

Non-Ischemic Myocardial Fibrosis in End-Stage Kidney Disease Patients: A New Perspective
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
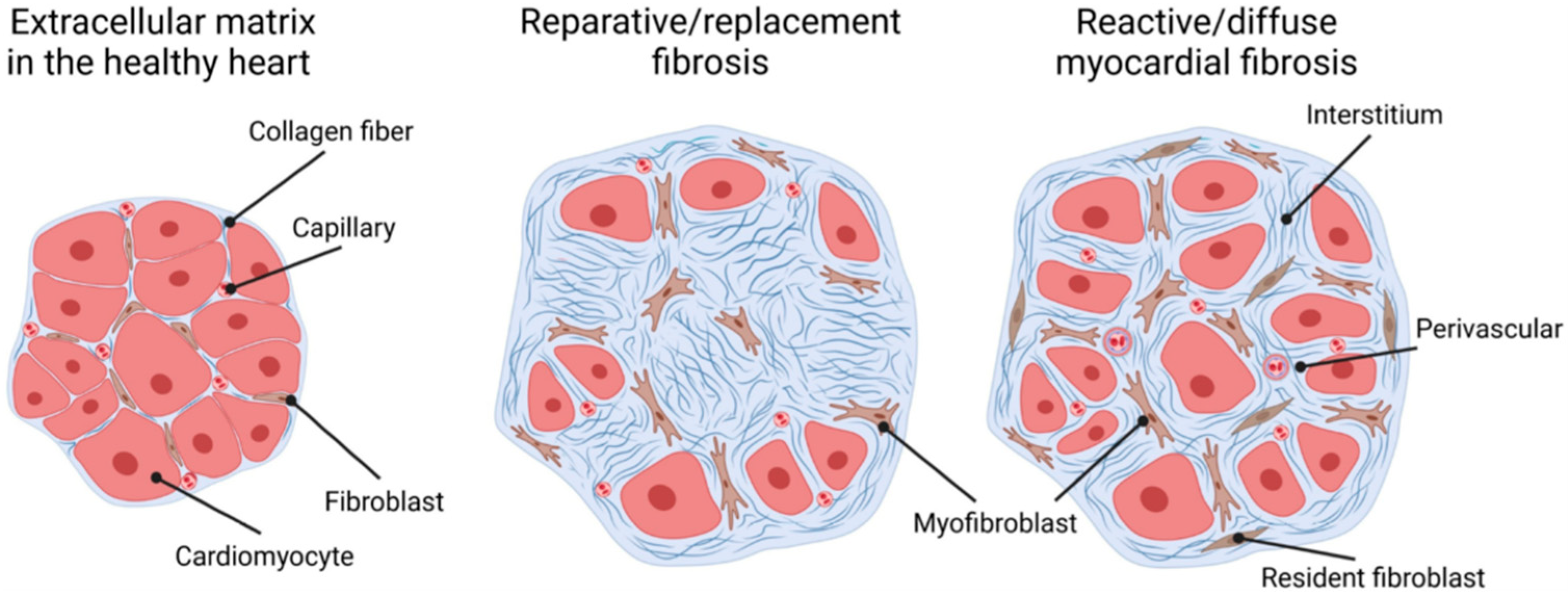
2. Myocardial Fibrosis in CKD
3. Mechanism of Reactive Myocardial Interstitial Fibrosis
4. Detection of Myocardial Fibrosis in a Clinical Setting
5. Potential Means of Preventing Cardiac Fibrosis
6. Future Directions
7. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sarnak, M.J.; Levey, A.S.; Schoolwerth, A.C.; Coresh, J.; Culleton, B.; Hamm, L.L.; McCullough, P.A.; Kasiske, B.L.; Kelepouris, E.; Klag, M.J.; et al. Kidney disease as a risk factor for development of cardiovascular disease: A statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 2003, 108, 2154–2169. [Google Scholar] [CrossRef]
- Ronco, C.; McCullough, P.; Anker, S.D.; Anand, I.; Aspromonte, N.; Bagshaw, S.M.; Bellomo, R.; Berl, T.; Bobek, I.; Cruz, D.N.; et al. Cardio-renal syndromes: Report from the consensus conference of the acute dialysis quality initiative. Eur. Heart J. 2010, 31, 703–711. [Google Scholar] [CrossRef]
- Rangaswami, J.; Bhalla, V.; Blair, J.E.A.; Chang, T.I.; Costa, S.; Lentine, K.L.; Lerma, E.V.; Mezue, K.; Molitch, M.; Mullens, W.; et al. Cardiorenal Syndrome: Classification, Pathophysiology, Diagnosis, and Treatment Strategies: A Scientific Statement from the American Heart Association. Circulation 2019, 139, e840–e878. [Google Scholar] [CrossRef]
- Nauta, S.T.; van Domburg, R.T.; Nuis, R.J.; Akkerhuis, M.; Deckers, J.W. Decline in 20-year mortality after myocardial infarction in patients with chronic kidney disease: Evolution from the prethrombolysis to the percutaneous coronary intervention era. Kidney Int. 2013, 84, 353–358. [Google Scholar] [CrossRef]
- Bae, E.H.; Lim, S.Y.; Cho, K.H.; Choi, J.S.; Kim, C.S.; Park, J.W.; Ma, S.K.; Jeong, M.H.; Kim, S.W. GFR and cardiovascular outcomes after acute myocardial infarction: Results from the Korea Acute Myocardial Infarction Registry. Am. J. Kidney Dis. 2012, 59, 795–802. [Google Scholar] [CrossRef]
- Massy, Z.A.; de Zeeuw, D. LDL cholesterol in CKD—To treat or not to treat? Kidney Int. 2013, 84, 451–456. [Google Scholar] [CrossRef]
- Wanner, C.; Krane, V.; Marz, W.; Olschewski, M.; Mann, J.F.; Ruf, G.; Ritz, E. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N. Engl. J. Med. 2005, 353, 238–248. [Google Scholar] [CrossRef]
- Fellstrom, B.C.; Jardine, A.G.; Schmieder, R.E.; Holdaas, H.; Bannister, K.; Beutler, J.; Chae, D.W.; Chevaile, A.; Cobbe, S.M.; Gronhagen-Riska, C.; et al. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N. Engl. J. Med. 2009, 360, 1395–1407. [Google Scholar] [CrossRef]
- Zanoli, L.; Lentini, P.; Briet, M.; Castellino, P.; House, A.A.; London, G.M.; Malatino, L.; McCullough, P.A.; Mikhailidis, D.P.; Boutouyrie, P. Arterial Stiffness in the Heart Disease of CKD. J. Am. Soc. Nephrol. 2019, 30, 918–928. [Google Scholar] [CrossRef]
- London, G.M. Cardiovascular disease in chronic renal failure: Pathophysiologic aspects. Semin. Dial. 2003, 16, 85–94. [Google Scholar] [CrossRef]
- London, G.M. Left ventricular alterations and end-stage renal disease. Nephrol. Dial. Transpl. 2002, 17 (Suppl. S1), 29–36. [Google Scholar] [CrossRef] [PubMed]
- Hayer, M.K.; Price, A.M.; Liu, B.; Baig, S.; Ferro, C.J.; Townend, J.N.; Steeds, R.P.; Edwards, N.C. Diffuse Myocardial Interstitial Fibrosis and Dysfunction in Early Chronic Kidney Disease. Am. J. Cardiol. 2018, 121, 656–660. [Google Scholar] [CrossRef]
- Edwards, N.C.; Moody, W.E.; Yuan, M.; Hayer, M.K.; Ferro, C.J.; Townend, J.N.; Steeds, R.P. Diffuse interstitial fibrosis and myocardial dysfunction in early chronic kidney disease. Am. J. Cardiol. 2015, 115, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Aoki, J.; Ikari, Y.; Nakajima, H.; Mori, M.; Sugimoto, T.; Hatori, M.; Tanimoto, S.; Amiya, E.; Hara, K. Clinical and pathologic characteristics of dilated cardiomyopathy in hemodialysis patients. Kidney Int. 2005, 67, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, K.; Ichimura, K.; Reddy, S.; Haddad, F.; Spiekerkoetter, E. Cardiac Fibrosis in the Pressure Overloaded Left and Right Ventricle as a Therapeutic Target. Front. Cardiovasc. Med. 2022, 9, 886553. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Kovacic, J.C. Extracellular Matrix in Ischemic Heart Disease, Part 4/4: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 2219–2235. [Google Scholar] [CrossRef] [PubMed]
- Diez, J.; Gonzalez, A.; Kovacic, J.C. Myocardial Interstitial Fibrosis in Nonischemic Heart Disease, Part 3/4: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 2204–2218. [Google Scholar] [CrossRef] [PubMed]
- van Hoeven, K.H.; Factor, S.M. A comparison of the pathological spectrum of hypertensive, diabetic, and hypertensive-diabetic heart disease. Circulation 1990, 82, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Umeda, K.; Sugihara, N.; Yoshio, H.; Ino, H.; Takeda, R.; Okada, Y.; Nakanishi, I. Collagen remodelling in myocardia of patients with diabetes. J. Clin. Pathol. 1993, 46, 32–36. [Google Scholar] [CrossRef]
- Treibel, T.A.; Lopez, B.; Gonzalez, A.; Menacho, K.; Schofield, R.S.; Ravassa, S.; Fontana, M.; White, S.K.; DiSalvo, C.; Roberts, N.; et al. Reappraising myocardial fibrosis in severe aortic stenosis: An invasive and non-invasive study in 133 patients. Eur. Heart J. 2018, 39, 699–709. [Google Scholar] [CrossRef]
- Shirani, J.; Pick, R.; Roberts, W.C.; Maron, B.J. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J. Am. Coll. Cardiol. 2000, 35, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Nitta, K.; Goto, S.; Masakane, I.; Hanafusa, N.; Taniguchi, M.; Hasegawa, T.; Nakai, S.; Wada, A.; Hamano, T.; Hoshino, J.; et al. Annual dialysis data report for 2018, JSDT Renal Data Registry: Survey methods, facility data, incidence, prevalence, and mortality. Ren. Replace. Ther. 2020, 6, 41. [Google Scholar] [CrossRef]
- Boenink, R.; Astley, M.E.; Huijben, J.A.; Stel, V.S.; Kerschbaum, J.; Ots-Rosenberg, M.; Asberg, A.A.; Lopot, F.; Golan, E.; Castro de la Nuez, P.; et al. The ERA Registry Annual Report 2019: Summary and age comparisons. Clin. Kidney J. 2022, 15, 452–472. [Google Scholar] [CrossRef] [PubMed]
- Di Lullo, L.; Gorini, A.; Russo, D.; Santoboni, A.; Ronco, C. Left Ventricular Hypertrophy in Chronic Kidney Disease Patients: From Pathophysiology to Treatment. Cardiorenal Med. 2015, 5, 254–266. [Google Scholar] [CrossRef]
- Paoletti, E.; De Nicola, L.; Gabbai, F.B.; Chiodini, P.; Ravera, M.; Pieracci, L.; Marre, S.; Cassottana, P.; Luca, S.; Vettoretti, S.; et al. Associations of Left Ventricular Hypertrophy and Geometry with Adverse Outcomes in Patients with CKD and Hypertension. Clin. J. Am. Soc. Nephrol. 2016, 11, 271–279. [Google Scholar] [CrossRef]
- Tonelli, M.; Karumanchi, S.A.; Thadhani, R. Epidemiology and Mechanisms of Uremia-Related Cardiovascular Disease. Circulation 2016, 133, 518–536. [Google Scholar] [CrossRef]
- Izumaru, K.; Hata, J.; Nakano, T.; Nakashima, Y.; Nagata, M.; Fukuhara, M.; Oda, Y.; Kitazono, T.; Ninomiya, T. Reduced Estimated GFR and Cardiac Remodeling: A Population-Based Autopsy Study. Am. J. Kidney Dis. 2019, 74, 373–381. [Google Scholar] [CrossRef]
- Toblli, J.E.; Cao, G.; Rivas, C.; Giani, J.F.; Dominici, F.P. Intravenous iron sucrose reverses anemia-induced cardiac remodeling, prevents myocardial fibrosis, and improves cardiac function by attenuating oxidative/nitrosative stress and inflammation. Int. J. Cardiol. 2016, 212, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Amann, K.; Breitbach, M.; Ritz, E.; Mall, G. Myocyte/capillary mismatch in the heart of uremic patients. J. Am. Soc. Nephrol. 1998, 9, 1018–1022. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Lee, J.K.T.; Franzone, A.; Lanz, J.; Siontis, G.C.M.; Stortecky, S.; Grani, C.; Roost, E.; Windecker, S.; Pilgrim, T. Early Detection of Subclinical Myocardial Damage in Chronic Aortic Regurgitation and Strategies for Timely Treatment of Asymptomatic Patients. Circulation 2018, 137, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Schnee, J.M.; Hsueh, W.A. Angiotensin II, adhesion, and cardiac fibrosis. Cardiovasc. Res. 2000, 46, 264–268. [Google Scholar] [CrossRef]
- Leask, A. Getting to the heart of the matter: New insights into cardiac fibrosis. Circ. Res. 2015, 116, 1269–1276. [Google Scholar] [CrossRef]
- Cowling, R.T.; Kupsky, D.; Kahn, A.M.; Daniels, L.B.; Greenberg, B.H. Mechanisms of cardiac collagen deposition in experimental models and human disease. Transl. Res. 2019, 209, 138–155. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, L.; Haukilahti, A.; Vahatalo, J.; Kentta, T.; Appel, H.; Kiviniemi, A.; Pakanen, L.; Huikuri, H.V.; Myerburg, R.J.; Junttila, J. Electrocardiographic associations with myocardial fibrosis among sudden cardiac death victims. Heart (Br. Card. Soc.) 2020, 106, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Matsukane, A.; Hayashi, T.; Tanaka, Y.; Iwasaki, M.; Kubo, S.; Asakawa, T.; Takahashi, Y.; Imamura, Y.; Hirahata, K.; Joki, N.; et al. Usefulness of an Upright T-Wave in Lead aVR for Predicting the Short-Term Prognosis of Incident Hemodialysis Patients: A Potential Tool for Screening High-Risk Hemodialysis Patients. Cardiorenal Med. 2015, 5, 267–277. [Google Scholar] [CrossRef]
- Sato, Y.; Hayashi, T.; Joki, N.; Fujimoto, S. Association of Lead aVR T-wave Amplitude With Cardiovascular Events or Mortality Among Prevalent Dialysis Patients. Ther. Apher. Dial. 2017, 21, 287–294. [Google Scholar] [CrossRef]
- Arcari, L.; Engel, J.; Freiwald, T.; Zhou, H.; Zainal, H.; Gawor, M.; Buettner, S.; Geiger, H.; Hauser, I.; Nagel, E.; et al. Cardiac biomarkers in chronic kidney disease are independently associated with myocardial edema and diffuse fibrosis by cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2021, 23, 71. [Google Scholar] [CrossRef] [PubMed]
- Querejeta, R.; Varo, N.; Lopez, B.; Larman, M.; Artinano, E.; Etayo, J.C.; Martinez Ubago, J.L.; Gutierrez-Stampa, M.; Emparanza, J.I.; Gil, M.J.; et al. Serum carboxy-terminal propeptide of procollagen type I is a marker of myocardial fibrosis in hypertensive heart disease. Circulation 2000, 101, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Klappacher, G.; Franzen, P.; Haab, D.; Mehrabi, M.; Binder, M.; Plesch, K.; Pacher, R.; Grimm, M.; Pribill, I.; Eichler, H.G.; et al. Measuring extracellular matrix turnover in the serum of patients with idiopathic or ischemic dilated cardiomyopathy and impact on diagnosis and prognosis. Am. J. Cardiol. 1995, 75, 913–918. [Google Scholar] [CrossRef]
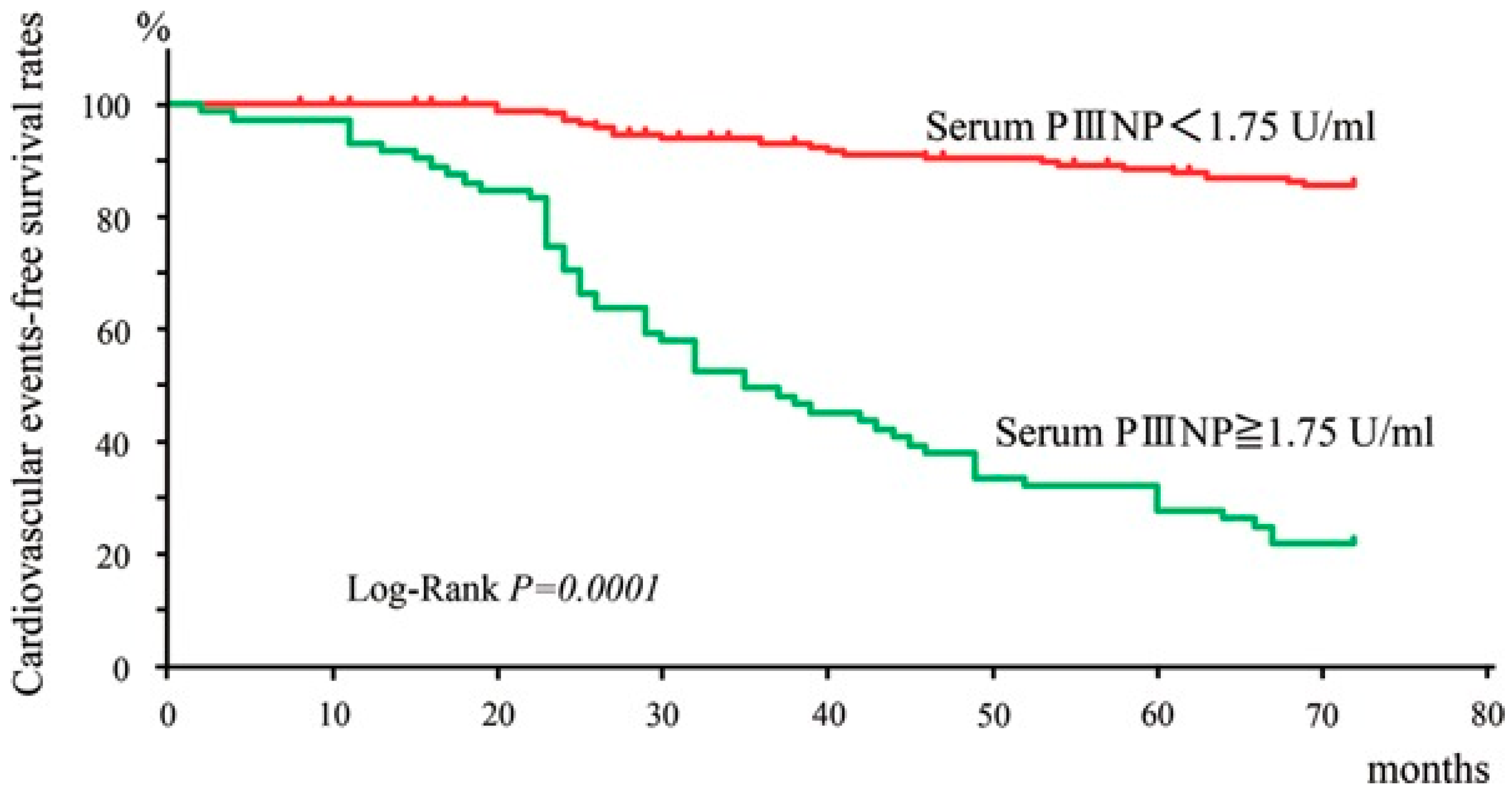
- Nishimura, M.; Tokoro, T.; Takatani, T.; Sato, N.; Hashimoto, T.; Kobayashi, H.; Ono, T. Circulating Aminoterminal Propeptide of Type III Procollagen as a Biomarker of Cardiovascular Events in Patients Undergoing Hemodialysis. J. Atheroscler. Thromb. 2019, 26, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Ge, Y.; Singh, A.; Grani, C.; Kwong, R.Y. Multimodality Imaging Assessment of Myocardial Fibrosis. JACC Cardiovasc. Imaging 2021, 14, 2457–2469. [Google Scholar] [CrossRef] [PubMed]
- Scully, P.R.; Bastarrika, G.; Moon, J.C.; Treibel, T.A. Myocardial Extracellular Volume Quantification by Cardiovascular Magnetic Resonance and Computed Tomography. Curr. Cardiol. Rep. 2018, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- Schelbert, E.B.; Hsu, L.Y.; Anderson, S.A.; Mohanty, B.D.; Karim, S.M.; Kellman, P.; Aletras, A.H.; Arai, A.E. Late gadolinium-enhancement cardiac magnetic resonance identifies postinfarction myocardial fibrosis and the border zone at the near cellular level in ex vivo rat heart. Circ. Cardiovasc. Imaging 2010, 3, 743–752. [Google Scholar] [CrossRef]
- Kim, R.J.; Fieno, D.S.; Parrish, T.B.; Harris, K.; Chen, E.L.; Simonetti, O.; Bundy, J.; Finn, J.P.; Klocke, F.J.; Judd, R.M. Relationship of MRI delayed contrast enhancement to irreversible injury, infarct age, and contractile function. Circulation 1999, 100, 1992–2002. [Google Scholar] [CrossRef]
- Azevedo, C.F.; Nigri, M.; Higuchi, M.L.; Pomerantzeff, P.M.; Spina, G.S.; Sampaio, R.O.; Tarasoutchi, F.; Grinberg, M.; Rochitte, C.E. Prognostic significance of myocardial fibrosis quantification by histopathology and magnetic resonance imaging in patients with severe aortic valve disease. J. Am. Coll. Cardiol. 2010, 56, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.C.; Reed, E.; Sheppard, M.N.; Elkington, A.G.; Ho, S.Y.; Burke, M.; Petrou, M.; Pennell, D.J. The histologic basis of late gadolinium enhancement cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2004, 43, 2260–2264. [Google Scholar] [CrossRef]
- Puntmann, V.O.; Voigt, T.; Chen, Z.; Mayr, M.; Karim, R.; Rhode, K.; Pastor, A.; Carr-White, G.; Razavi, R.; Schaeffter, T.; et al. Native T1 mapping in differentiation of normal myocardium from diffuse disease in hypertrophic and dilated cardiomyopathy. JACC Cardiovasc. Imaging 2013, 6, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Iles, L.M.; Ellims, A.H.; Llewellyn, H.; Hare, J.L.; Kaye, D.M.; McLean, C.A.; Taylor, A.J. Histological validation of cardiac magnetic resonance analysis of regional and diffuse interstitial myocardial fibrosis. Eur. Heart J. Cardiovasc. Imaging 2015, 16, 14–22. [Google Scholar] [CrossRef]
- Hayer, M.K.; Radhakrishnan, A.; Price, A.M.; Liu, B.; Baig, S.; Weston, C.J.; Biasiolli, L.; Ferro, C.J.; Townend, J.N.; Steeds, R.P.; et al. Defining Myocardial Abnormalities Across the Stages of Chronic Kidney Disease: A Cardiac Magnetic Resonance Imaging Study. JACC Cardiovasc. Imaging 2020, 13, 2357–2367. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, E.; Talle, M.A.; Mangion, K.; Bell, E.; Rauhalammi, S.M.; Roditi, G.; McComb, C.; Radjenovic, A.; Welsh, P.; Woodward, R.; et al. Defining myocardial tissue abnormalities in end-stage renal failure with cardiac magnetic resonance imaging using native T1 mapping. Kidney Int. 2016, 90, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Brilla, C.G.; Funck, R.C.; Rupp, H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation 2000, 102, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Diez, J.; Querejeta, R.; Lopez, B.; Gonzalez, A.; Larman, M.; Martinez Ubago, J.L. Losartan-dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients. Circulation 2002, 105, 2512–2517. [Google Scholar] [CrossRef]
- Izawa, H.; Murohara, T.; Nagata, K.; Isobe, S.; Asano, H.; Amano, T.; Ichihara, S.; Kato, T.; Ohshima, S.; Murase, Y.; et al. Mineralocorticoid receptor antagonism ameliorates left ventricular diastolic dysfunction and myocardial fibrosis in mildly symptomatic patients with idiopathic dilated cardiomyopathy: A pilot study. Circulation 2005, 112, 2940–2945. [Google Scholar] [CrossRef]
- Suzuki, H.; Kanno, Y.; Sugahara, S.; Ikeda, N.; Shoda, J.; Takenaka, T.; Inoue, T.; Araki, R. Effect of angiotensin receptor blockers on cardiovascular events in patients undergoing hemodialysis: An open-label randomized controlled trial. Am. J. Kidney Dis. 2008, 52, 501–506. [Google Scholar] [CrossRef]
- Iseki, K.; Arima, H.; Kohagura, K.; Komiya, I.; Ueda, S.; Tokuyama, K.; Shiohira, Y.; Uehara, H.; Toma, S.; Olmesartan Clinical Trial in Okinawan Patients Under OKIDS (OCTOPUS) Group. Effects of angiotensin receptor blockade (ARB) on mortality and cardiovascular outcomes in patients with long-term haemodialysis: A randomized controlled trial. Nephrol. Dial. Transpl. 2013, 28, 1579–1589. [Google Scholar] [CrossRef]
- Hasegawa, T.; Nishiwaki, H.; Ota, E.; Levack, W.M.; Noma, H. Aldosterone antagonists for people with chronic kidney disease requiring dialysis. Cochrane Database Syst. Rev. 2021, 2, CD013109. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Belohlavek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Claggett, B.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; et al. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N. Engl. J. Med. 2022, 387, 1089–1098. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Bohm, M.; Brunner-La Rocca, H.P.; Choi, D.J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Habibi, J.; Aroor, A.R.; Sowers, J.R.; Jia, G.; Hayden, M.R.; Garro, M.; Barron, B.; Mayoux, E.; Rector, R.S.; Whaley-Connell, A.; et al. Sodium glucose transporter 2 (SGLT2) inhibition with empagliflozin improves cardiac diastolic function in a female rodent model of diabetes. Cardiovasc. Diabetol. 2017, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Lopez, B.; Querejeta, R.; Gonzalez, A.; Sanchez, E.; Larman, M.; Diez, J. Effects of loop diuretics on myocardial fibrosis and collagen type I turnover in chronic heart failure. J. Am. Coll. Cardiol. 2004, 43, 2028–2035. [Google Scholar] [CrossRef]
- Lopez, B.; Gonzalez, A.; Beaumont, J.; Querejeta, R.; Larman, M.; Diez, J. Identification of a potential cardiac antifibrotic mechanism of torasemide in patients with chronic heart failure. J. Am. Coll. Cardiol. 2007, 50, 859–867. [Google Scholar] [CrossRef]
- Veeraveedu, P.T.; Watanabe, K.; Ma, M.; Thandavarayan, R.A.; Palaniyandi, S.S.; Yamaguchi, K.; Suzuki, K.; Kodama, M.; Aizawa, Y. Comparative effects of torasemide and furosemide in rats with heart failure. Biochem. Pharmacol. 2008, 75, 649–659. [Google Scholar] [CrossRef]
- Veeraveedu, P.T.; Watanabe, K.; Ma, M.; Palaniyandi, S.S.; Yamaguchi, K.; Suzuki, K.; Kodama, M.; Aizawa, Y. Torasemide, a long-acting loop diuretic, reduces the progression of myocarditis to dilated cardiomyopathy. Eur. J. Pharmacol. 2008, 581, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Trippel, T.D.; Van Linthout, S.; Westermann, D.; Lindhorst, R.; Sandek, A.; Ernst, S.; Bobenko, A.; Kasner, M.; Spillmann, F.; Gonzalez, A.; et al. Investigating a biomarker-driven approach to target collagen turnover in diabetic heart failure with preserved ejection fraction patients. Effect of torasemide versus furosemide on serum C-terminal propeptide of procollagen type I (DROP-PIP trial). Eur. J. Heart Fail. 2018, 20, 460–470. [Google Scholar] [CrossRef]
- Lopez, B.; Gonzalez, A.; Hermida, N.; Laviades, C.; Diez, J. Myocardial fibrosis in chronic kidney disease: Potential benefits of torasemide. Kidney Int. Suppl. 2008, 74, S19–S23. [Google Scholar] [CrossRef] [PubMed]
- Kraft, L.; Erdenesukh, T.; Sauter, M.; Tschope, C.; Klingel, K. Blocking the IL-1beta signalling pathway prevents chronic viral myocarditis and cardiac remodeling. Basic Res. Cardiol. 2019, 114, 11. [Google Scholar] [CrossRef]
- Edgley, A.J.; Krum, H.; Kelly, D.J. Targeting fibrosis for the treatment of heart failure: A role for transforming growth factor-beta. Cardiovasc. Ther. 2012, 30, e30–e40. [Google Scholar] [CrossRef] [PubMed]
- Shinde, A.V.; Su, Y.; Palanski, B.A.; Fujikura, K.; Garcia, M.J.; Frangogiannis, N.G. Pharmacologic inhibition of the enzymatic effects of tissue transglutaminase reduces cardiac fibrosis and attenuates cardiomyocyte hypertrophy following pressure overload. J. Mol. Cell. Cardiol. 2018, 117, 36–48. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakata, K.; Joki, N. Non-Ischemic Myocardial Fibrosis in End-Stage Kidney Disease Patients: A New Perspective. Kidney Dial. 2023, 3, 311-321. https://doi.org/10.3390/kidneydial3030027
Nakata K, Joki N. Non-Ischemic Myocardial Fibrosis in End-Stage Kidney Disease Patients: A New Perspective. Kidney and Dialysis. 2023; 3(3):311-321. https://doi.org/10.3390/kidneydial3030027
Chicago/Turabian StyleNakata, Kenji, and Nobuhiko Joki. 2023. "Non-Ischemic Myocardial Fibrosis in End-Stage Kidney Disease Patients: A New Perspective" Kidney and Dialysis 3, no. 3: 311-321. https://doi.org/10.3390/kidneydial3030027