
Sonochemical Synthesis of Silica-Supported Iron Oxide Nanostructures and Their Application as Catalysts in Fischer–Tropsch Synthesis
 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Silica Nanospheres by the Stober Process
2.3. Sonochemical Synthesis of Iron Oxide on the Silica Nanoparticle Support
2.4. Characterizations of Silica and Si-Fe Hybrid Catalyst
2.5. Study of the F–T Synthesis
3. Results and Discussions
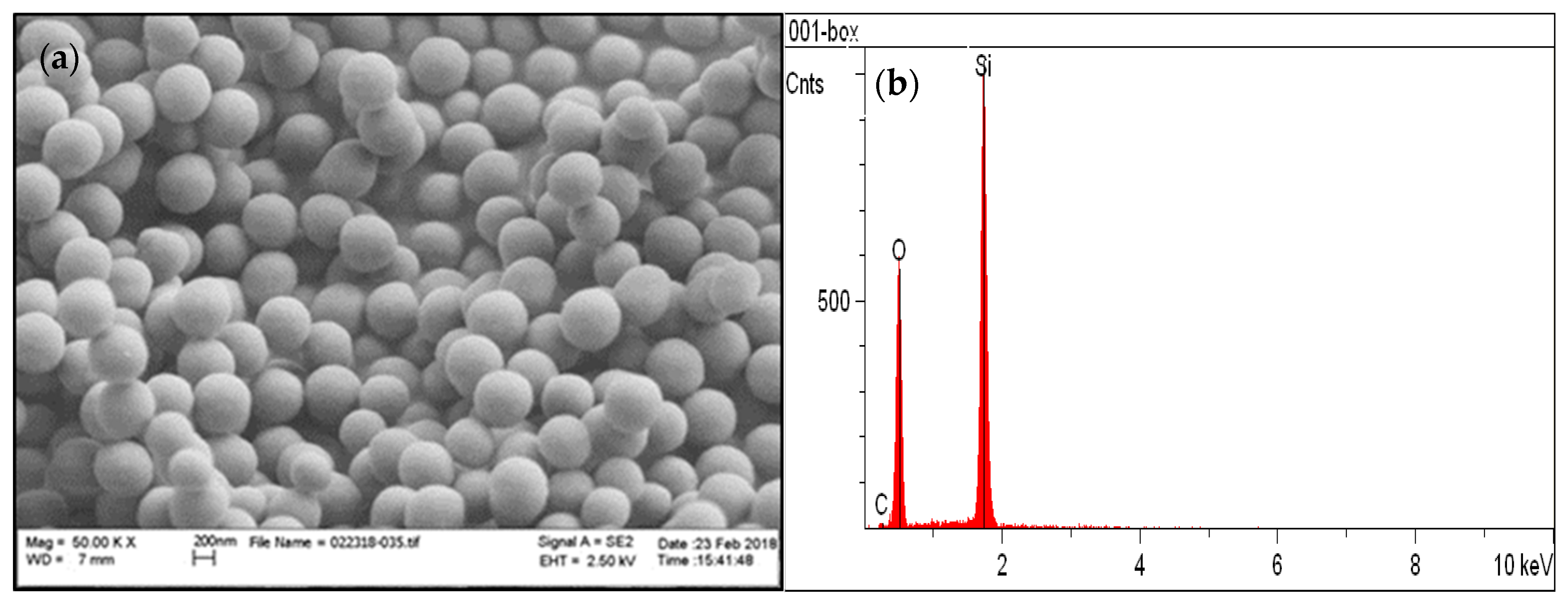
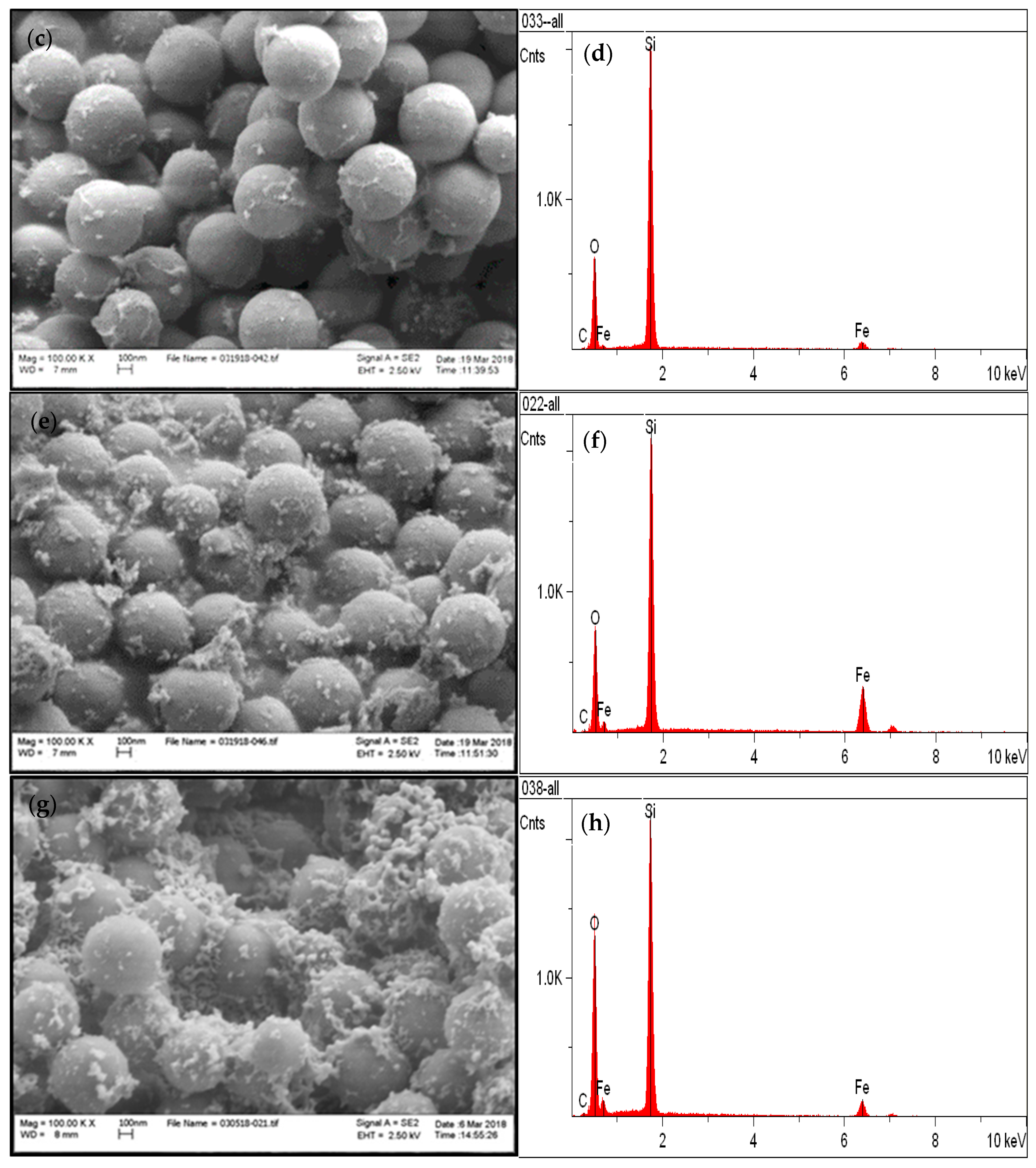
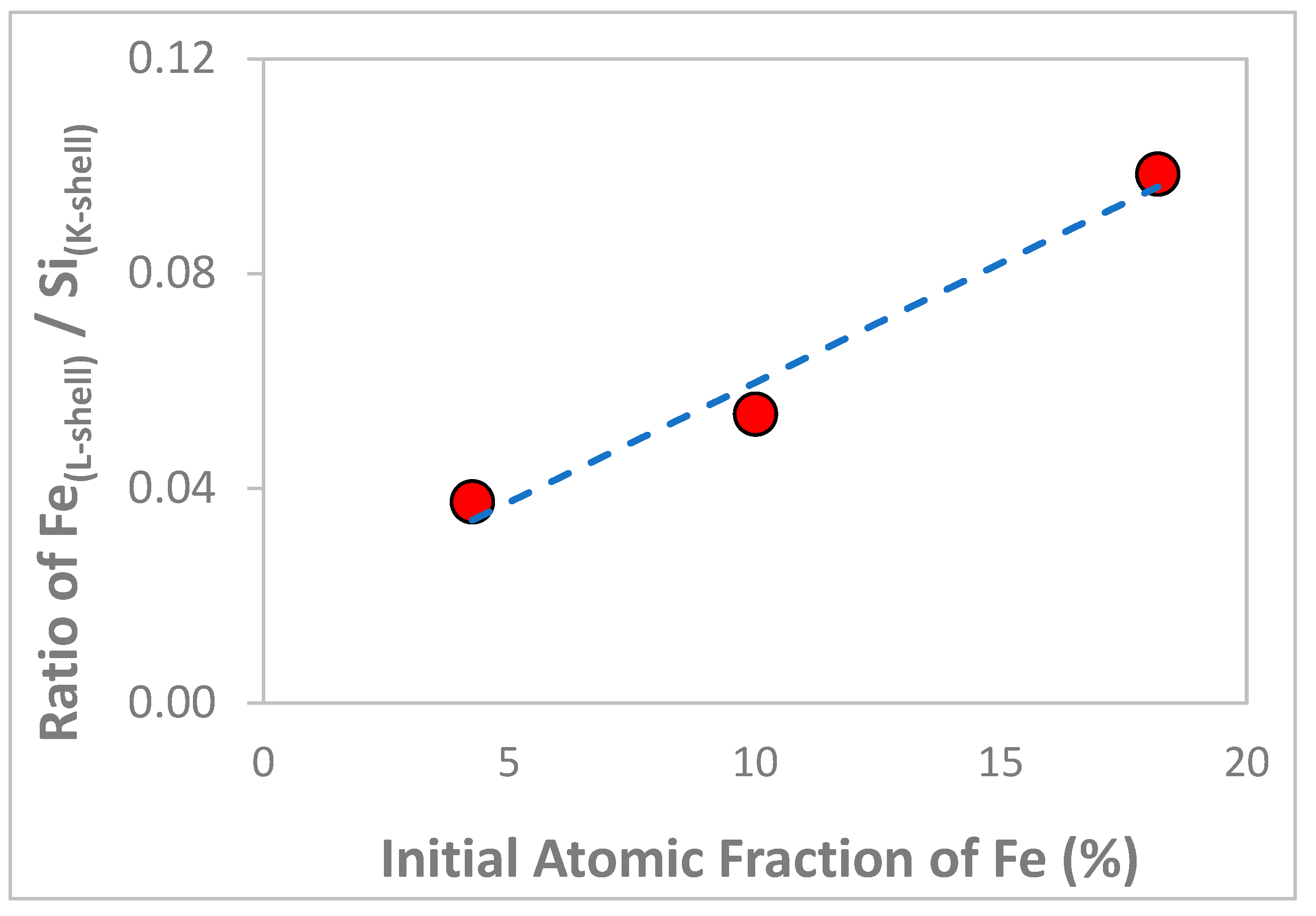
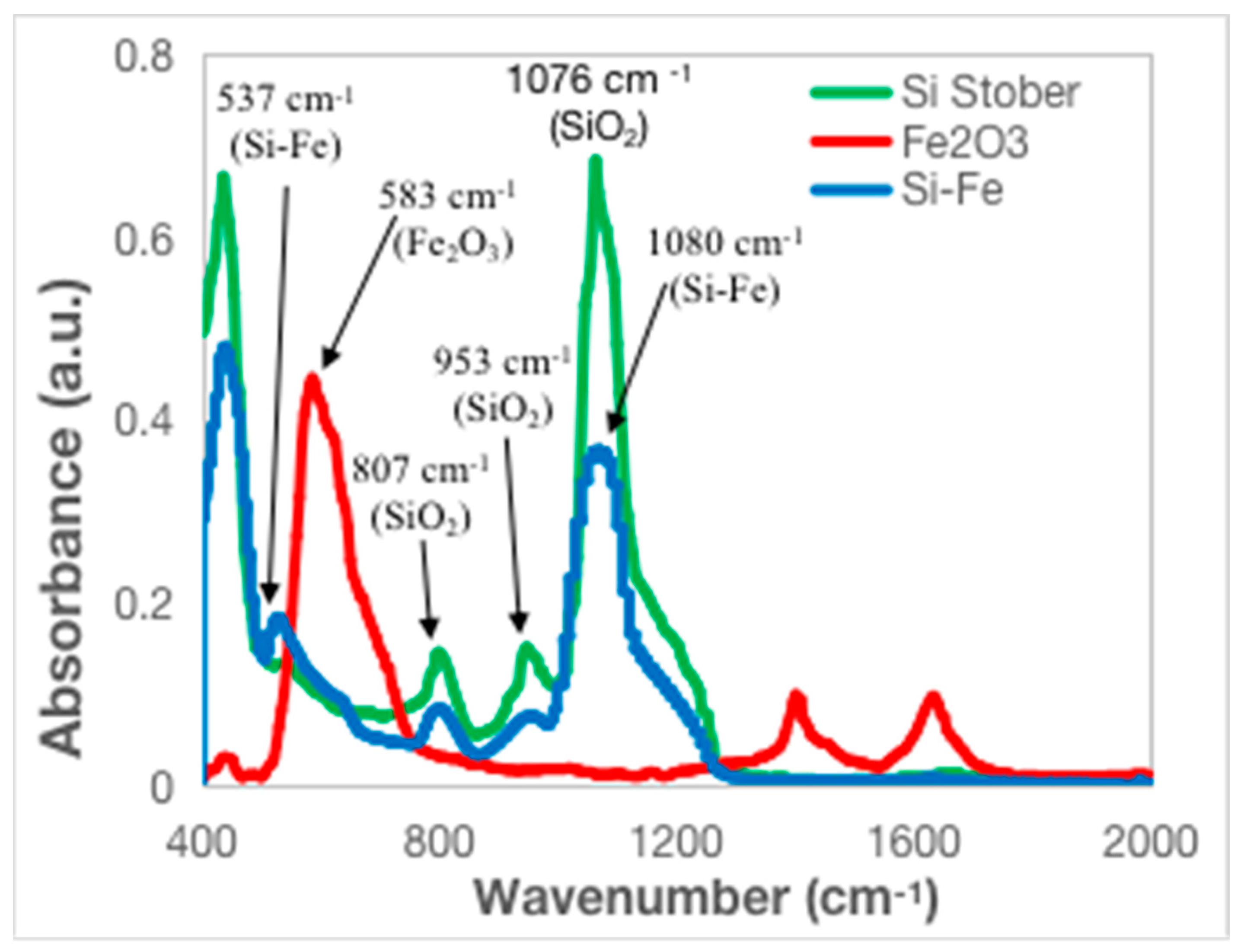
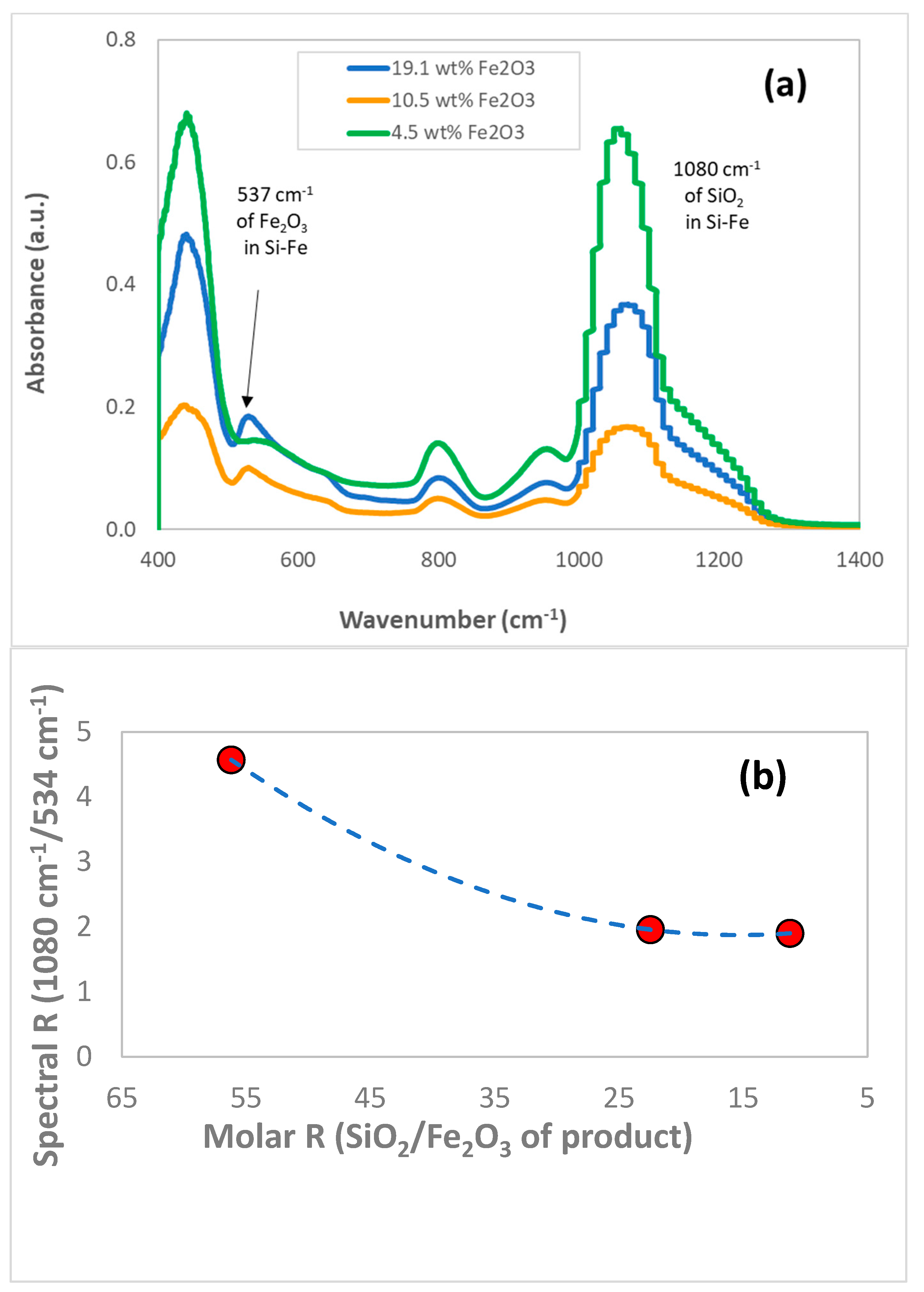
3.1. Chemical and Structural Characterization of the Si-Fe Catalyst
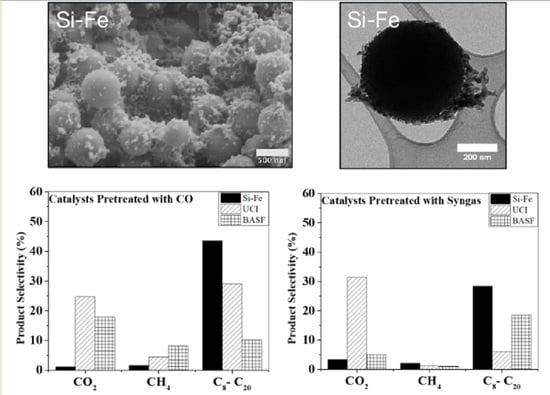
3.2. Comparison of F–T Synthesis Products
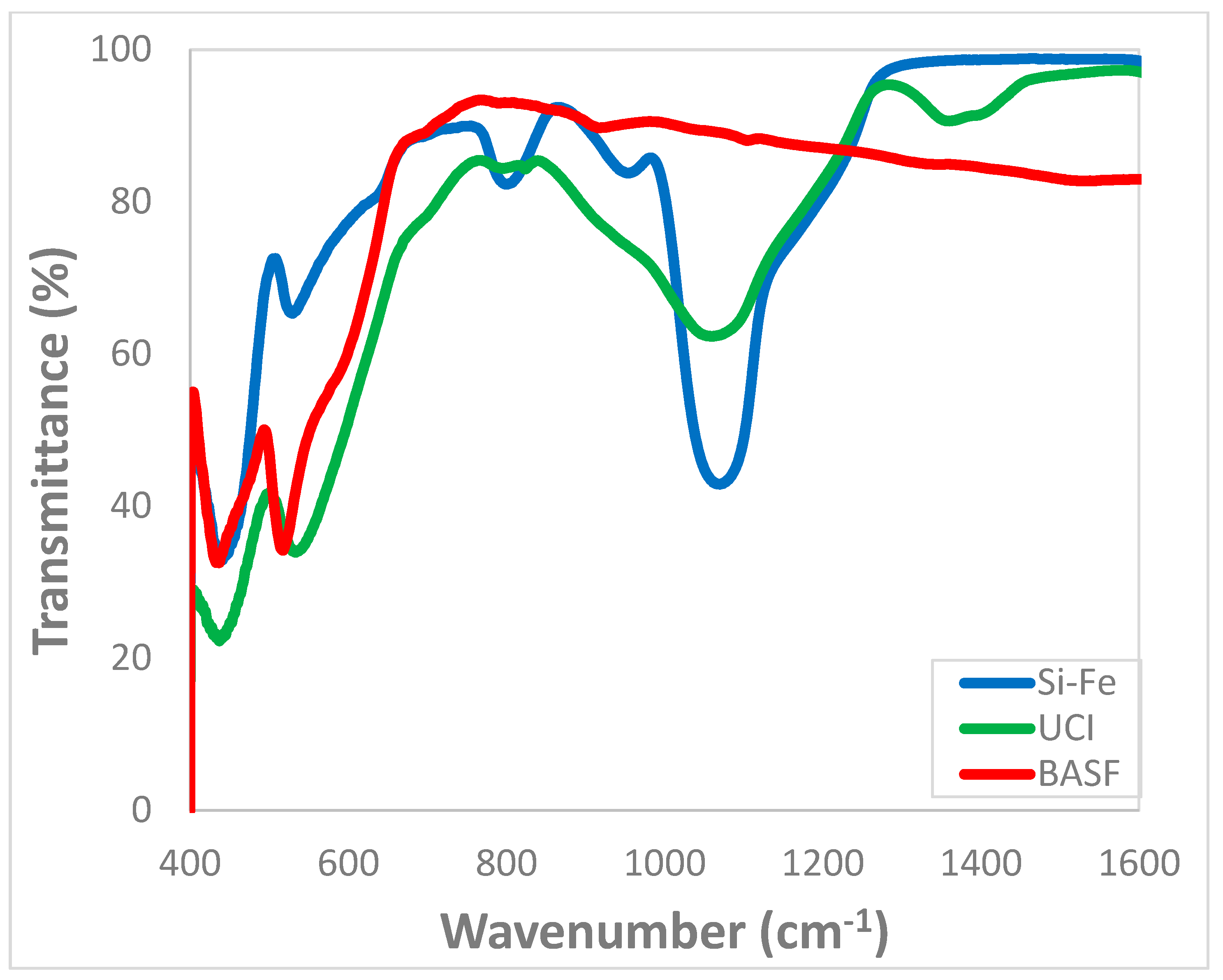
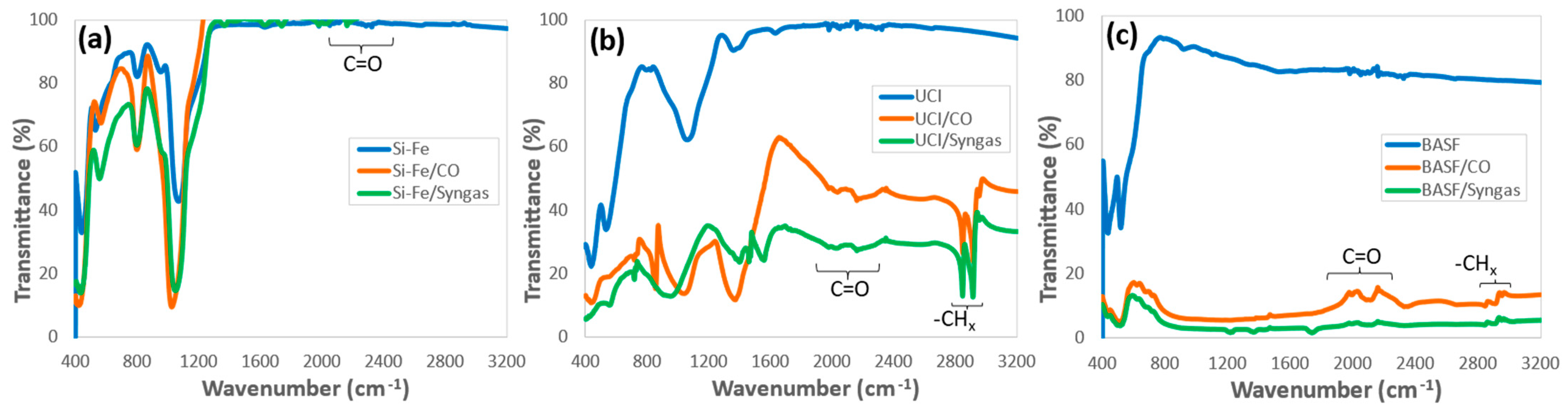
3.3. Morphology and Chemical Structure of the Catalysts after the F–T Synthesis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Schulz, H. Short history and present trends of Fischer–Tropsch synthesis. Appl. Catal. A Gen. 1999, 186, 3–12. [Google Scholar] [CrossRef]
- Dry, M.E. Practical and theoretical aspects of the catalytic Fischer-Tropsch process. Appl. Catal. A Gen. 1996, 138, 319–344. [Google Scholar] [CrossRef]
- Mahajan, D.; Gütlich, P.; Ensling, J.; Pandya, K.; Stumm, U. Vijayaraghavan Evaluation of nanosized iron in slurry-phase Fischer—Tropsch synthesis. Energy Fuels 2003, 17, 1210–1221. [Google Scholar] [CrossRef]
- Thomas, J.M.; Thomas, W.J. Principles and Practice of Heterogeneous Catalysis; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Mahajan, D.; Kobayashi, A.; Gupta, N. Fischer–Tropsch synthesis catalysed by ultrafine particles of iron: Cessation of water–gas shift activity. Chem. Commun. 1994, 7, 795–796. [Google Scholar] [CrossRef]
- Pour, A.N.; Housaindokht, M.R.; Tayyari, S.F.; Zarkesh, J. Kinetics of the water-gas shift reaction in Fischer-Tropsch synthesis over a nano-structured iron catalyst. J. Nat. Gas Chem. 2010, 19, 362–368. [Google Scholar] [CrossRef]
- Dlamini, H.; Motjope, T.; Joorst, G.; Stege, G.t.; Mdleleni, M. Changes in Physico-Chemical Properties of Iron-Based Fischer–Tropsch Catalyst Induced by SiO2 Addition. Catal. Lett. 2002, 78, 201–207. [Google Scholar] [CrossRef]
- Zhao, R.; Goodwin, J.G.; Oukaci, R. Attrition assessment for slurry bubble column reactor catalysts. Appl. Catal. A Gen. 1999, 189, 99–116. [Google Scholar] [CrossRef]
- Botes, F.G.; Niemantsverdriet, J.W.; van de Loosdrecht, J. A comparison of cobalt and iron-based slurry phase Fischer–Tropsch synthesis. Catal. Today 2013, 215, 112–120. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Pannell, R.B.; Butler, J.L. Support and crystallite size effects in CO hydrogenation on nickel. J. Catal. 1980, 65, 335–347. [Google Scholar] [CrossRef]
- Bond, G. Metal-support and Metal-additive Effects in Catalysis. Platin. Met. Rev. 1983, 27, 16–18. [Google Scholar]
- Bukur, D.B.; Lang, X.; Mukesh, D.; Zimmerman, W.H.; Rosynek, M.P.; Li, C. Binder/support effects on the activity and selectivity of iron catalysts in the Fischer-Tropsch synthesis. Ind. Eng. Chem. Res. 1990, 29, 1588–1599. [Google Scholar] [CrossRef]
- Wang, H.; Mao, D.; Qi, J.; Zhang, Q.; Wang, D. Hollow multishelled structure of heterogeneous Co3O4–CeO2− x nanocomposite for CO catalytic oxidation. Adv. Funct. Mater. 2019, 29, 1806588. [Google Scholar] [CrossRef]
- Li, Z.; Wang, H.; Zhao, W.; Xu, X.; Wang, D. Enhanced catalytic activity of Au-CeO2/Al2O3 monolith for low-temperature CO oxidation. Catal. Commun. 2019, 129, 105729. [Google Scholar] [CrossRef]
- Wang, Z.; Qi, J.; Yang, N.; Yu, R.; Wang, D. Core–shell nano/microstructures for heterogeneous tandem catalysis. Mater. Chem. Front. 2021, 5, 1126–1139. [Google Scholar] [CrossRef]
- Suslick, K. Homogeneous sonochemistry. In Ultrasound: It’s Chemical, Physical, Biological Effects; VCH Publishers: New York, NY, USA, 1988; pp. 123–163. [Google Scholar]
- Flint, E.B.; Suslick, K.S. The Temperature of Cavitation. Science 1991, 253, 1397. [Google Scholar] [CrossRef]
- Suslick, K.S.; Choe, S.-B.; Cichowlas, A.A.; Grinstaff, M.W. Sonochemical synthesis of amorphous iron. Nature 1991, 353, 414–416. [Google Scholar] [CrossRef]
- Suslick, K.S.; Hyeon, T.; Fang, M.; Cichowlas, A.A. Sonochemical synthesis of nanostructured catalysts. Mater. Sci. Eng. A 1995, 204, 186–192. [Google Scholar] [CrossRef]
- Suslick, K.S.; Hyeon, T.; Fang, M. Nanostructured Materials Generated by High-Intensity Ultrasound: Sonochemical Synthesis and Catalytic Studies. Chem. Mater. 1996, 8, 2172–2179. [Google Scholar] [CrossRef]
- Nozawa, K.; Gailhanou, H.; Raison, L.; Panizza, P.; Ushiki, H.; Sellier, E.; Delville, J.P.; Delville, M.H. Smart Control of Monodisperse Stöber Silica Particles: Effect of Reactant Addition Rate on Growth Process. Langmuir 2005, 21, 1516–1523. [Google Scholar] [CrossRef]
- Tannenbaum, R.; Goldberg, E.P.; Flenniken, C.L. Decomposition of Iron Carbonyls in Solid Polymer Matrices: Preparation of Novel Metal-Polymer Composites. In Metal-Containing Polymeric Systems; Sheats, J.E., Carraher, C.E., Pittman, C.U., Eds.; Springer US: Boston, MA, USA, 1985; pp. 303–339. [Google Scholar]
- Richmond, T.G.; Shi, Q.Z.; Trogler, W.C.; Basolo, F. Kinetics and mechanism of Lewis-base-induced disproportionation of vanadium hexacarbonyl and its phosphine-substituted derivatives. J. Am. Chem. Soc. 1984, 106, 76–80. [Google Scholar] [CrossRef]
- Pomogailo, A.D.; Rozenberg, A.S.; Dzhardimalieva, G.I. Controlled Pyrolysis of Metal-Containing Precursors as a Way for Synthesis of Metallopolymer Nanocomposites. In Metal–Polymer Nanocomposites; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004; pp. 75–122. [Google Scholar]
- Chen, L.; Sharma, S.; Darienzo, R.E.; Tannenbaum, R.J.M.R.E. Decoration of cellulose nanocrystals with iron oxide nanoparticles. Mater. Res. Express 2020, 7, 055003. [Google Scholar] [CrossRef]
- Beganskienė, A.; Sirutkaitis, V.; Kurtinaitienė, M.; Juškėnas, R.; Kareiva, A. FTIR, TEM and NMR investigations of Stöber silica nanoparticles. Mater. Sci. 2004, 10, 287–290. [Google Scholar]
- Suo, H.; Wang, S.; Zhang, C.; Xu, J.; Wu, B.; Yang, Y.; Xiang, H.; Li, Y.-W. Chemical and structural effects of silica in iron-based Fischer–Tropsch synthesis catalysts. J. Catal. 2012, 286, 111–123. [Google Scholar] [CrossRef]
- Al-Oweini, R.; El-Rassy, H. Synthesis and characterization by FTIR spectroscopy of silica aerogels prepared using several Si (OR)4 and R′′Si(OR′)3 precursors. J. Mol. Struct. 2009, 919, 140–145. [Google Scholar] [CrossRef]
- Belousov, Y.A. Radical chemistry of iron carbonyls. Russ. Chem Rev. 2007, 76, 41–58. [Google Scholar] [CrossRef]
- Luo, F.H.; Yang, S.R.; Li, C.S.; Duan, J.P.; Cheng, C.H. Kinetics and electron paramagnetic resonance evidence of an electron-transfer chain mechanism for PPh3 substitution of [Fe3(CO)12]. J. Chem. Soc. Dalton Trans. 1991, 9, 2435–2439. [Google Scholar] [CrossRef] [Green Version]
- Ragaini, F. Mechanistic Study of the Phase-Transfer-Catalyzed Reduction of Nitrobenzene to Aniline by Iron Carbonyl Complexes. Role of the Radical Anion [Fe3(CO)11]. Organometallics 1996, 15, 3572–3578. [Google Scholar] [CrossRef]
- Belousov, Y.A.; Brin, E.F. Investigation of the kinetics of the chain-radical process in the system iron carbonyl–Lewis base. Polyhedron 2001, 20, 2765–2769. [Google Scholar] [CrossRef]
- Sivakumar, M.; Gedanken, A. Insights into the sonochemical decomposition of Fe(CO)5: Theoretical and experimental understanding of the role of molar concentration and power density on the reaction yield. Ultrason. Sonochem. 2004, 11, 373–378. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Y.; Wang, Y.; Liu, S.; Deng, Y. Sonochemical formation of iron oxide nanoparticles in ionic liquids for magnetic liquid marble. Phys. Chem. Chem. Phys. 2012, 14, 5132–5138. [Google Scholar] [CrossRef]
- Mogorosi, R.P.; Fischer, N.; Claeys, M.; van Steen, E. Strong-metal–support interaction by molecular design: Fe–silicate interactions in Fischer–Tropsch catalysts. J. Catal. 2012, 289, 140–150. [Google Scholar] [CrossRef]
- Hauchard, C.; Rowntree, P.A. Low-energy electron-induced decarbonylation of Fe(CO)5 films adsorbed on Au(111) surfaces. Can J. Chem. 2011, 89, 1163–1173. [Google Scholar] [CrossRef]
- Alexandrescu, R.; Morjann, I.; Crunteanu, A.; Cojocaru, S.; Petcu, S.; Teodorescu, V.; Huisken, F.; Koh, B.; Ehbrecht, M. Iron-oxide-based nanoparticles produced by pulsed infrared laser pyrolysis of Fe(CO)5. Mater. Chem. Phys. 1998, 55, 115–121. [Google Scholar] [CrossRef]
- Suslick, K.S.; Goodale, J.W.; Schubert, P.F.; Wang, H.H. Sonochemistry and sonocatalysis of metal carbonyls. J. Am. Chem. Soc. 1983, 105, 5781–5785. [Google Scholar] [CrossRef]
- Einhiuser, T.J. ICP-OES and SEM-EDX Analysis of Dust and Powder Produced by the Laser-Processing of a Cr-Ni-Steel Alloy. Mikrochim. Acta 1997, 127, 265–268. [Google Scholar] [CrossRef]
- Nakamichi, H.; Sato, K.; Miyata, Y.; Kimura, M.; Masamura, K. Quantitative analysis of Cr-depleted zone morphology in low carbon martensitic stainless steel using FE-(S) TEM. Corr. Sci. 2008, 50, 309–315. [Google Scholar] [CrossRef]
- Wang, Y.; Muramatsu, A.; Sugimoto, T. FTIR analysis of well-defined α-Fe2O3 particles. Coll. Surf. A Physicochem. Eng. Asp. 1998, 134, 281–297. [Google Scholar] [CrossRef]
- Kayani, Z.N.; Arshad, S.; Riaz, S.; Naseem, S. Synthesis of Iron Oxide Nanoparticles by Sol–Gel Technique and Their Characterization. IEEE Trans. Magnet. 2014, 50, 2200404. [Google Scholar] [CrossRef]
- Mahajan, D.; Papish, E.T.; Pandya, K. Sonolysis induced decomposition of metal carbonyls: Kinetics and product characterization. Ultrason Sonochem. 2004, 11, 385–392. [Google Scholar] [CrossRef]
- Ojeda, M.; Nabar, R.; Nilekar, A.U.; Ishikawa, A.; Mavrikakis, M.; Enrique, E. CO activation pathways and the mechanism of Fischer–Tropsch synthesis. J. Catal. 2010, 272, 287–297. [Google Scholar] [CrossRef]
- Mahmoudi, H.; Mahmoudi, M.; Doustdar, O.; Jahangiri, H.; Tsolakis, A.; Gu, S.; Miroslaw, L.M. A review of Fischer Tropsch synthesis process, mechanism, surface chemistry and catalyst formulation. Biofuels Eng. 2017, 2, 11–31. [Google Scholar] [CrossRef]
- Peña, D.; Cognigni, A.; Neumayer, T.; van Beek, W.; Jones, D.S.; Quijada, M.; Rønning, M. Identification of carbon species on iron-based catalysts during Fischer-Tropsch synthesis. Appl. Catal. A 2018, 554, 10–23. [Google Scholar] [CrossRef]
- van der Laan, G.P.; Beenackers, A.A.C.M. Kinetics and selectivity of the Fischer–Tropsch synthesis: A literature review. Catal. Rev. 1999, 41, 255–318. [Google Scholar] [CrossRef]
- Ma, W.; Jacobs, G.; Sparks, D.E.; Klettlinger, J.L.S.; Yen, C.H.; Davis, B.H. Fischer-Tropsch synthesis and water gas shift kinetics for a precipitated iron catalyst. Catal. Today 2016, 275, 49–58. [Google Scholar] [CrossRef]
- Davidson, G. Spectroscopic Properties of Inorganic and Organometallic Compounds; RSC Publishing: Cambridge, UK, 2007; Volume 39. [Google Scholar]
- Benziger, J.B.; Larson, L.R. An infrared spectroscopy study of the adsorption of CO on Fe/MgO. J. Catal. 1982, 77, 550–553. [Google Scholar] [CrossRef]
- Sheline, R.K.; Pitzer, K.S. The Infrared Spectra and Structures of the Iron Carbonyls. J. Am. Chem. Soc. 1950, 72, 1107–1112. [Google Scholar] [CrossRef]
- Bian, G.; Oonuki, A.; Kobayashi, Y.; Koizumi, N.; Yamada, M. Syngas adsorption on precipitated iron catalysts reduced by H2, syngas or CO and on those used for high-pressure FT synthesis by in situ diffuse reflectance FTIR spectroscopy. Appl. Catal. A Gen. 2001, 219, 13–24. [Google Scholar] [CrossRef]
- Dey, S.; Sun, S.; Mehta, N.S. Carbon monoxide catalytic oxidation over various iron-based nanoparticles at ambient conditions: A Review. Carbon Capture Sci. Technol. 2021, 1, 100013. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Weight and Molar Composition of Reactants | Reactant Ratios | ||||
|---|---|---|---|---|---|
| SiO2 wt | SiO2 Moles | Fe(CO)5 wt | Fe(CO)5 Moles | ||
| 2.00 g | 0.0333 | 0.29 g | 0.0015 | 22.5 | 6.9 |
| 2.00 g | 0.0333 | 0.73 g | 0.0037 | 9.0 | 2.8 |
| 2.00 g | 0.0333 | 1.45 g | 0.0074 | 4.5 | 1.4 |
| Reactant Composition and Ratio | Products Ratios | |||
|---|---|---|---|---|
| SiO2 Moles | Fe(CO)5 Moles | I1080/I534 | ||
| 0.0333 | 0.0015 | 22.5 | 4.573 | 56.2 |
| 0.0333 | 0.0037 | 9.0 | 1.958 | 22.5 |
| 0.0333 | 0.0074 | 4.5 | 1.902 | 11.2 |
| Catalyst | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Conversion (%) | UCI | BASF | Si-Fe | ||||||
| 493 K | 513 K | 533 K | 493 K | 513 K | 533 K | 493 K | 513 K | 553 K | |
| Total (H2/CO) | 83.7 | 75.3 | 72.2 | 77.6 | 81.7 | 85.3 | 55.8 | 68.4 | 60.0 |
| CO | 95.9 | 96.0 | 93.5 | 81.9 | 89.8 | 76.1 | 54.6 | 67.1 | 61.1 |
| Selectivity (C mole %) | UCI | BASF | Si-Fe | ||||||
| CO2 | 24.8 | 22.0 | 27.4 | 15.2 | 19.8 | 18.7 | 1.6 | 0.5 | 1.1 |
| CH4 | 3.2 | 3.6 | 6.6 | 6.7 | 8.9 | 8.7 | 3.9 | 0.5 | 0.2 |
| C8–C20 hydrocarbons | 10.1 | 13.0 | 7.5 | 12.3 | 43.6 | 31.4 | 17.4 | 58.3 | 54.9 |
| Catalyst | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Conversion (%) | UCI | BASF | Si-Fe | ||||||
| 493 K | 513 K | 533 K | 493 K | 513 K | 533 K | 493 K | 513 K | 533 K | |
| Total (H2/CO) | 62.1 | 75.0 | 75.7 | 54.7 | 54.1 | 63.0 | 28.7 | 25.9 | 35.3 |
| CO | 73.0 | 95.5 | 95.1 | 53.0 | 55.8 | 69.1 | 25.1 | 19.8 | 28.9 |
| Selectivity (C mole%) | UCI | BASF | Si-Fe | ||||||
| CO2 | 27.4 | 34.4 | 32.3 | 3.2 | 4.2 | 7.7 | 3.5 | 1.9 | 4.6 |
| CH4 | 0.7 | 1.3 | 1.4 | 0.6 | 0.7 | 1.5 | 1.4 | 2.5 | 2.5 |
| C8–C20 hydrocarbons | 6.7 | 5.4 | 6.0 | 23.6 | 13.4 | 18.6 | 21.2 | 39.3 | 24.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Costa, E.; Kileti, P.; Tannenbaum, R.; Lindberg, J.; Mahajan, D. Sonochemical Synthesis of Silica-Supported Iron Oxide Nanostructures and Their Application as Catalysts in Fischer–Tropsch Synthesis. Micro 2022, 2, 632-648. https://doi.org/10.3390/micro2040042
Chen L, Costa E, Kileti P, Tannenbaum R, Lindberg J, Mahajan D. Sonochemical Synthesis of Silica-Supported Iron Oxide Nanostructures and Their Application as Catalysts in Fischer–Tropsch Synthesis. Micro. 2022; 2(4):632-648. https://doi.org/10.3390/micro2040042
Chicago/Turabian StyleChen, Lyufei, Emily Costa, Pradheep Kileti, Rina Tannenbaum, Jake Lindberg, and Devinder Mahajan. 2022. "Sonochemical Synthesis of Silica-Supported Iron Oxide Nanostructures and Their Application as Catalysts in Fischer–Tropsch Synthesis" Micro 2, no. 4: 632-648. https://doi.org/10.3390/micro2040042