1. Introduction
Infertility, defined by the World Health Organization (WHO) as “the failure to achieve a pregnancy after 12 months or more of regular unprotected sexual intercourse” remains a significant concern for couples of reproductive age. Male-related factors contribute to approximately 50% of infertility cases [
1,
2], underscoring the essential need for a precise evaluation of semen quality to gauge male fertilization potential [
3]. While sperm count and motility stand as primary parameters assessed in semen quality analysis, the structure of spermatozoa plays an equally vital and intricate role in determining the fertilization potential of male reproductive cells [
4]. Specifically, teratozoospermia is characterized by a lower percentage of normally shaped sperm compared to established reference limits. The definition of “normal” has evolved significantly over time, transitioning from 50% in 1980 [
5] to 4% in the WHO classification published in 2010 [
6]. This condition encompasses a spectrum of morphological deviations impacting diverse components of sperm structure, including the head, neck, midpiece, and tail [
7,
8]. Beyond presenting a wide array of sperm irregularities, teratozoospermia also exists across varying degrees of severity that directly influence male fertilization capacity [
8]. In general, the morphological features of sperm cells result from highly intricate cellular transformations that occur during spermatogenesis [
9] and intriguingly, aberrant sperm morphology has been linked to increased indicators of sperm damage, such as DNA fragmentation [
2] and overproduction of reactive oxygen species (ROS) [
10,
11].
Despite notable advancements in exploring teratozoospermia, a comprehensive understanding of the molecular mechanisms responsible for this male infertility condition remains elusive. In broad terms, unraveling the molecular origins of male infertility presents a substantial hurdle as more than 4000 genes are involved in the spermatogenesis process [
12]. However, recently, various genes have been associated with teratozoospermia.
The Aurora Kinase C gene (
AURKC), located on chromosome 19q13.43, encodes a member of a family of highly conserved serine/threonine kinases that are crucial for chromosome segregation during both mitosis and meiosis [
13]. The two other family members,
AURKA and
AURKCB, are highly expressed in many cancer types and act as oncogenes [
14]. Limited information is available regarding the involvement of
AURKC in oncogenesis but it is also found to be overexpressed in certain cancers [
15]. Concerning male infertility,
AURKC is expressed in meiotic cells, and pathogenic mutations in this gene can disrupt the protein’s function, leading to improper mitotic spindle formation and subsequently causing male infertility [
16]. To this day, various mutations of
AURKC have been discovered, affecting protein function and resulting in a specific form of teratozoospermia known as macrozoospermia or large-headed spermatozoa [
16,
17,
18,
19,
20,
21].
SPATA16 (spermatogenesis-associated protein 16), located on chromosome 3q26.31 and formerly recognized as
NYD-SP12, exhibits high expression in the human testes, particularly during puberty, where it plays a significant role in its development [
22]. It features a conserved tetratricopeptide repeat (TPR) domain, known for its role in facilitating protein–protein interactions [
23]. This protein localizes within the Golgi apparatus and proacrosomal vesicles, which merge during spermiogenesis to form the acrosome [
24]. Several studies have identified mutations within
SPATA16 that lead to a distinct form of teratozoospermia known as globozoospermia [
25,
26]. Globozoospermia is characterized by the presence of round-headed spermatozoa that lack an acrosome. Furthermore, research conducted in mice underscores the significance of
SPATA16 in the process of sperm formation [
27]. Mutations in
SPATA16 have also been linked to other types of male infertility [
28].
SUN5 is a gene located on chromosome 20q11.21, encoding for a transmembrane protein consisting of an N-terminal nucleoplasmic section, a coiled-coil region, a transmembrane helical domain, and a SUN domain segment [
29]. It is a testis-specific gene [
30] and its encoded protein localizes to the junction between the sperm head and tail [
29,
31]. SUN5 belongs to the family of SUN domain proteins, which play a role in tethering the centrosome to the nuclear membrane [
32]. SUN5 is a relatively recent addition to the SUN family, and while limited information is available regarding its function [
31], it is suggested that it may be involved in nuclear envelope reconstitution and nuclear migration [
29]. Notably, studies conducted in mice have shown that Sun5
−/− mice are infertile, and in the absence of functional SUN5, the sperm head-to-tail coupling apparatus becomes detached from the nucleus during spermatid elongation [
31]. Additionally, several mutations in this gene have been identified, which are associated with acephalic spermatozoa syndrome, a severe form of teratozoospermia [
29,
33,
34,
35].
The aforementioned studies confirm the pivotal role of the mentioned genes in the pathogenesis of teratozoospermia and underscore the significance of specific mutations in its etiology. Nevertheless, the number of mutations associated with male infertility remains relatively limited. Single nucleotide polymorphisms (SNPs) are the most prevalent type of genetic mutation, occurring in the genome approximately every 100 to 300 base pairs [
36]. While mutations in coding regions are typically linked to the development of various diseases due to alterations in the amino acid sequence, research suggests that SNPs located in non-coding regions are more likely to contribute to the pathogenesis of most genetic disorders [
37]. More specifically, variants found in non-coding regions can exert various regulatory functions within the genome, including disruption of interactions with transcription factors (TFs), microRNAs (miRNAs), and the creation or disruption of splice sites, etc. [
38]. Consequently, variants in non-coding regions may impact protein function by reducing protein solubility or destabilizing protein structure [
39]. Notably, SNPs in the 3′ untranslated regions (3′ UTR) are of particular significance, as they serve as primary binding sites for miRNAs. miRNAs play a crucial role in gene expression regulation and their interactions with the 3′ UTR lead to gene silencing after transcription and translation suppression [
40]. Moreover, in the context of male infertility, several studies have demonstrated differential expression of miRNAs between fertile and infertile males. These miRNAs hold the potential to unveil the molecular mechanisms underlying infertility and may serve as noninvasive biomarkers for diagnosing this condition [
41]. Likewise, SNPs within the 5′ untranslated region (5′ UTR) hold significant importance and can contribute to the development of various diseases. Specifically, 5′ UTRs play a pivotal role in influencing both mRNA stability and translation efficiency [
42,
43]. Additionally, functional elements such as the internal ribosome entry site (IRES), upstream open reading frames (uORFs), and iron-responsive element (IRE) within the 5′ UTR play a crucial role in precisely modulating protein expression in alignment with the specific requirements of the cell [
42]. Consequently, SNPs have the potential to disrupt the smooth translation of mRNA or compromise the stability of the mRNA molecule, making it more susceptible to degradation. Disruption of the aforementioned functional elements can also lead to alterations in gene expression. Ultimately, the irregular gene expression resulting from mutations in the 5′ UTR can significantly contribute to the progression and manifestation of a spectrum of diseases.
Therefore, in the present day, while coding region SNPs have garnered significant attention in candidate gene studies due to their critical regulatory roles, there has been notably less emphasis on the functional analysis of non-coding SNPs [
44]. The continuous evolution of SNP discovery technologies and the dynamic annotation of the genome have resulted in the accumulation of an overwhelming amount of information and a large number of SNPs that are challenging to study experimentally [
45]. Consequently, computational methods are becoming increasingly indispensable in genomic research for SNP selection and the prediction of their functional consequences in disease development [
46].
Today, bioinformatics tools play a crucial role in prioritizing SNPs with functional significance from the vast pool of neutral non-risk variants [
47]. These tools assess the potential functional impacts of SNPs across five key levels: splicing, transcription, translation, post-translation, and protein stability. While most existing bioinformatics tools focus on evaluating SNP effects with respect to a single biological function, others offer a comprehensive analysis of SNP function by integrating various algorithms, data sources, etc. [
44,
45,
46].
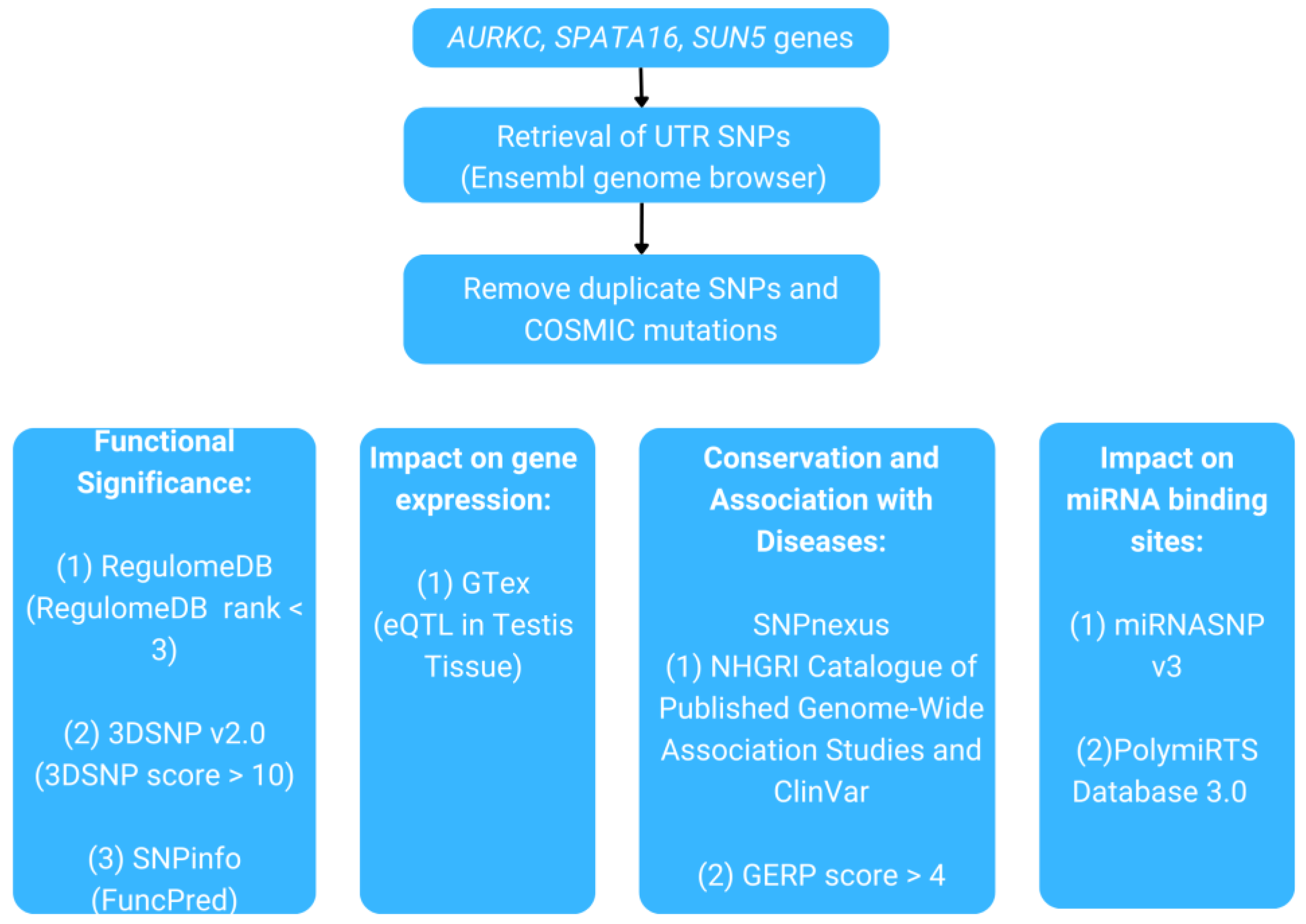
The objective of the present study was to analyze UTR variants in the AURKC, SPATA16, and SUN5 genes using computational methods, given the significance of UTR variants in numerous studies and their association with various diseases. These genes are well known for their role in teratozoospermia. Therefore, the SNPs identified in their UTR were prioritized based on several criteria, including their functional significance, association with expression quantitative trait loci (eQTL) and diseases, presence within evolutionarily conserved regions, and their impact on the creation or disruption of miRNA binding sites. As a result, this study involves a rigorous process of filtering through a list of SNPs to identify SNPs that are most likely to be associated with teratozoospermia. To the best of our knowledge, this is the first comprehensive computational analysis of UTR SNPs in the AURKC, SPATA16, and SUN5 genes. It provides a valuable foundation for future research, listing candidate variants that may be linked to teratozoospermia, thereby contributing to a deeper understanding of the molecular mechanisms underlying male infertility. Furthermore, this research can facilitate the development of biomarkers to enhance assisted reproductive technology (ART) and improve the diagnosis and prognosis of male infertility, especially teratozoospermia.
4. Discussion
AURKC,
SPATA16, and
SUN5 are pivotal genes known to play critical roles in the intricate processes of spermatogenesis and meiosis [
7,
8,
31,
66]. Numerous studies have provided evidence of the association between specific SNPs within these genes and teratozoospermia [
16,
17,
18,
19,
20,
21,
26,
28,
29,
33,
34,
35]. However, the sheer volume of SNPs within these genes poses a formidable challenge for comprehensive analysis. Herein, the indispensable role of bioinformatics tools comes into play, enabling the judicious selection of a limited number of prioritized variants. These selected variants hold the potential to significantly contribute to our understanding of teratozoospermia’s pathogenesis and pave the way for future genetic screening endeavors. By pinpointing these key genetic factors, researchers can unravel the intricate molecular mechanisms underlying teratozoospermia, offering invaluable insights into both diagnosis and potential therapeutic strategies for this complex reproductive disorder.
Building upon this foundation, it is important to note that SNPs residing within the untranslated regions (UTRs) of genes are frequently overlooked but hold significant potential implications in the context of various pathologies [
67]. In this study, we harnessed a diverse array of bioinformatics tools to thoroughly assess the impact of UTR variants within
AURKC,
SPATA16, and
SUN5. The primary objective was to discern and prioritize these variants, thereby assembling a comprehensive catalog of SNPs that possess the promise of being instrumental in forthcoming investigations.
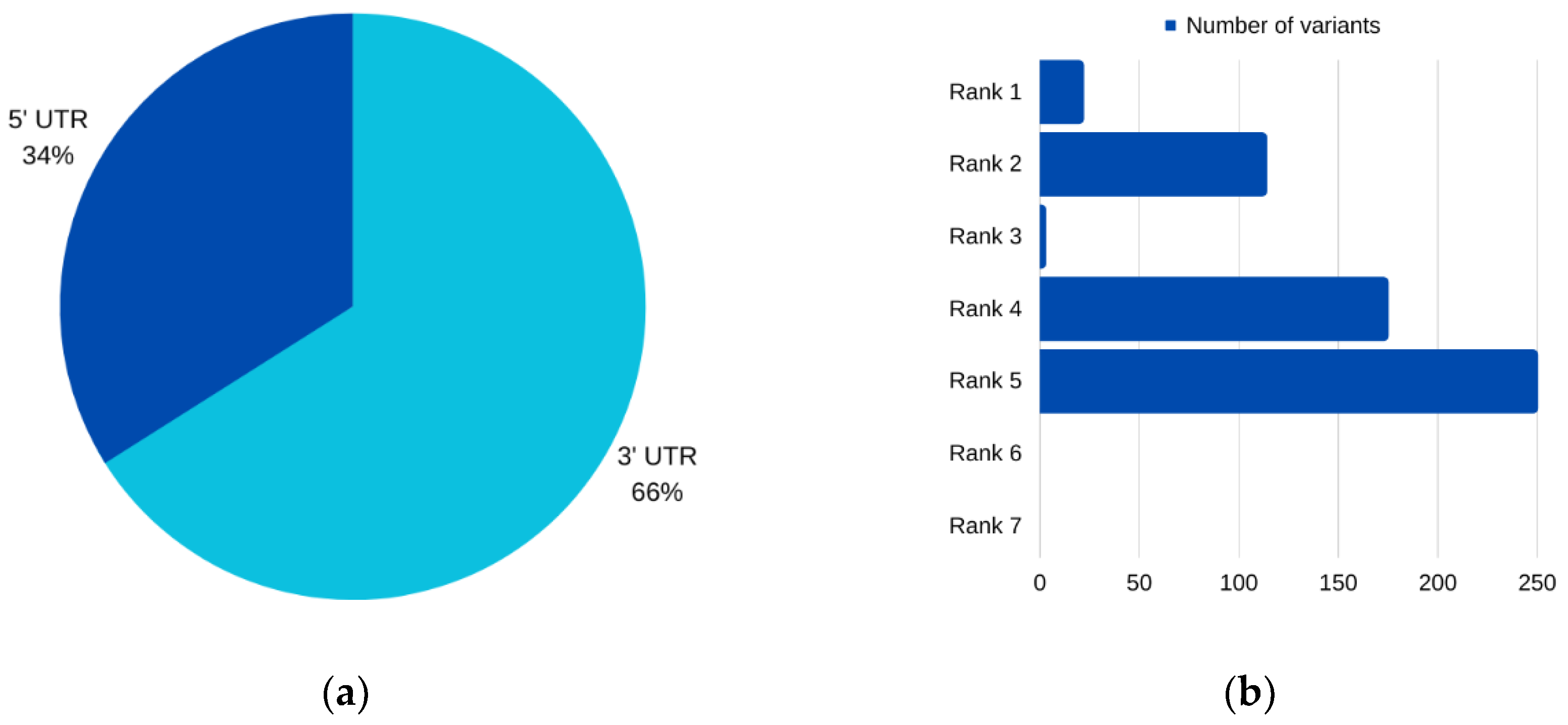
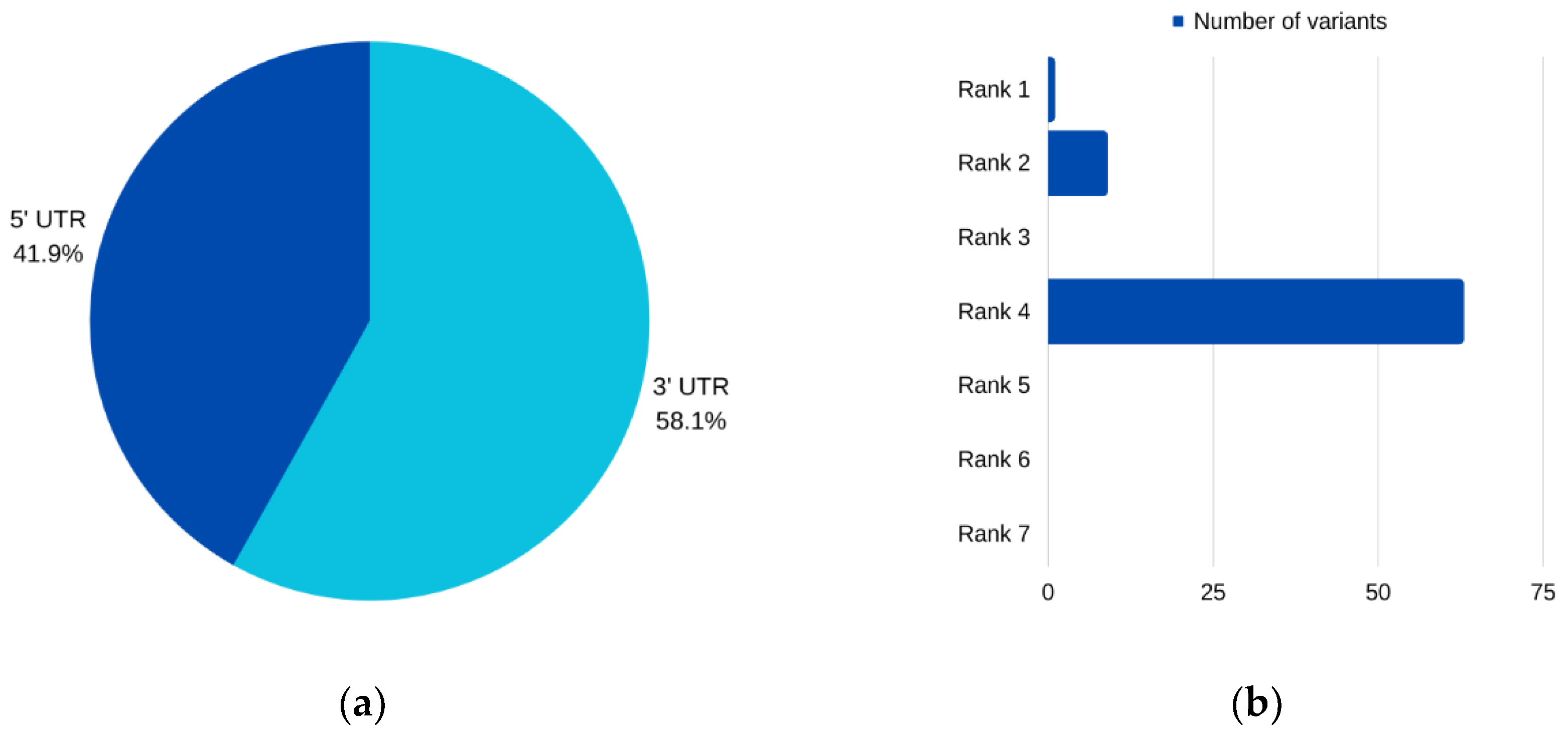
For
AURKC, six SNPs emerged as prime candidates for potential pathogenicity, as corroborated by multiple analytical tools. Among these, three were situated within the 5′ UTR, while two resided in the 3′ UTR. Intriguingly, one SNP was characterized as impacting both the 3′ and 5′ UTRs for different transcripts of the
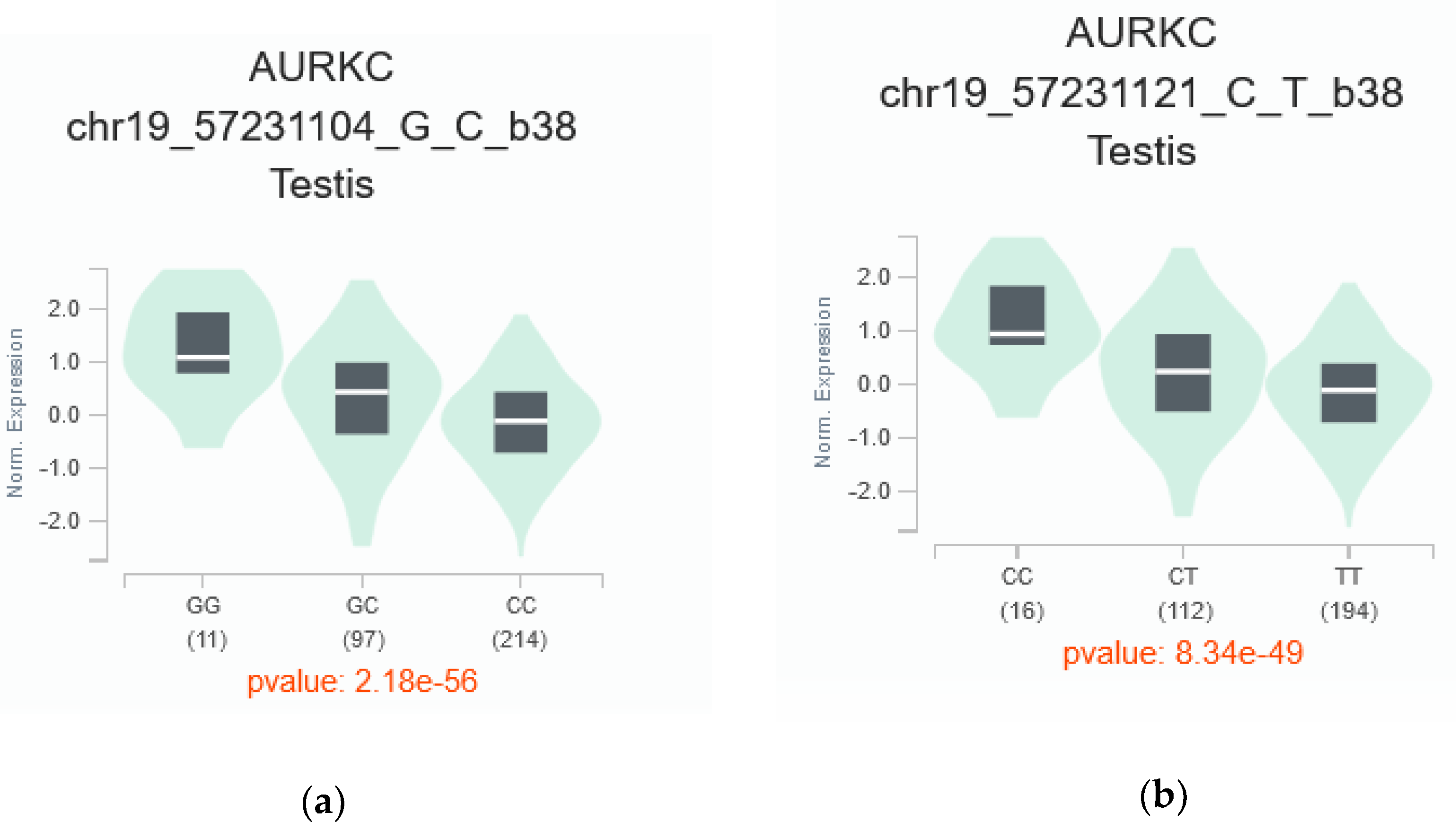
AURKC gene. Notably, two of these prioritized SNPs, rs11084490 and rs58264281, were found to significantly affect
AURKC expression in testis tissue. This observation aligns with SNPinfo (FuncPred) [
52] predictions, which indicated that these variants perturbed transcription-factor binding sites. Existing evidence underscores the robust associations between nucleotide sequences within transcription-factor binding sites (TFBSs) and gene expression levels [
68]. TFBS polymorphisms have garnered substantial attention, constituting 31% of trait-associated polymorphisms identified by genome-wide association studies (GWAS), underscoring their pivotal role in disease development [
69]. According to ClinVar [
60], these two SNPs are also linked to spermatogenic failure, particularly infertility associated with multi-tailed spermatozoa and excessive DNA, albeit being classified as benign. Nonetheless, our study findings advocate for their further exploration, given their potential regulatory role in teratozoospermia. Thus, subsequent investigations in a large sample of infertile males are suggested. Additionally, rs533889458 and rs2361127 earned prioritization based on their functional significance, with rs2361127 notably identified as a TFBS polymorphism, prompting the need for future studies elucidating its impact on
AURKC expression levels. rs55710619 is another prioritized variant in
AURKC that is associated with multi-tailed spermatozoa and excessive DNA and is characterized as likely benign according to ClinVar [
60]. SNPinfo (FuncPred) [
52] also ascribes functional significance to this variant. Special attention should be accorded to rs35582299, which exerts an impact on miRNA binding sites, as affirmed by several tools, including miRNASNP v3 [
64], PolymiRTS Database 3.0 [
63], and SNPinfo (FuncPred) [
52]. More specifically, the above tools demonstrate that rs35582299 causes the loss or gain of sites affecting 29 miRNAs. miRNAs, small RNA molecules, are pivotal in gene expression regulation, and studies have revealed their differential expression between fertile and infertile males [
41]. miRNAs fine-tune genes involved in sperm production and maturation, and dysregulation can disrupt this balance, culminating in abnormalities in sperm morphology and reduced fertility [
70,
71,
72]. Thus, future investigations should delve into the list of miRNAs identified in this study as affected by SNPs, as they hold the potential to modulate
AURKC expression. Intriguingly, none of the miRNAs that are affected by these SNPs have been previously implicated in male infertility. Similarly, the above prioritized variants are reported for the first time as potentially involved in teratozoospermia and further exploration of their role is required.
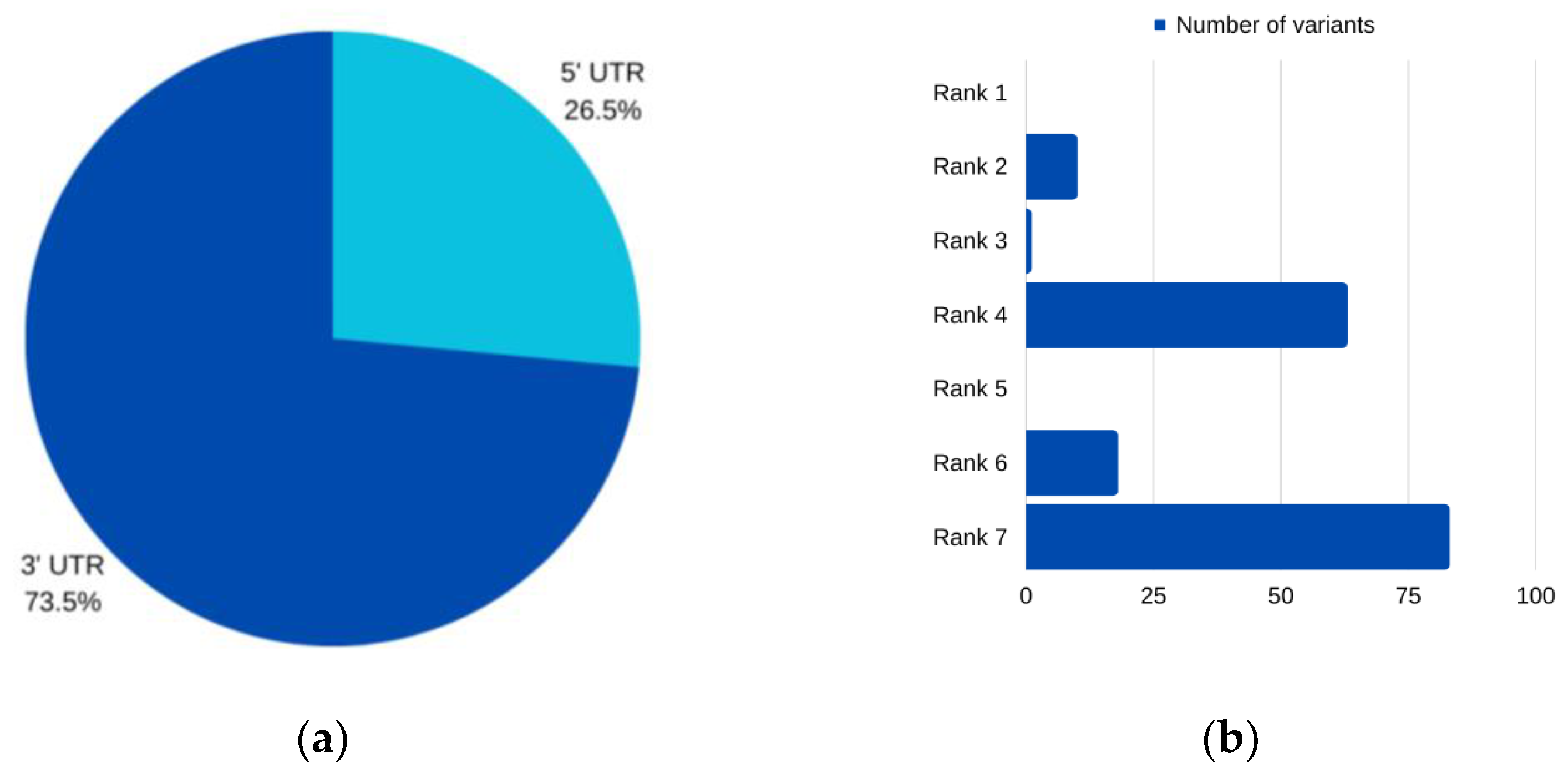
For
SPATA16, a gene with a crucial role in sperm production and testicular development [
73], we identified two 3′ UTR variants through analysis with various tools. Among these prioritized SNPs, rs146640459 is indicated as a variant with functional significance according to 3DSNP v2.0 [
51] and RegulomeDB [
50]. Meanwhile, rs148085657 affects miRNA binding sites according to miRNASNP v3 [
64] and PolymiRTS Database 3.0 [
63]. More specifically, rs148085657 causes gain and loss of target sites, affecting six miRNAs (hsa-miR-5092, hsa-miR-205-5p, hsa-miR-5586-5p, hsa-miR-4267, hsa-miR-6512-3p, hsa-miR-6720-5p). Some of these miRNAs have been associated with different types of cancer [
74,
75,
76], but none of them have shown any association with spermatogenesis or other aspects of male fertility. Similarly to
AURKC, these two variants have not been previously associated with male infertility either.
For
SUN5, three 3′ UTR variants were identified, all of which were characterized as having functional significance according to the RegulomeDB [
50]. Simultaneously, these variants were found to impact miRNA binding sites according to miRNASNP v3 [
64]. Specifically, two SNPs (rs1485087675 and rs1478197315) resulted in the loss of binding sites for the same miRNA, hsa-miR-7155-5p. The third SNP (rs762026146) not only disrupted the binding site of hsa-miR-7155-5p (resulting in target loss) but also created a binding site for hsa-miR-7162-3p. It is worth noting that there is limited research on these miRNAs, with only one publication suggesting that hsa-miR-7162-3p may play a role in the repair of endometrial stromal cell injury [
77]. Furthermore, there are no available studies for the three variants prioritized and no association with male reproduction.
The present study has yielded a wealth of data and identified numerous SNPs in AURKC, SPATA16, and SUN5 genes that hold promise for future investigations into the molecular mechanisms of teratozoospermia. To guide future research efforts, functional experiments can be designed to validate the roles of these SNPs in teratozoospermia. These experiments may focus on assessing their functional impact on mRNA–miRNA interactions and exploring how these SNPs influence the expression of AURKC, SPATA16, and SUN5 genes, particularly in tissues relevant to male fertility and reproductive organs, such as the testes. Additionally, conducting large-scale GWAS studies in cohorts of individuals with and without male infertility can provide valuable insights by determining whether these SNPs are more prevalent in the affected group, thus establishing a link between these genetic variants and male infertility risk.
As the variants identified in this study may significantly contribute to teratozoospermia, it is imperative to discuss the role of UTR variants in gene regulation and their potential impact on disease pathogenesis, particularly in the context of male infertility. Variants situated within the 3′ UTRs of genes can exert profound effects on gene expression and subsequent cellular functions [
78,
79,
80]. More specifically, these alterations can affect mRNA stability, thus influencing the half-life of the messenger RNA and ultimately modulating protein expression levels [
81]. Additionally, disruptions in the 3′ UTR can intricately perturb post-transcriptional regulatory mechanisms, encompassing RNA processing, transport, localization, and degradation, consequently leading to dysregulation of essential cellular processes and pathways. Furthermore, they might influence mRNA localization within the cell, thereby impacting local protein synthesis and altering various cellular activities [
78,
79,
80,
81]. Similarly, variations in the 5′ UTRs can disrupt the efficiency of translation initiation, thereby affecting ribosomal binding and subsequent translation processes, ultimately resulting in variations in protein synthesis levels. Moreover, they can interfere with the binding sites for specific transcription factors or regulatory proteins, potentially influencing gene transcription and leading to dysregulation of downstream cellular processes [
42,
78].
As the UTR variants identified in this study are within AURKC, SPATA16, and SUN5, they may disrupt the regulatory mechanisms of these genes, potentially contributing to male infertility due to the crucial roles of the above genes in spermatogenesis. Specifically, AURKC regulates chromosomal segregation during meiosis, ensuring the production of genetically balanced gametes [
13], while SPATA16 is essential for sperm function and fertilization, participating in various processes critical for normal sperm development and function, including sperm–egg interaction and fusion [
23,
24]. Similarly, SUN5, belonging to the SUN domain family, is indispensable for sperm head shaping and nuclear membrane remodeling during spermatogenesis [
31,
34]. Thus, any UTR variants in these genes can potentially disrupt gene expression through the mechanisms described earlier, consequently impacting the intricate process of spermatogenesis. It is also worth noting that given the significant role of these genes in fertilization, UTR variants may extensively disturb the processes involved in sperm production, affecting other crucial sperm parameters such as motility or count. Therefore, further investigations, including functional experiments, are imperative to elucidate the precise mechanisms of action of the reported variants and their specific impacts on additional sperm parameters beyond morphology and teratozoospermia.
Furthermore, given the pivotal role of miRNAs in various cellular processes, it is highly promising to delve deeper into the broader miRNA interaction network involving the SNPs reported in this study. This exploration can help identify other miRNAs that may be affected by these SNPs, unveiling potential overlapping or synergistic effects on gene regulation. Notably, miRNAs altered by these SNPs may exhibit differential expression between fertile and infertile males, offering the potential for their use as biomarkers for assessing male infertility risk or as therapeutic targets. These avenues of research align with the primary goal of our study, which was to provide a prioritized list of SNPs and miRNAs to catalyze future investigations in the field of teratozoospermia.
Finally, it is important to acknowledge that while our study was an in-depth analysis employing an extensive array of bioinformatics tools and stringent criteria, it is an in silico study with inherent limitations. As such, further research is imperative to validate and expand upon the findings presented here.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}