell 2005, 9, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Carmi, I.; Kopczynski, J.B.; Meyer, B.J. The Nuclear Hormone Receptor SEX-1 Is an X-Chromosome Signal That Determines Nematode Sex. Nature 1998, 396, 168–173. [Google Scholar] [CrossRef]
- Carmi, I.; Meyer, B.J. The Primary Sex Determination Signal of Caenorhabditis elegans. Genetics 1999, 152, 999–1015. [Google Scholar] [CrossRef]
- Nicoll, M.; Akerib, C.C.; Meyer, B.J. X-Chromosome-Counting Mechanisms That Determine Nematode Sex. Nature 1997, 388, 200–204. [Google Scholar] [CrossRef]
- Farboud, B.; Nix, P.; Jow, M.M.; Gladden, J.M.; Meyer, B.J. Molecular Antagonism between X-Chromosome and Autosome Signals Determines Nematode Sex. Genes Dev. 2013, 27, 1159–1178. [Google Scholar] [CrossRef]
- Gladden, J.M.; Meyer, B.J. A ONECUT Homeodomain Protein Communicates X Chromosome Dose to Specify Caenorhabditis elegans Sexual Fate by Repressing a Sex Switch Gene. Genetics 2007, 177, 1621–1637. [Google Scholar] [CrossRef] [PubMed]
- Farboud, B.; Novak, C.S.; Nicoll, M.; Quiogue, A.; Meyer, B.J. Dose-Dependent Action of the RNA Binding Protein FOX-1 to Relay X-Chromosome Number and Determine C. Elegans Sex. eLife 2020, 9, e62963. [Google Scholar] [CrossRef]
- Rhind, N.R.; Miller, L.M.; Kopczynski, J.B.; Meyer, B.J. Xol-1 Acts as an Early Switch in the C. Elegans Male/Hermaphrodite Decision. Cell 1995, 80, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.C.; Nabeshima, K.; Csankovszki, G. The C. Elegans Dosage Compensation Complex Mediates Interphase X Chromosome Compaction. Epigenetics Chromatin 2014, 7, 31. [Google Scholar] [CrossRef]
- Csankovszki, G.; Collette, K.; Spahl, K.; Carey, J.; Snyder, M.; Petty, E.; Patel, U.; Tabuchi, T.; Liu, H.; McLeod, I.; et al. Three Distinct Condensin Complexes Control C. Elegans Chromosome Dynamics. Curr. Biol. 2009, 19, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Chuang, P.-T.; Albertson, D.G.; Meyer, B.J. DPY-27: A Chromosome Condensation Protein Homolog That Regulates C. Elegans Dosage Compensation through Association with the X Chromosome. Cell 1994, 79, 459–474. [Google Scholar] [CrossRef]
- Csankovszki, G.; McDonel, P.; Meyer, B.J. Recruitment and Spreading of the C. Elegans Dosage Compensation Complex Along X Chromosomes. Science 2004, 303, 1182–1185. [Google Scholar] [CrossRef]
- Ercan, S.; Dick, L.L.; Lieb, J.D. The C. Elegans Dosage Compensation Complex Propagates Dynamically and Independently of X Chromosome Sequence. Curr. Biol. CB 2009, 19, 1777–1787. [Google Scholar] [CrossRef]
- Blauwkamp, T.A.; Csankovszki, G. Two Classes of Dosage Compensation Complex Binding Elements along Caenorhabditis elegans X Chromosomes. Mol. Cell. Biol. 2009, 29, 2023–2031. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jimenez, D.S.; Ragipani, B.; Zhang, B.; Street, L.A.; Kramer, M.; Albritton, S.E.; Winterkorn, L.H.; Morao, A.K.; Ercan, S. Condensin DC Loads and Spreads from Recruitment Sites to Create Loop-Anchored TADs in C. elegans. eLife 2022, 11, e68745. [Google Scholar] [CrossRef] [PubMed]
- Kruesi, W.S.; Core, L.J.; Waters, C.T.; Lis, J.T.; Meyer, B.J. Condensin Controls Recruitment of RNA Polymerase II to Achieve Nematode X-Chromosome Dosage Compensation. eLife 2013, 2, e00808. [Google Scholar] [CrossRef]
- Anderson, E.C.; Frankino, P.A.; Higuchi-Sanabria, R.; Yang, Q.; Bian, Q.; Podshivalova, K.; Shin, A.; Kenyon, C.; Dillin, A.; Meyer, B.J. X Chromosome Domain Architecture Regulates Caenorhabditis elegans Lifespan but Not Dosage Compensation. Dev. Cell 2019, 51, 192–207.e6. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.B.; Jash, E.; Chawla, B.; Haines, R.A.; Tushman, L.E.; Troll, R.; Csankovszki, G. Dual Roles for Nuclear RNAi Argonautes in Caenorhabditis elegans Dosage Compensation. Genetics 2022, 221, iyac033. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of tethering pathways for heterochromatin and telomeric chromatin.
Figure 1.
Schematic representation of tethering pathways for heterochromatin and telomeric chromatin.

Figure 2.
Timeline of chromatin reorganization during embryogenesis.


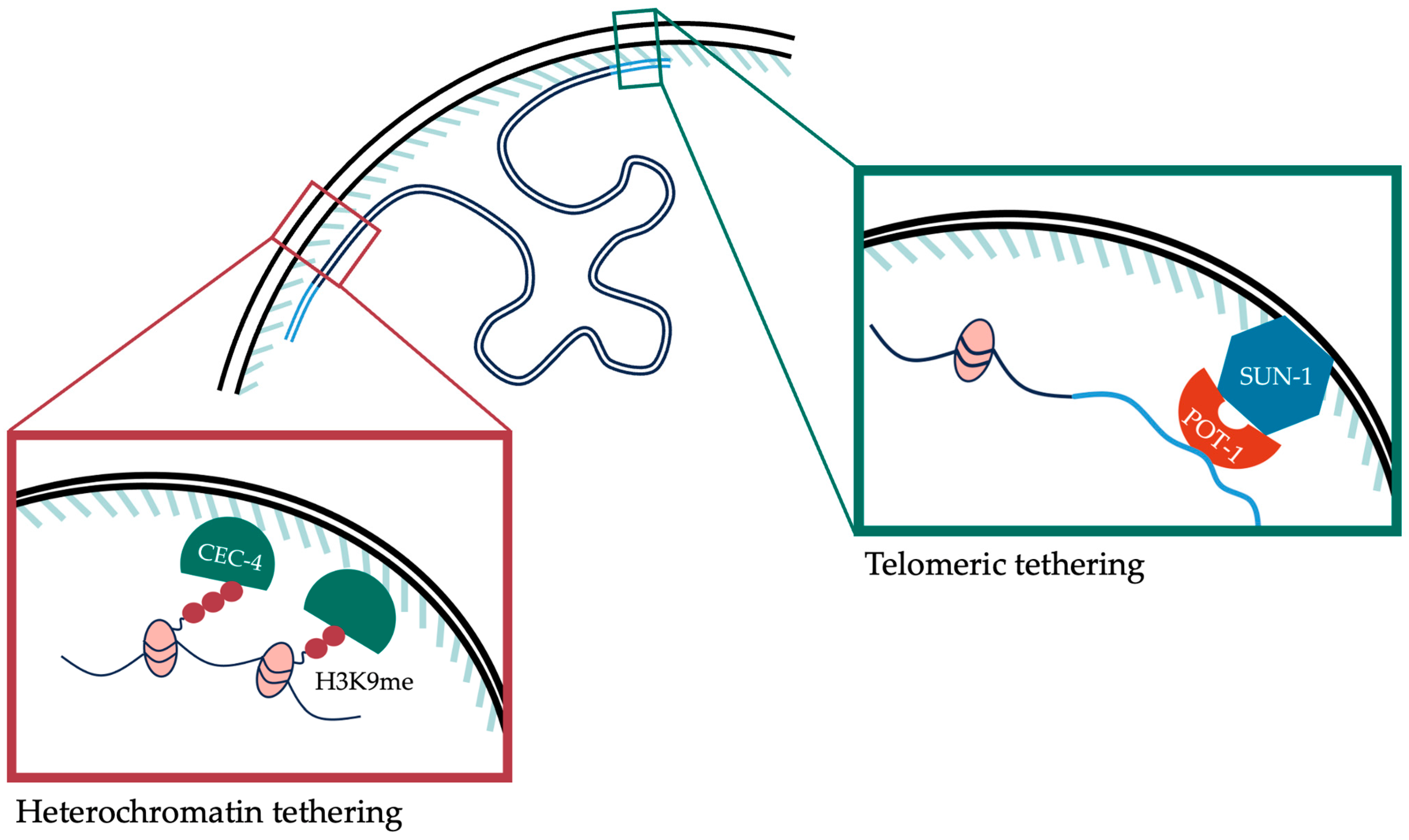{kind=link}
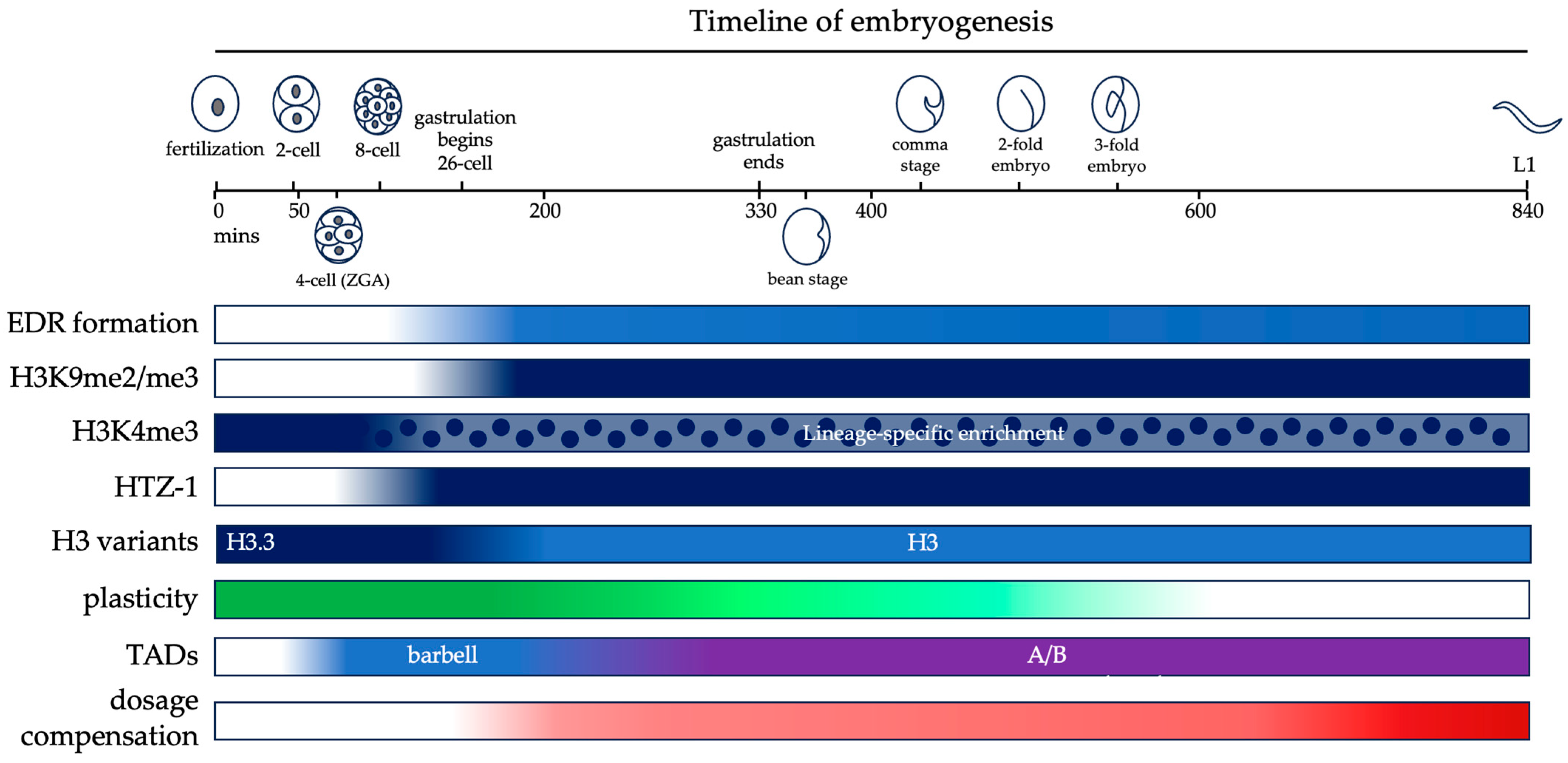{kind=link}
Table 1.
Histone variants, associated PTMs and functions in embryogenesis.
| Histone Variant | Histone Marks | Active/Repressive | Localization and Function | Ref |
|---|---|---|---|---|
| H3 | H3K9me2/me3, H3K27me2/me3 | Repressive | Canonical histone H3 that favors the deposition of repressive histone marks. Depleted from chromatin in early embryos. Transcribed in embryos from 2-cell stage and accumulates on chromatin throughout early embryogenesis on all cells except P-lineage cells. Promotes termination of developmental plasticity. | [39,42] |
| H3.3 | H3K4me3, H3K36me2 | Active | Histone H3 variant that favors the deposition of active histone marks. Inherited from germ cells in early embryos in 2-cell to 50-cell stage. Depleted from chromatin during embryo development, except in P-lineage cells. | [38,39,41,42] |
| H1.1 | H3K9me2 | Repressive | Linker histone H1 that promotes the accumulation of repressive histone marks. Rapidly translocated from the cytoplasm into the nucleus after fertilization. Associated with chromatin in all embryonic cells except Z2 and Z3 PGCs. Promotes the silencing of heterochromatic loci. | [44,45] |
| HTZ-1 | Active | Histone H2A variant enriched upstream of transcribed genes required for development where it influences PolII engagement. Incorporated into chromatin starting at 4-cell stage and required for appropriate embryonic development. | [58] |
Table 2.
Histone modifications and their functions during embryogenesis.
| Histone Modification | Histone Methyl/Acetyl Transferase | Histone Demethylase/Deacetylase | Active/Repressive | Function | Ref |
|---|---|---|---|---|---|
| H3K9me2 | met-2 | jmjd-1.2 | Repressive | Dynamically increases during gastrulation. Repressive mark enriched on heterochromatin. High levels of H3K9me2 promotes developmental plasticity. | [9,14,19,20,25,36,61] |
| H3K9me3 | set-25 | Repressive | Dynamically increases during gastrulation. Repressive mark enriched on heterochromatin. Linked to heterochromatin formation, tethering of the chromatin to nuclear periphery, TAD formation and establishment of dosage compensation on the hermaphrodite X chromosome. | [14,16,18,20,21,25,62,63,64] | |
| H3K27me3 | mes-2 | utx-1, jmjd-3.1, jmjd-3.2, jmjd-3.3 | Repressive | Inherited from maternal and paternal germ cells and dynamically enriched in embryonic cells in a lineage-specific manner. Repressive mark enriched on heterochromatin and LADs. Promotes termination of developmental plasticity. | [25,27,28,29,30,62,65] |
| H3K23me2 | set-32 | jmjd-1.2 | Repressive | Repressive mark enriched on heterochromatin. | [24,25,36] |
| H4K20me1/me2 | set-4 deposits H4K20me2 | dpy-21 converts H4K20me2 to H4K20me1 on the X chromosome | Repressive | H4K20me1 is selectively enriched on the X chromosome by dpy-21. Repressive mark required for the establishment of dosage compensation in hermaphrodite embryos. | [66,67,68,69,70] |
| H3K4me2 | ash-2, set-16 | Active | Active mark inherited from paternal and maternal germ cells, generally enriched over gene bodies where it permits transcription at genomic loci. Enriched uniformly in all cells in early embryos, enriched specifically on PGCs in late embryos. | [2,47,50,51,52,54,56,57] | |
| H3K4me3 | ash-2, set-2, set-16 | Active | Active mark inherited from paternal and maternal germ cells, generally enriched at TSS where it permits transcription at genomic loci. Enriched uniformly in all cells in early embryos. Enriched in a lineage-specific manner starting at the eight-cell stage. | [2,49,50,51,52,54,57] | |
| H3K36me3 | mes-4, met-1 | Active | Active mark enriched on euchromatin. Required for expression of germline-specific genes in early embryos. | [17,28] | |
| H3K9ac | Active | Active mark excluded from heterochromatin by MET-2 nuclear foci. | [19] | ||
| H3K27ac | Active | Active mark excluded from heterochromatin by MET-2 nuclear foci. | [19] | ||
| H4K16ac | Active | Active mark inherited from maternal germ cells and selectively depleted from the X chromosome. | [69,70] | ||
| H3K56me3 | Repressive | Repressive mark enriched on heterochromatin. | [37] | ||
| H3K79me2 | Active | Active mark inherited from paternal and maternal germ cells. Depleted on chromatin in 1–4 cell embryos, then enriched after 16-cell stage. | [48] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jash, E.; Csankovszki, G. Chromatin Organization during C. elegans Early Development. DNA 2024, 4, 64-83. https://doi.org/10.3390/dna4010004
AMA Style
Jash E, Csankovszki G. Chromatin Organization during C. elegans Early Development. DNA. 2024; 4(1):64-83. https://doi.org/10.3390/dna4010004
Chicago/Turabian StyleJash, Eshna, and Györgyi Csankovszki. 2024. "Chromatin Organization during C. elegans Early Development" DNA 4, no. 1: 64-83. https://doi.org/10.3390/dna4010004